Abstract
Occupational injuries and toxicant exposures lead to the development of neuroinflammation by activating distinct mechanistic signaling cascades that ultimately culminate in the disruption of neuronal function leading to neurological and neurodegenerative disorders. The entry of toxicants into the brain causes the subsequent activation of glial cells, a response known as ‘reactive gliosis’. Reactive glial cells secrete a wide variety of signaling molecules in response to neuronal perturbations and thus play a crucial role in the progression and regulation of central nervous system (CNS) injury. In parallel, the roles of protein phosphorylation and cell signaling in eliciting neuroinflammation are evolving. However, there is limited understanding of the molecular underpinnings associated with toxicant- or occupational injury-mediated neuroinflammation, gliosis, and neurological outcomes. The activation of signaling molecules has biological significance, including the promotion or inhibition of disease mechanisms. Nevertheless, the regulatory mechanisms of synergism or antagonism among intracellular signaling pathways remain elusive. This review highlights the research focusing on the direct interaction between the immune system and the toxicant- or occupational injury-induced gliosis. Specifically, the role of occupational injuries, e.g., trips, slips, and falls resulting in traumatic brain injury, and occupational toxicants, e.g., volatile organic compounds, metals, and nanoparticles/nanomaterials in the development of neuroinflammation and neurological or neurodegenerative diseases are highlighted. Further, this review recapitulates the recent advancement related to the characterization of the molecular mechanisms comprising protein phosphorylation and cell signaling, culminating in neuroinflammation.
Keywords:
Alzheimer’s disease; amyotrophic lateral sclerosis; astrocytes; cell signaling; gliosis; hydrocarbons; inflammation; immune response; metals; microglia; multiple sclerosis; nanoparticles; neuroinflammation; neurological disorders; neurodegenerative diseases; occupational injury; Parkinson’s disease; traumatic brain injury; workplace toxicants 1. Introduction
Neuroinflammation is characterized by a complex inflammatory process in the CNS, a biological response of the neuroimmune system elicited by various chemical, biological, and physical agents. However, little is known about how neurons, glia, and resident macrophages interact during the inflammatory cascade, thereby inducing disease pathogenesis. Remarkably, inflammation is an evolutionarily conserved process during the activation of the immune and non-immune cells against an antigen or tissue injury [1,2,3]. It has been speculated that acute or intermittent inflammation is critical for survival during injury and infection, but chronic systemic inflammation can be counterproductive [3,4,5].
There is scientific consensus that certain environmental and social influences, as well as lifestyle factors, can contribute to chronic systemic inflammation, which leads to several disease outcomes such as autoimmune, cardiovascular, hepatic, renal, cancer, diabetes, as well as neurodegenerative disorders, all of which are significant global public health issues [3,4,5].
An inflammatory response occurs following infection or after an injury caused by physical, chemical, or metabolic insults. Inflammation elicited by systemic infection is mediated by pattern recognition receptors expressed on innate immune cells, which recognize conserved molecular motifs on microbes/pathogens, called pathogen-associated molecular patterns (PAMPs) [3]. Similarly, inflammation triggered by physical, chemical, or metabolic insults are mediated by damage-associated molecular patterns (DAMPs), also known as alarmins, which are endogenous distress molecules released by stressed, injured, damaged, and necrotic cells [3]. Like PAMPs, DAMPs can also activate innate immune responses. Evidence from preclinical studies suggests that acute and chronic inflammatory outcomes are associated with the release of proinflammatory cytokines and chemokines, which modulate the secretion or release of various hormones (e.g., gonadotropin-releasing, follicle-stimulating, and luteinizing) and neurotransmitters (e.g., norepinephrine, dopamine, and acetylcholine) [6]. However, the unresolved issue is the determination of how inflammatory mediators exert their effects on the brain, and if there are region-specific differences in the inflammatory profile, response, and injury outcomes.
An interesting aspect of a recent study is the recognition that the brain parenchyma lacks memory T-cells, which makes the inflammatory process quite different from the inflammation occurring in other organs of the body. Outside the brain, memory T-cells, the antigen-specific T cells that persist for long periods after infection, are central to orchestrating antigen-specific responses to repeated infectious exposures [7]. A subgroup of circulating memory T-cells has been shown to enhance inflammation by migrating to the sites of injury or by making their entry into lymph nodes to initiate cellular immune responses, including the production of additional effector T-cells, in lymphoid tissues [7]. Infectious and non-infectious agents, as well as cellular damage signals, can activate inflammatory cells through the nuclear factor kappa B (NF-κB), janus kinase-signal transducer and activator of transcription (JAK-STAT), and the mitogen-activated protein kinase (MAPK), signaling pathways [4].
However, within the brain, “microglia,” the resident macrophages, play a vital role in the occurrence and development of neurological dysfunction, including multiple sclerosis (MS), ischemic brain injuries, and Alzheimer’s disease (AD) [8,9,10]. Several studies have indicated that microglial polarization can activate the astrocytic function, which produces proinflammatory markers [11,12]. Microglial polarization into either M1 (pro-inflammatory) or M2 (anti-inflammatory) phenotype, which is termed classical activation and alternative activation, respectively, occurs due to perturbation in the microglial micro-environment [13]. Microglia are key regulators of neurogenesis and are known to control the number of neuronal precursor cells [14], as well as participate in the formation and elimination of neuronal synapses [15]. In the CNS, microglia serve as the main antigen-presenting cells and mediate autoimmune effector T-cell infiltration to the brain, as seen in MS.
When microglia are activated, their cell body enlarges, their processes become shorter or withdrawn, and they assume a round or amoebic shape, which allows for their migration to sites of injury and gain phagocytic abilities [9,16]. Activated microglia express microglial receptors, including the triggering receptor expressed on myeloid cells-2 (TREM2), low-density lipoprotein receptor-related protein 1 (LRP1), toll-like receptors 2 and 4 (TLR2 and TLR4), cholinergic receptor nicotinic alpha 7 subunit (CHRNA7; also known as alpha 7 subtype of nACHR/α7nACHR), and the calcium-sensing receptor (CASR) [10]. It is now known that astrocytes are not well equipped with receptors recognizing pathogens compared to microglia [17,18]. When activated by polarized microglia, they become reactive, release inflammatory mediators, and modulate inflammation.
Industrial chemicals such as aldehydes, polycyclic aromatic hydrocarbons, phenols, phthalates, per- and polyfluoroalkyl- substances, pesticides, metals, and others, can cause inflammation via multiple mechanisms [3,19] including endocrine disruptors or cytotoxic agents. These chemicals have been associated with diverse diseases such as hormone-dependent cancers, metabolic syndrome, type 2 diabetes, hypertension, cardiovascular disorders, respiratory diseases, autoimmune diseases, and neurodegenerative disorders [3]. Additionally, metal toxicants such as aluminum (Al), copper (Cu), zinc (Zn), and iron (Fe) deposits have been reported within the core and periphery of senile plaques and colocalize with beta-amyloid, suggesting their role in neurodegenerative diseases such as AD [20].
This review aims to summarize the recent research highlighting the role of occupational toxicants in neurodegenerative diseases, with a specific focus on gliosis, including its genetic, molecular, signaling, and neuroinflammatory features.
2. Occupational Brain Injuries
An occupational injury describes any injury or illness to a worker as related to their specific work/occupational demands or requirement [21]. It is often a life-altering event that is also considered a form of disability [22], which is covered under the provisions of the 1990 Americans with Disabilities Act. The National Safety Council identified slips, trips, and falls are among the top three leading causes of work-related injuries [23]. Slips or trips causing a fall are prevalent work-related injuries accounting for 20% to 40% of disabling occupational injuries [21,24].
The National Institute for Occupational Safety and Health (NIOSH) has studied slips, trips, and falls in healthcare settings, and wholesale/retail trade establishments to identify risk factors [25,26]. The common risk factors that can cause slips, trips, and falls at the workplace include liquid spills, ice, snow, rain, loose flooring, carpets, mats or rugs, boxes/containers, poor or improper lighting, work pressure, age, fatigue, failing eyesight, and inappropriate/poorly fitting footwear [25,26,27]. Traumatic workplace injuries can result in protracted disability and other health outcomes, which are the primary causes for a prolonged delay in return to work of the injured worker [28].
In a variety of workplaces, slips, trips, and falls can be one of the primary causes associated with traumatic brain injury (TBI). Notably, falls account for almost half of the emergency department visits for TBIs. Severe blunt forces, e.g., bumps and blows, which cause hits against the skull, can also perturb brain function, causing damage [5]. The injury can be penetrating as caused by a sharp object or non-penetrating when hit by objects/projectiles. TBI can also occur through high-velocity probes or projectiles such as gunshots and shrapnel. An explosion can cause concussions and TBI. Different types and grades of TBI common in occupational injuries include mild, moderate, severe, uncomplicated, complicated, closed, open, and non-traumatic. Specific occupations likely to have a higher risk of TBIs include construction and manufacturing, healthcare, hospitality and service industry, military, and law enforcement [29,30].
In addition to slips and falls, chemical exposures are also occupational hazards. For example, volatile organic compounds (VOC) that are generally released from paints, varnishes, waxes, and solvents used in the paint, automobile, furniture, and electronic manufacturing industries have been shown to cause neurological problems. They are organic compounds with a variety of functional groups and include xylenes, toluene, formaldehyde, and benzene, all of which have been reported to be environmental hazards and cause serious occupational injuries [19]. VOC have been shown to interact with nitrogen oxides (NOx), forming ozone and peroxyacetyl nitrate, which are known occupational and environmental toxicants [19]. Little is known about the mechanism by which VOC induces inflammation. Nevertheless, VOC are known to impact the cellular machinery, including transcriptional regulation of CD4 T-cell related genes, demethylation, the release of cytokines, activation of various T-helper cells (Th1, Th2, Th17, and Treg), the release of adhesion molecules (CTLA4, CD40L, and CD70), upregulation of cell cycle proteins (CDKN1A), and triggering autoimmune diseases [31]. Notably, VOC found in the air as pollutants have been linked to neurological diseases, including stroke, PD, and AD because of their ability to easily cross the blood-brain barrier (BBB) and activate the CNS response [19].
Another prominent industrial chemical that is known to enter the CNS and exert its toxic actions is 1,2-Dichloroethane (DCE). DCE is a synthetic chemical widely exploited in the production of vinyl chloride, which in turn is the starting material for a variety of plastic and vinyl products. DCE-based vinyl products are extensively used in furniture and automobile upholstery, wall coverings, housewares, and automobile parts. Clinical investigations and post-mortem examinations of the brains of workers exposed to DCE revealed that brain edema was the primary pathological change leading to death among DCE-exposed workers [32,33]. Similarly, in DCE-intoxicated mice, astroglial and microglial cross-talk caused inflammation and brain edema [34]. DCE is known to easily cross the BBB and target glial cells, particularly the astroglia and oligodendroglia [35]. Recently, DCE has been shown to inhibit the expression of aquaporin 4, a brain-specific water channel protein, in astrocytes, and reduce myelin basic protein in oligodendrocytes leading to demyelination [36].
3. Reactive Gliosis in Neural Injury and Neurodegeneration
Gliosis, a “reactive” state of glia, is a pathological hallmark of all types of central nervous system (CNS) injuries [37,38]. The activation of astrocytes and microglia is the universal component of the neuroinflammatory response. It is implicated in toxicant-induced neurotoxicity and progression of neurodegeneration after ischemia, seizure, AD, PD, MS, and ALS, to name a few [37,39,40]; see Table 1 and Table 2.

Table 1.
Astroglial activation associated with occupational toxicants, brain injuries, and neurological disorders.
Table 2.
Microglial activation associated with occupational toxicants, brain injuries, and neurological disorders.
Responses to injury and disease in the CNS involve the interaction of various neural and non-neural cells to maintain cellular homeostasis, neuronal integrity, neuronal viability, and neuronal function [126].
Some fundamental issues must be considered to fully understand the involvement of glial response in toxicant-induced neurotoxicity when measuring the expression of glial markers. First, the number of glial cells (astroglia and microglia) can vary across brain regions; second, glial cells from different brain regions exhibit a differential time course of expression; and third, the magnitude of glial response to an insult can vary [37,127]. Another consideration is that the glial response to toxicant exposure can vary depending on the injury stage or disease progression in which they occur.
Astrocytes and microglia adopt an activated phenotype following an insult, resulting in two polarization states, the pro-inflammatory phenotypes (A1 and M1, respectively) and an anti-inflammatory phenotype (A2 and M2) [128]. Accumulated scientific evidence also suggests that astrocytes and microglia express endogenous pattern recognition receptors including a variety of DAMPs, which can result in a molecular signaling cascade toward inflammation and disease propagation [39,129]. Activation of DAMP-mediated cellular signaling is known to induce the secretion of numerous pro-inflammatory mediators including adipokines, cytokines (for example, IL1B, IL6, TNFA), and chemokines (C-C motif chemokine ligand 2, CCL2/MCP1), as well as activating various cell types such as adipocytes, endothelial cells, and resident immune cells [3]. In the pathological brain, stretch injury to the astrocyte causes the release of vasoactive factors such as endothelins and isoprostanes, upregulation of the inositol triphosphate signaling, and enhancement of neural cell sensitivity to extracellular glutamate and inflammatory cytokines [130].
Microglia are the primary resident sentinel immune cells that are thought to maintain homeostasis and one of the significant non-neuronal cell types that contribute to neurodegeneration [4,39,131,132], see Box 1. An acute insult can instigate microglial activation, causing elongation of its processes and increasing the expression of marker proteins such as AIF1/IBA1 and integrin subunit alpha M (ITGAM/CD11B) [133]. Olfactomedin-like protein 3 (OLFML3) has also been determined as microglia-specific gene that is involved in early developmental patterning, while sialic acid binding Ig-like lectin H (Siglec-H) is associated with innate immune cell differentiation [132,134,135]. During the presentation of cues such as foreign agents/particles, cell debris, or toxicants, the homeostatic resident microglia transform and attain a reactive state (see Box 1).
Box 1. Homeostatic and Inflammatory Microglial Markers.
Homeostatic microglial markers
- Purinergic Receptor P2Y12 (P2RY12)
- Transmembrane protein 119 (TMEM119)
- Olfactomedin-Like Protein 3 (OLFML3)
- Fc Receptor-Like 2 (FCRLS)
- Spalt Like Transcription Factor 1 (SALL1)
- C-X3-C Motif Chemokine Receptor (CX3CR1)
- G Protein-Coupled Receptor 34 (GPR34)
Inflammatory microglial markers
- Galactose-Specific Lectin 3/Galectin 3 (LGALS3)
- C-Type Lectin Domain Family 7 member A (CLEC7A)
- Allograft Inflammatory Factor 1/Ionized Calcium-Binding Adapter Molecule 1 (AIF1/IBA1)
- Transmembrane Immune Signaling Adaptor TYROBP/TYRO Protein Tyrosine Kinase Binding Protein (TYROBP)
- Tumor necrosis factor alpha (TNFA)
- C-C Motif Chemokine Ligand 2 (CCL2/MCP1)
- Integrin Subunit Alpha M (ITGAM)
3.1. Gliosis in Brain Injury
TBI is characterized as one of the leading causes of death and disabling occupational injuries [136,137,138], with a high incidence rate in both the military and civilian populations [139]. Nearly 5.3 million people live with TBI in the U.S. [136,137,140,141,142], including work-related TBI. Approximately one in four mild TBI (mTBI) cases in adults are considered work-related [28]. However, neuroinflammatory and neurological outcomes of work-related TBI have not been thoroughly investigated. The U.S. Centers for Disease Control and Prevention (CDC) has disseminated the case definitions for TBI using the Ninth and Tenth Revisions of the International Classification of Disease, Clinical Modification (ICD-9-CM and ICD-10-CM codes), commonly used in injury research [140,143]. Similarly, the Occupational Injury and Illness Classification System were developed by the U.S. Bureau of Labor Statistics to characterize work-related injuries and illnesses.
All-encompassing, the terms concussion, mild head injury, mTBI, and cerebral trauma, are used interchangeably to describe the physical damage and the ensuing symptomatic consequences arising from impairment of the brain structure or function [5,144]. Concussions are usually caused by a blow, bump or jolt to the head. A hit to the body that causes the head and brain to rush back and forth can also cause a concussion [145].
Immune cell activation and inflammatory responses are the cardinal features of traumatic injury. However, poor or failed resolution of acute inflammation, and compromise of the defense mechanisms, can contribute to chronic inflammation or an exaggerated systemic inflammatory response syndrome (SIRS) [138]. SIRS occurs shortly after trauma or traumatic injury when inflammatory cytokines enlist peripheral leukocytes to the site of the inflamed or injured tissue [138,146].
Several lines of evidence point to a role for inflammation in the clinical and functional outcomes seen in TBI [5]. Inflammatory mediators, e.g., cytokines and chemokines released during traumatic injury can augment the immune response by engaging several immune cells. Both human and animal studies have shown an increase in the expression of high mobility group box 1 protein (HMGB1) following brain injury [5], which binds to TLR4 and initiates the inflammatory cascade. The deposition of oxidized phospholipids and oxygenated free-fatty acids has also been shown to cause acute brain injury and induce an inflammatory response [147]. In addition to changes in the lipid profile, several other genes are also upregulated following mild and severe TBI. These include antigen-presenting factors (MHC-II, CD86, CD74), cytokines (IL6, IL12, IL10, IL1B, TGFB, IFNG), phagocytosis factors (FCGR, FCGR4, C3, C4), chemotaxis factors (CCL2, CCL4, CXCL1, CXCL4), and astrocytic proteins (GFAP, AQP4) [5].
Head injury is a significant risk factor for the initiation and progression of dementia, characterized by the aggregation of beta-amyloid (Aβ) plaques [148]. In addition, repeated insults/injuries to the brain heighten the glial response by enhancing microglial or astrocytic cell density and release of inflammatory mediators [149,150]. Evidence obtained from human post-mortem tissues revealed that repeat injuries generate a more robust glial response, demonstrating that phosphorylated tau and CD68 cell density can be predictors of repeated head injury [151].
The primary pathological outcomes associated with TBI have been reported to include focal intracranial hemorrhage, epidural and subdural hematoma, and axonal injury [139]. Computed tomography studies also provided evidence of meningeal vascular leakage in approximately 50% of concussed patients who were otherwise considered clinically normal [152]. Such vascular changes can potentially lead to secondary lesions that can progressively culminate in neurological or neurodegenerative outcomes.
Following TBI, activated microglia/macrophages exhibit different phenotypic features [153], commonly referred to as classically activated or M1 (pro-inflammatory) and alternatively activated or M2 (anti-inflammatory) microglia (Figure 1). The inflammatory or anti-inflammatory glial response to TBI is governed by numerous cell-signaling events that are selectively activated depending on the form and severity of the neural insult [126,154,155]. Thus, microglia become rapidly activated in response to CNS injury caused by TBI.
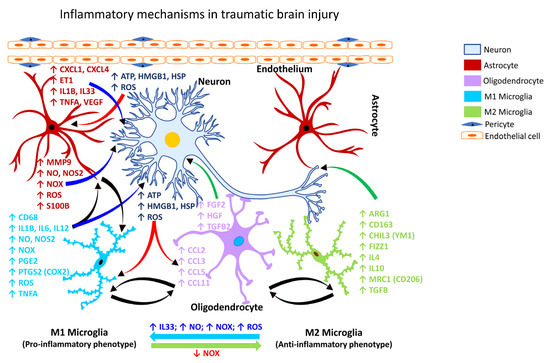
Figure 1.
Inflammatory mechanism in traumatic brain injury. Schematic representation of the molecular mechanisms associated with glia-mediated functional interactions and systematic perturbations within the CNS to induce neuroinflammation in traumatic brain injury. The endothelial cells form the inner lining of the blood vessel with pericytes enveloping the surface of the vasculature forming tight junctions to maintain the BBB integrity. Upon insult, neurons release danger/damage signals that cause activation of neighboring glial cells. M1 microglia (proinflammatory phenotype, neurotoxic) release various proinflammatory mediators including free radicals, cytokines, and chemokines that further stimulate other glial cells and collectively contribute to exacerbating the neuronal injury/damage. M2 microglial cells (anti-inflammatory phenotype, neuroprotective) can polarize to an M1 state and release proinflammatory mediators in the presence of increased levels of NOX, ROS, NO, and IL33 released by M1 microglia and/or astrocytes and thereby augment the neuroinflammatory and neuronal injury process leading to synaptic dysfunction, neuronal injury, and neuronal death. Astrocytes respond by releasing proinflammatory mediators including free radicals, cytokines, and chemokines, which further contribute to enhancing the endothelial permeability, disrupting BBB integrity, and allowing for infiltration of peripheral immune cells, events that further intensify inflammation and neuronal injury. Feedback regulation of NOX or its inhibition causes M1 microglia to polarize to the M2 state (anti-inflammatory phenotype), which downregulates M1 functions and promotes regulation of neuroinflammation and neurorepair by releasing anti-inflammatory mediators, e.g., cytokines, neurotrophic, and growth factors. Mediators released by specific neural cell types (neuron, astrocyte, microglia, or oligodendrocyte) are listed adjacent to each cell type in similar colored text. Curved arrows indicate the direction of signal flow between various neural cells for the inflammation activation process. Red curved arrows show the directional flow of danger/damage signals from neurons to glial cells (astroglia, microglia, oligodendroglia); blue curved arrows show the flow of proinflammatory signals from astrocytes and M1 microglia towards distressed neurons; green curved arrows show the flow of neurotrophic signals from M2 microglia and oligodendroglia towards the distressed neurons as a neuroprotective/neurorescue endeavor; black curved arrows show the directional crosstalk among various glial cells to mount a glial response to neuronal injury/damage. ↑, increase; ↓, decrease; ARG1, arginase 1; ATP, adenosine triphosphate; BBB, blood-brain barrier; CCL2, C-C motif chemokine ligand 2 (also referred to as MCP1, monocyte chemoattractant protein 1); CCL3, C-C motif chemokine ligand 3; CCL5, C-C motif chemokine ligand 5; CCL11, C-C motif chemokine ligand 11; CD68, CD68 molecule; CD163, CD163 molecule; CHIL3, chitinase-like protein 3 (also referred to as YM1); CXCL1, C-X-C motif chemokine ligand 1; CXCL4, C-X-C motif chemokine ligand 4; ET1, endothelin 1; FGF2, fibroblast growth factor 2; FIZZ1, found in inflammatory zone 1; HGF, hepatocyte growth factor; HMGB1, high-mobility group box 1; HSP, heat shock proteins; IL1B, interleukin 1 beta; IL4, interleukin 4; IL6, interleukin 6; IL10, interleukin 10; IL12, interleukin 12; IL33, interleukin 33; MMP9, matrix metalloproteinase 9; MRC1, mannose receptor C-type 1 (also referred to as CD206); NO, nitric oxide; NOS2, nitric oxide synthase 2 (inducible nitric oxide synthase); NOX, NADPH oxidase 1; PGE2, prostaglandin E2; PTGS2, prostaglandin-endoperoxide synthase 2 (also referred to as COX2, cyclooxygenase 2); ROS, reactive oxygen species; S100B, calcium binding protein B; TGFB, transforming growth factor beta; TGFB2, transforming growth factor beta 2; TNFA, tumor necrosis factor alpha; VEGF, vascular endothelial growth factor.
In animal models of concussion brain injury, microglial activation, robust upregulation of inflammatory cytokines, and impairment of white matter have been shown to be the key pathological features [153]. Mounting evidence points to a progressive neuroinflammatory process after a head injury that is persistent even after the resolution of the acute injury response [156], which can potentially contribute to cognitive and behavioral deficits.
The caveats in TBI studies arise due to the complex immunological mechanisms and the interindividual differences in the pathological response among humans, which creates inconsistencies in translating findings from animal research into clinical applications. There are analytical constraints in identifying the precise mechanisms for primary and secondary injuries linked to TBI in experimental models, which need to be singled out to target-specific subgroups for better translation toward injury diagnosis and disease intervention in humans [5]. The secondary inflammation associated with TBI can be slower in development ranging from months to years, which could further impede the translational applications. TBI can be driven by several cellular signaling pathways involving free radicals, membrane damage, immune activation, and excitotoxicity due to excess glutamate, among others [5,137,141,147].
3.2. Gliosis Following Chemical-Induced Neural Injury
3.2.1. Gliosis Associated with Hydrocarbon Exposure
The rapid industrialization and urbanization have brought with it an unprecedented rise in human exposure to an ever-expanding list of hazardous chemicals and pollutants [157]. Every year, more than 2000 new chemicals are introduced into the environment, and humans can potentially come in to contact with such chemicals either at the workplace or through daily consumption or use, including via food, cosmetics, pharmaceuticals, cleaning agents, and herbicides/pesticides. In 2008, the Toxicology in the 21st Century (Tox21) Consortium [158], a collaboration of federal agencies, including the U.S. Environmental Protection Agency (EPA), the National Institute of Environmental Health Sciences (NIEHS)-National Toxicology Program (NTP), the National Center for Advancing Translational Sciences (NCATS), and the Food and Drug Administration (FDA) came into effect to evaluate and understand the adverse human health risks of commercial chemicals, including pesticides, food additives/contaminants, and medical products [159,160]. Some of the chemicals of interest to the Tox21 program are polycyclic aromatic hydrocarbons, phthalates, per- and poly-fluoroalkyl substances, bisphenols, and flame retardants [160], all of which have potential for adverse human exposures in occupational settings given the consumption demand and high production volumes.
Exposure to aliphatic hydrocarbons, particularly n-hexane and halogenated compounds, has been shown to cause a widespread dopaminergic neuronal loss in the substantia nigra and depletion of tyrosine hydroxylase immunoreactivity in the striatum, which was associated with severe gliosis [161,162]. The effects of several natural and synthetic compounds, including halogenated aromatic hydrocarbons (e.g., biphenyls, dioxins, furans) and polycyclic aromatic hydrocarbons (PAH; e.g., benzo[a]pyrene, benzanthracenes, 3-methylcholanthrene) are mediated by the aryl hydrocarbon receptor (AHR/AhR). The AHR is a ligand-dependent transcription factor that integrates various metabolic cues from the environment, food chain, or microorganisms to regulate transcription in a cell- and ligand-specific manner [163]. Increased AHR immunoreactivity is associated with activated microglia in the middle cerebral artery occlusion (MCAO) model of brain ischemia [164]. Evaluation of AHR immunoreactivity in human hippocampal post-mortem tissue and its association with reactive astrocytes revealed their greater participation in the development of neurodegenerative diseases, including AD [165], suggesting that occupational and environmental exposure to PAH may contribute to the neuropathogenesis seen in many neurodegenerative disorders.
Deliberate inhalation of certain volatile hydrocarbons for their mood-altering effects is prevalent among humans. Volatile hydrocarbons are predominantly found in glues, solvents, lighter fluids, gasoline, and paints. High exposure to PAH has been shown to reduce subcortical volume and cause cortical thinning in older people, primarily affecting the parietal, temporal, and insular regions in men, while in women, the frontal and parietal cortical lobes appear to be affected more severely [166]. Additionally, PAH formed during the incomplete combustion of organic matter poses a significant risk for firefighters at fire sites [167], indicating a potential occupational health risk.
Toluene is an aromatic hydrocarbon widely used in various occupational settings, including the paint and adhesive industry; rubber and lumber sector; dry cleaning; automobile and aviation manufacturing; and chemical industries. The highly lipophilic nature of toluene [168] can facilitate its entry into the brain after inhalation exposure and target the myelin sheath given it is made up of 70–75% lipids. Thus, the effect of toluene on glial cells, particularly on Schwann cells, oligodendrocytes, and astrocytes are more significant than on the neuronal population, as axonal integrity is relatively well preserved in toluene leukoencephalopathy [168]. Toluene also disrupts the differentiation of astrocyte precursors and decreases ATPase activity in astrocytes [169,170]. Indeed, altered astrocytic function and reactive astrogliosis have been documented in human cases of toluene abuse [171]. Toluene is also known to dose-dependently inhibit N-methyl-D-aspartate (NMDA)-mediated excitatory postsynaptic currents (EPSCs) and induce a delayed but persistent reduction in evoked or spontaneous alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)-mediated EPSCs [172]. In addition, toluene also inhibits the nicotinic acetylcholine receptor, which has a critical role in brain development [173]. In rats inhaling toluene, loss of hippocampal neurons and cerebellar Purkinje cells is reported [174], with the latter also causing ‘thinning’ of the cerebellar white matter [174]. Chronic or repeated toluene exposure has been shown to induce pro-oxidants, reduce antioxidants, and cause memory impairment [175].
3.2.2. Gliosis Associated with Metal Exposure
With rapid industrialization, there is an increasing demand for metals. The high-volume production and use of metals are of significant concern for occupational safety due to potential worker exposure and consequent adverse health effects. Chronic workplace exposure to high-production volume metals such as iron (Fe), aluminum (Al), and manganese (Mn) are believed to be associated with an increased risk of neurodegeneration. Fe translocation to the brain is mediated by the transferrin receptor and the solute carrier family 11 member 2 (SLC11A2; also known as divalent metal transporter 1/DMT1 [176]. Its prooxidant characteristics elicit ROS generation via the Fenton reaction and Haber-Weiss reaction [177], which can subsequently mediate oxidative stress. Free Fe has been known to induce fibrillation and aggregation of alpha-synuclein (αSYN; encoded by the gene SNCA/PARK1) in a dose- and time-dependent manner in cultured cells [178]. Emerging evidence also shows that Fe accumulation in the brain accelerates disease progression in AD, but the mechanism through which this occurs is not known [179]. Further, the study demonstrated the occurrence of Fe-accumulating microglia that exhibited a dystrophic morphological state. This subset of Fe-accumulating microglia showed augmented expression of the iron storage protein ferritin light chain (FTL) and allograft inflammatory factor 1 (AIF1; also known as ionized calcium-binding adaptor protein 1/IBA1) while downregulating the expression of transmembrane protein 119 (TMEM119) and the purinergic receptor P2Y12 (P2RY12) [179].
Al is a potent neurotoxic element involved in the etiology of occupational neurodegenerative disorders. Al causes oxidative stress leading to the deposition of intracellular reactive oxygen species [180]. However, the evidence suggesting the role of Al in the development of AD is inconsistent. Chronic oral administration of Al has been shown to increase Aβ levels in the cortex and hippocampus of rats [181]. Evidence shows that acute and chronic Al intoxication induces astrogliosis in the motor cortex and the hippocampus [180]. Exposure of Tg2576 mice, a model for AD, to Al resulted in increased expression of miRNAs (miR146a and miR125b) demonstrating a proinflammatory response like that seen in the brain of patients with AD [182,183,184].
Mn has been implicated in regulating cellular homeostasis and maintaining physiological functions. While Mn is an essential trace element for normal brain development and function, excess brain Mn is known to be neurotoxic. Occupational exposure to elevated airborne levels of Mn in mining and ferroalloy industries has been reported to cause neurological disorders [185,186,187,188,189,190,191], and has been linked to the slowed movement of upper extremities, poor balance and gait coordination, neuropsychological abnormalities, disruption of sleep, cognitive deficits, as well as parkinsonism [192,193,194]. Sustained exposure to low concentrations of Mn has been found to cause Mn-induced parkinsonism [195]. Microglial activation has been observed in the substantia nigra pars reticulata (SNpr) and substantia nigra pars compacta (SNpc) of cynomolgus macaques chronically exposed to Mn [196].
While the exact mechanisms of Mn transport and neurotoxicity are unclear, several studies suggest that Mn influx to the brain is carrier-mediated [197,198,199,200]. Other studies suggest that divalent metal transporters and L-type calcium channels may potentially be involved in the influx of Mn ions across the BBB [201,202]. Additionally, brain accumulation and toxicity of Mn is thought to be influenced by its elemental speciation. The oxidation state of Mn appears to be critical for its solubility, function, transport, retention, and toxicity [203,204]. Mn aerosol in ferroalloy industries is reported to exist in mixed oxidation states [205] including Mn (0), Mn (II), Mn (III) and Mn (IV). Of these, Mn (II) and Mn (III) are thought to be the predominant forms transported to the brain, where they potentially accumulate [206].
Welding fumes (WF) are a complex mixture of toxic metals and gases arising due to the burning of welding electrodes during welding. WF aerosols contain Mn, which is implicated in the development of PD-like neurological dysfunction seen among welders. Particles and aerosols inhaled through the nose and air passages can translocate to the brain via retrograde transport across olfactory neurons and accumulate in deeper brain areas [207,208,209,210]. WF may likely follow the same portals for entry into the brain. Alternatively, following deposition in the pulmonary targets, WF particles or soluble metal components of the WF may enter the systemic circulation and be transported to the brain following permeation through the BBB or the circumventricular organs (pituitary gland, median eminence, area postrema, choroid plexus), areas of the brain that are devoid of BBB. Indeed, there is evidence linking Mn and WF exposure to manganism and PD-like manifestations, including neuropsychological and neuropsychiatric disturbances [192,193,211,212,213,214,215,216,217,218,219]. Further, experimental studies have also shown that WF causes dopaminergic neurotoxicity [57,104,220,221].
3.2.3. Gliosis Associated with Nanoparticles/Nanomaterials
In recent years, accumulating experimental evidence suggests the potential role of nanoparticles (NPs) in neuroinflammation and brain injury [222,223,224,225]. The toxicological effects of nanoparticles depend on their physicochemical properties such as size, shape, and surface charge. The underlying mechanisms of their toxicity are not fully realized, given this is an emerging area of research. Nonetheless, it is thought that much of their effects may be related to their physical interaction with cellular membranes, likely causing membrane disruption, eliciting inflammation, and generating free radicals, including reactive oxygen species (ROS) [226].
Carbon-Based Nanomaterials
Carbon nanotubes (CNTs) have the structure of tubes made of carbon-based nanomaterials. Carbon nanotubes find application in manufacturing nanocomposites and semiconductors because of their remarkable physicochemical properties, such as high flexibility, good thermal conductivity, low density, and high chemical stability [227]. Carbon nanomaterials exhibit a wide variety of toxic effects including inflammatory effects on dendritic cells, ROS generation, DNA damage, pulmonary macrophage activation and inflammation, lysosomal damage, mitochondrial dysfunction, and apoptosis or necrosis-mediated cell death [228]. Engineered carbon nanomaterials such as single-walled carbon nanotubes, double-walled carbon nanotubes, and multi-walled carbon nanotubes (MWCNT) have the potential to elicit neurotoxicity due to their small size, ability to aerosolize, and bio-persistence [229]. Inhaled MWCNT is distributed to various organs, including the brain [230]. Research has shown that MWCNT exhibits spatial association with Aβ fibrils in the brains of mice [231]. Further, long-term inhalation of MWCNT has been shown to cause neurological effects, including ROS production, lipid peroxidation, cytochrome c release, and mitochondrial swelling [224].
Metal Oxide Nanoparticles
Metal oxide nanoparticles (NPs), e.g., copper oxide NP (nano-CuO), iron oxide NP (nano-FeO), silica dioxide NP (nano-SiO2), titanium dioxide NP (nano-TiO2), silver (nano-Ag), selenium NP (nano-Se), and zinc oxide NP (nano-ZnO) are high production volume metal NPs that find application in a variety of industrial processes, manufacturing, agriculture, and nanomedicine. However, due to their small size, large surface area, and potential prooxidant capabilities, there are concerns regarding their toxicologic potential.
Exposure of mice to nano-TiO2 has been shown to cause oxidative stress, gliosis, alteration in the expression of genes associated with memory and cognition, as well as the death of hippocampal neurons [222]. Inhalation of nano-TiO2 has been shown to augment BBB permeability and inflammatory cytokine production in the brain of aged rats [223]. Nano-Ag has been shown to upregulate the expression of inflammatory and antioxidant genes such as interleukin 1 (IL1), C-X-C motif chemokine 13 (CXCL13), macrophage receptor with collagenous structure (MARCO), and glutathione synthetase (GSS) [184,232]. Exposure to nano-CuO has been shown to decrease the spontaneous excitatory postsynaptic currents (sEPSCs) and miniature EPSCs (mEPSCs) and diminish pre-synaptic and post-synaptic glutamate neurotransmission, indicating reduced long-term potentiation (LTP) and cognitive dysfunction [233]. Exposure to nano-Se has been shown to cause enhanced Ca2+ signaling selectively within the astrocytes, increase lactate release, suppress hyperexcitation of neural networks, and activate A2-type astrocytes [234].
3.3. Gliosis in Neurological Disease States
Microglia play a critical role in regulating CNS physiology in the healthy brain. They are highly ramified structures that maintain direct contact with the dendritic spines, axons, and synapses of neurons, suggesting they are key participants in activity-dependent processes such as the regulation of the synaptic structure and function [235,236,237,238]. Like microglia, astrocytes also regulate brain signaling by modulating synapses, regulating homeostasis, transporting nutrients, and maintaining structural support [239,240].
3.3.1. Gliosis in AD
It is estimated that by the year 2050, more than 150 million people worldwide will be impacted by some form of dementia, with AD accounting for nearly 70% of such dementia cases [240,241]. Chronic neuroinflammation is a prominent feature of AD pathology [242] manifested by reactive gliosis and robust expression of proinflammatory mediators, associated with synapse loss [243,244]. Aβ causes functional and morphological changes in the neighboring astrocytes, eliciting an astroglial response (Figure 2; Table 3).
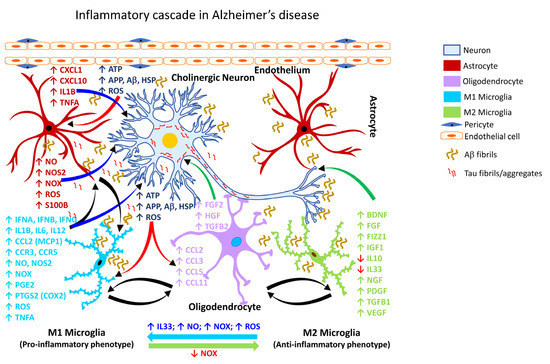
Figure 2.
Inflammatory cascade in Alzheimer’s disease. Schematic representation of the molecular mechanisms associated with glia-mediated functional interactions and systematic perturbations within the CNS to induce neuroinflammation in Alzheimer’s disease. The endothelial cells form the inner lining of the blood vessel with pericytes enveloping the surface of the vasculature forming tight junctions to maintain the BBB integrity. Upon insult, the augmented production and release, as well as impaired clearance of Aβ and Tau fibrils sustains chronic activation of the primed microglia resulting in the production and release of proinflammatory mediators including free radicals, cytokines and chemokines, thereby affecting the resident CNS cells (astrocytes, oligodendrocytes, and neurons) and leading to Aβ and Tau aggregation. M1 microglia (proinflammatory phenotype, neurotoxic) release various proinflammatory mediators including free radicals, cytokines, and chemokines that further stimulate other glial cells and collectively contribute to exacerbating the neuronal injury/damage. M2 microglial cells (anti-inflammatory phenotype, neuroprotective) can polarize to an M1 state and release proinflammatory mediators in the presence of increased levels of NOX, ROS, NO, and IL33 released by M1 microglia and/or astrocytes and thereby augment the neuroinflammatory and neuronal injury process leading to synaptic dysfunction, neuronal injury, and neuronal death. Astrocytes respond by releasing proinflammatory mediators including free radicals, cytokines, and chemokines, which further contribute to enhancing the endothelial permeability, disrupting BBB integrity, and allowing for infiltration of peripheral immune cells, events that further intensify inflammation and neuronal injury. Feedback regulation of NOX or its inhibition causes M1 microglia to polarize to the M2 state (anti-inflammatory phenotype), which downregulates M1 functions and promotes regulation of neuroinflammation and neurorepair by releasing anti-inflammatory mediators such as cytokines, neurotrophic, and growth factors. Mediators released by specific neural cell types (neuron, astrocyte, microglia, or oligodendrocyte) are listed adjacent to each cell type in similar colored text. Curved arrows indicate the direction of signal flow between various neural cells for the inflammation activation process. Red curved arrows show the directional flow of danger/damage signals from neurons to glial cells (astroglia, microglia, oligodendroglia); blue curved arrows show the flow of proinflammatory signals from astrocytes and M1 microglia towards distressed neurons; green curved arrows show the flow of neurotrophic signals from M2 microglia and oligodendroglia towards the distressed neurons as a neuroprotective/neurorescue endeavor; black curved arrows show the directional crosstalk among various glial cells to mount a glial response to neuronal injury/damage. ↑, increase; ↓, decrease; Aβ, beta amyloid; APP, amyloid precursor protein; ATP, adenosine triphosphate; BBB, blood-brain barrier; BDNF, brain-derived neurotrophic factor; CCL2, C-C motif chemokine ligand 2 (also referred to as MCP1, monocyte chemoattractant protein 1); CCL3, C-C motif chemokine ligand 3; CCL5, C-C motif chemokine ligand 5; CCL11, C-C motif chemokine ligand 11; CCR3, C-C motif chemokine receptor 3; CCR5, C-C motif chemokine receptor 5; 1; CXCL1, C-X-C motif chemokine ligand 1; CXCL10, C-X-C motif chemokine ligand 10; FGF, fibroblast growth factor; FGF2, fibroblast growth factor 2; FIZZ1, found in inflammatory zone 1; HGF, hepatocyte growth factor; HSP, heat shock proteins; IFNA, interferon alpha; IFNB, interferon beta; IFNG, interferon gamma; IGF1, insulin-like growth factor 1; IL1B, interleukin 1 beta; IL6, interleukin 6; IL10, interleukin 10; IL12, interleukin 12; IL33, interleukin 33; NGF, nerve growth factor; NO, nitric oxide; NOS2, nitric oxide synthase 2 (inducible nitric oxide synthase); NOX, NADPH oxidase 1; PDGF, platelet-derived growth factor; PGE2, prostaglandin E2; PTGS2, prostaglandin-endoperoxide synthase 2 (also referred to as COX2, cyclooxygenase 2); ROS, reactive oxygen species; S100B calcium binding protein B; TGFB1, transforming growth factor beta 1; TGFB2, transforming growth factor beta 2; TNFA, tumor necrosis factor alpha; VEGF, vascular endothelial growth factor.
Table 3.
Glial inflammatory mediators associated with Alzheimer’s disease.
Aβ-mediated microglial activation (Figure 2; Table 3) can also downregulate homeostatic genes, such as C-X-3-C motif chemokine receptor (CX3CR1), P2RY12, and genes involved in cell adhesion, lipid signaling, and G protein-coupled receptor (GPCR) pathways [273,274]. Complement factors have also been implicated in the synaptic neurotoxicity and neurodegeneration associated with AD. For example, pharmacological inhibition of the complement pathway has been shown to ameliorate synapse loss and neurodegeneration in murine models of AD [244,275].
Recently, a bidirectional interaction of the nervous and immune systems has been demonstrated [276]. The study showed that a systemic inflammatory event could elicit selective neuronal activation within the insular lobe of the cortex, and a subsequent insult of this immune-imprinted neuronal population recapitulated the primary inflammatory episode in the peripheral target [276]. It is also well-recognized that the breakdown of BBB facilitates the infiltration of toxicants and immune cells into the brain, causing neuroinflammation and subsequent activation of downstream cascades associated with neural injury and neurodegeneration [277].
Post-mortem analysis of brains from patients with AD has revealed abnormal accumulation of metal ions such as Fe, copper (Cu), and zinc (Zn), suggesting a role for dysregulated redox metals in the pathogenesis of AD. Specifically, deposits of metals, e.g., Fe, Cu, and Zn have been found in the rim and core of senile plaques and appear to co-localize with Aβ aggregates [278,279]. Fe causes lipid peroxidation via iron-dependent oxidases such as lipoxygenase, which activates ferroptosis and AD [280]. Zn in the synaptic cleft has been shown to be neurotoxic because it inhibits NMDAR and increases α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR)-mediated toxicity [281], thus affecting memory regulation in AD [282]. Zn is also known to interact with Aβ and aggravate pathogenesis due to Zn dyshomeostasis in AD [282].
Among the several proposed mechanisms of neuroinflammation in AD, a recent study has shown that the soluble epoxide hydroxylase is aberrantly elevated in the brains of patients with AD, and in transgenic or knock-in mouse models of AD [283]. Specifically, increased soluble epoxide hydrolase levels were reported to occur within the astrocytes. Functionally, the soluble epoxide hydrolase is known to rapidly scavenge the anti-inflammatory arachidonic acid derivatives, thereby facilitating the progression of the inflammatory process [283].
3.3.2. Gliosis in PD
PD is a chronic and progressive neurodegenerative disease characterized by motor and non-motor dysfunction, with the former being attributable to the loss of nigrostriatal dopaminergic neurons. The clinical motor dysfunction phenotype comprises of a complex of symptoms including muscle rigidity, resting tremor bradykinesia, and postural instability, collectively termed parkinsonism. The non-motor dysfunction typically involves neurons outside of the dopaminergic pathway, and the clinical phenotype is characterized by sleep disruption, cognitive impairment, and depression [284,285]. Numerous transgenic animal models have been exploited to study PD, including SNCA (PARK1), parkin RBR E3 ubiquitin protein ligase (PRKN/PARK2/Parkin), PTEN-induced kinase 1 (PINK1/PARK6), parkinsonism-associated deglycase (DJ1/PARK7), and leucine-rich repeat kinase 2 (LRRK2/PARK8). These models capture many of the salient features of PD, but none solely recapitulate all the cardinal features of dopaminergic neurodegeneration [286]. For example, while exogenous αSYN (SNCA) leads to mitochondrial dysfunction, the overexpression of PRKN rescues mitochondrial dysfunction [287]. The degeneration of the nigrostriatal pathway is always associated with extra-nigral dysfunction involving the dorsal motor nucleus of the glossopharyngeal and vagal nerves, thalamic sub-nuclei, amygdala, and the neocortex [274,288]. The pathology in PD evolves in stages, beginning with lesions of the dorsal IX/X motor nuclei in the medulla oblongata (stage 1), progressing to the gigantocellular reticular nucleus and caudal raphe nuclei in the medulla oblongata and pontine tegmentum (stage 2), substantia nigra pars compacta/SNpc in the midbrain (stage 3), and the cortical areas, mesocortex (stage 4) and neocortex (stages 5 and 6) [289]. Further, the non-motor symptoms often precede motor dysfunction by several years or decades [289]. Experimental neurotoxic models of PD also include the use of chemical agents, e.g., 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), 6-hydroxydopamine (6-OHDA), as well as pesticides and herbicides, e.g., rotenone and paraquat [290].
Multiple lines of investigations have shown that reactive microgliosis is associated with PD (Table 4).
Table 4.
Glial inflammatory mediators associated with Parkinson’s disease.
Reactive microgliosis occurs in the midbrain, striatum (primarily the caudate and putamen), hippocampal formation, and cortical regions of post-mortem brains obtained from patients with PD [313], following recreational administration of the dopaminergic neurotoxicant MPTP [314], or in experimental models of PD using MPTP [315,316,317,318]. Microglial inflammation-related activation of the aldosterone and metabolic pathways generating ROS also appears to be altered in PD, besides synaptic activity, neurotransmission, and neuronal injury or rescue [319].
Recently, it has been shown that αSYN (SNCA) can promote neurotoxic astrocyte activation (Figure 3; Table 4), and receptor-interacting serine/threonine kinase 1 (RIPK1; also known as receptor-interacting protein/RIP) signaling can regulate glial cell biology and neuroinflammation [320]. The role of αSYN (SNCA) in astrocyte activation was previously unknown until it was shown that that pre-formed αSYN (SNCA) fibrils can induce A1 and A2 activation states in astrocytes isolated from the human midbrain [320].
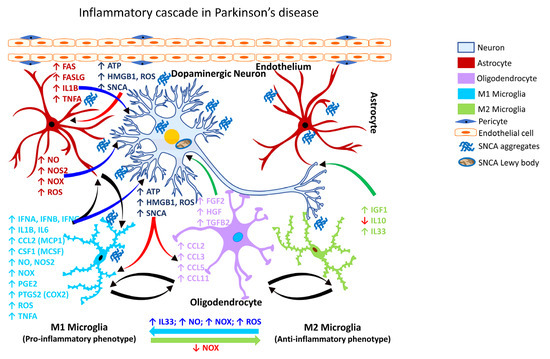
Figure 3.
Inflammatory cascade in Parkinson’s disease. Schematic representation of the molecular mechanisms associated with glia-mediated functional interactions and systematic perturbations within the CNS to induce neuroinflammation in Parkinson’s disease. The endothelial cells form the inner lining of the blood vessel with pericytes enveloping the surface of the vasculature forming tight junctions to maintain the BBB integrity. Upon insult, the augmented production and release, as well as impaired clearance of αSYN (SNCA) sustains chronic activation of the primed microglia resulting in the production and release of proinflammatory mediators including free radicals, cytokines and chemokines, thereby affecting the resident CNS cells (astrocytes, oligodendrocytes, and neurons) and leading to αSYN (SNCA) aggregation, neuronal injury and Lewy body formation. M1 microglia (proinflammatory phenotype, neurotoxic) release various proinflammatory mediators, including free radicals, cytokines, and chemokines that further stimulate other glial cells and collectively contribute to exacerbating neuronal injury/damage. M2 microglial cells (anti-inflammatory phenotype, neuroprotective) can polarize to an M1 state and release proinflammatory mediators in the presence of increased levels of NOX, ROS, NO, and IL33 released by M1 microglia and/or astrocytes and thereby augment the neuroinflammatory and neuronal injury process leading to synaptic dysfunction, neuronal injury, and neuronal death. Astrocytes respond by releasing proinflammatory mediators, including free radicals, cytokines, and chemokines, which further contribute to enhancing endothelial permeability, disrupting BBB integrity, and allowing for infiltration of peripheral immune cells, events that further intensify inflammation and neuronal injury. Feedback regulation of NOX or its inhibition causes M1 microglia to polarize to the M2 state (anti-inflammatory phenotype), which downregulates M1 functions and promotes regulation of neuroinflammation and neurorepair by releasing anti-inflammatory mediators such as cytokines, neurotrophic, and growth factors. Mediators released by specific neural cell types (neuron, astrocyte, microglia, or oligodendrocyte) are listed adjacent to each cell type in similar colored text. Curved arrows indicate the direction of signal flow between various neural cells for the inflammation activation process. Red curved arrows show the directional flow of danger/damage signals from neurons to glial cells (astroglia, microglia, oligodendroglia); blue curved arrows show the flow of proinflammatory signals from astrocytes and M1 microglia towards distressed neurons; green curved arrows show the flow of neurotrophic signals from M2 microglia and oligodendroglia towards the distressed neurons as a neuroprotective/neurorescue endeavor; black curved arrows show the directional crosstalk among various glial cells to mount a glial response to neuronal injury/damage. ↑, increase; ↓, decrease; ATP, adenosine triphosphate; BBB, blood-brain barrier; CCL2, C-C motif chemokine ligand 2 (also referred to as MCP1, monocyte chemoattractant protein 1); CCL3, C-C motif chemokine ligand 3; CCL5, C-C motif chemokine ligand 5; CCL11, C-C motif chemokine ligand 11; CSF1, colony-stimulating factor 1; FAS, Fas cell surface death receptor; FASLG, Fas ligand; FGF2, fibroblast growth factor 2; HGF, hepatocyte growth factor; HMGB1, high-mobility group box 1; IFNA, interferon alpha; IFNB, interferon beta; IFNG, interferon gamma; IGF1, insulin-like growth factor 1; IL1B, interleukin 1 beta; IL6, interleukin 6; IL10, interleukin 10; IL33, interleukin 33;NO, nitric oxide; NOS2, nitric oxide synthase 2 (inducible nitric oxide synthase); NOX, NADPH oxidase 1; PGE2, prostaglandin E2; PTGS2, prostaglandin-endoperoxide synthase 2 (also referred to as COX2, cyclooxygenase 2); ROS, reactive oxygen species; αSYN/SNCA, alpha synuclein; TGFB2, transforming growth factor beta 2; TNFA, tumor necrosis factor alpha.
3.3.3. Gliosis in CJD
Creutzfeldt-Jackob disease (CJD) is characterized as a rapidly progressive, fatal, transmissible neurodegenerative disease linked to the accumulation of untreatable prion protein (PrPc) resulting in encephalopathy and neurodegenerative disorders in the CNS [321,322]. Early diagnosis of CJD remains a major clinical challenge because the manifestation of prion disease at onset are inconsistent and often nonspecific. CJD affects humans and many other mammalian species [237,323].
Broadly, CJD is categorized into three subtypes: sporadic, inherited, and acquired. The most interesting characteristics of sporadic CJD are pathological features implicated during the development of spongiform change in the cerebral gray matter, which is further associated with the deposition of abnormal forms of prion protein (PrPsc), resulting in cognitive and motor dysfunction [324]. While the etiology of sporadic CJD remains unknown, it is hypothesized that a somatic mutation in the prion protein (PRNP) or misfolding of PrPc into PrPsc [325] might underlie the etiopathogenesis of CJD. Rapidly progressive dementia is typical in CJD, and the patients generally die within one year after clinical onset [326].
Aside from the prion protein-related hypothesis, it is suggested that the cytokine profile in CJD can be pro-inflammatory and anti-inflammatory, with increases seen in the inflammatory cytokine interleukin 8 (IL8) and a decrease in transforming growth factor beta 2 (TGFB2) but no changes in the levels of interleukin 1 beta (IL1B), interleukin 12 (IL12), or tumor necrosis factor alpha (TNFA) [327]. The regulatory protein PU1, interleukin 34 (IL34), and CCAAT enhancer binding protein alpha (CEBPA) is also involved in microglial proliferation in CJD [237,328]. Additionally, prostaglandin-endoperoxide synthase 1 (PTGS1/COX1), prostaglandin-endoperoxide synthase 2 (PTGS2/COX2), and prostaglandin E2 (PGE2) have been reported to be elevated in CJD [329,330], supporting the involvement of inflammation. Further, it has been shown that the increased inflammatory response seen in CJD is associated with the activation of NFκB and STAT3 signaling pathways [330]. Numerous studies have also reported signs of oxidative stress [134,237,331,332], and activated microglia in the brains of patients with CJD [333].
3.3.4. Gliosis in ALS
Amyotrophic lateral sclerosis (also known as Lou Gehrig’s disease) is a progressive neurodegenerative disorder affecting the motor neurons in the cerebral cortex, brainstem, and spinal cord. While the underlying mechanism of the neuronal degeneration remains elusive, a pathological basis involving the ubiquitin-immunoreactive cytoplasmic inclusions has been suggested, which is associated with a robust inflammatory reaction [334]. In ALS, mutations in the TAR DNA binding protein (TARDBP) are usually rare despite ALS patients exhibiting cytoplasmic aggregates of TDP43 in the affected brain areas [335].
Recent studies have shown that a deficiency of the guanine nucleotide exchange factor C9orf72 (C9orf72) alters the homeostatic gene signature of microglia [336] and contributes to the loss of synaptic processes [336]. Another line of investigation demonstrated that pharmacological inhibition of the purinergic receptor P2X 7 (P2RX7) decreases microgliosis, inhibits the expression of NF-κB, and attenuates motor neuron death [274,337]. Further, studies of the spatiotemporal dynamics of microglial activation in SOD1G93A mice have shown that microglial dysfunction precedes the onset of the disease [338].
Cumulative evidence over the past two decades shows microglial activation is associated with the degeneration seen in patients with ALS, as assessed by PET analysis. One of the earliest studies revealed diffuse microglial activation in both the motor and non-motor regions of the cerebral cortex [274,335,339,340,341,342]. Additionally, activation of glia in ALS (Figure 4) is associated with marked elevation of ROS, and inflammatory mediators, e.g., PTGS2 (COX2), IL1B, interleukin 6 (IL6), and TNFA [334].
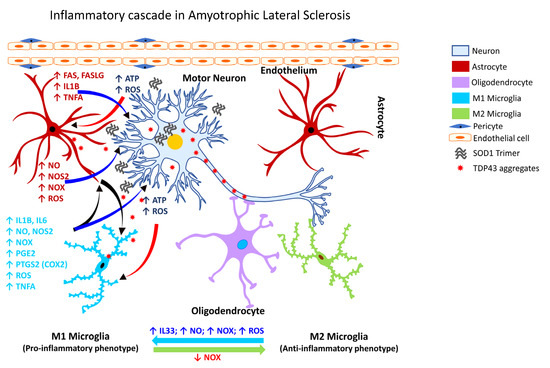
Figure 4.
Inflammatory cascade in Amyotrophic Lateral Sclerosis. Schematic representation of the molecular mechanisms associated with glia-mediated functional interactions and systematic perturbations within the CNS to induce neuroinflammation in Parkinson’s disease. The endothelial cells form the inner lining of the blood vessel with pericytes enveloping the surface of the vasculature forming tight junctions to maintain the BBB integrity. Upon insult, the augmented production and release, as well as impaired clearance of mutant SOD1 trimers, TDP43, and ubiquitin aggregates sustains chronic activation of the primed microglia resulting in the production and release of proinflammatory mediators, including free radicals, cytokines, and chemokines, thereby affecting the resident CNS cells (astrocytes, oligodendrocytes, and neurons) and leading to disruption of nuclear-cytoplasmic transport, ubiquitination and accumulation of TDP43 in the cytoplasm, aggregation of SODI trimers, and subsequent neuronal injury/damage. M1 microglia (proinflammatory phenotype, neurotoxic) release various proinflammatory mediators including free radicals, cytokines and chemokines that further stimulate other glial cells and collectively contribute to exacerbating the neuronal injury/damage. M2 microglial cells (anti-inflammatory phenotype, neuroprotective) can polarize to an M1 state and release proinflammatory mediators in the presence of increased levels of NOX, ROS, NO, and IL33 released by M1 microglia and/or astrocytes and thereby augment the neuroinflammatory and neuronal injury process leading to synaptic dysfunction, neuronal injury, and neuronal death. Astrocytes respond by releasing proinflammatory mediators, including free radicals, cytokines, and chemokines, which further contribute to enhancing the endothelial permeability, disrupting BBB integrity, and allowing for infiltration of peripheral immune cells, events that further intensify inflammation and neuronal injury. Feedback regulation of NOX or its inhibition causes M1 microglia to polarize to the M2 state (anti-inflammatory phenotype), which downregulates M1 functions and promotes regulation of neuroinflammation and neurorepair by releasing anti-inflammatory mediators, e.g., cytokines, neurotrophic, and growth factors. Mediators released by specific neural cell types (neuron, astrocyte, microglia or oligodendrocyte) are listed adjacent to each cell type in similar colored text. Curved arrows indicate the direction of signal flow between various neural cells for the inflammation activation process. Red curved arrows show the directional flow of danger/damage signals from neurons to glial cells (astroglia, microglia, oligodendroglia); blue curved arrows show the flow of proinflammatory signals from astrocytes and M1 microglia towards distressed neurons; green curved arrows show the flow of neurotrophic signals from M2 microglia and oligodendroglia towards the distressed neurons as a neuroprotective/neurorescue endeavor; black curved arrows show the directional crosstalk among various glial cells to mount a glial response to neuronal injury/damage. ↑, increase; ↓, decrease; ATP, adenosine triphosphate; BBB, blood-brain barrier; FAS, Fas cell surface death receptor; FASLG, Fas ligand; IL1B, interleukin 1 beta; IL6, interleukin 6; IL33, interleukin 33; NO, nitric oxide; NOS2, nitric oxide synthase 2 (inducible nitric oxide synthase); NOX, NADPH oxidase 1; PGE2, prostaglandin E2; PTGS2, prostaglandin-endoperoxide synthase 2 (also referred to as COX2, cyclooxygenase 2); ROS, reactive oxygen species; SOD1, superoxide dismutase 1; TNFA, tumor necrosis factor alpha.
3.3.5. Gliosis in MS
MS is a heterogeneous autoimmune, and complex inflammatory disease of the CNS characterized by demyelinating lesions. The inflammatory outcome in MS is attributed to microglia, however, the extent of their involvement in the disease process and the underlying molecular mechanisms through the mediation of neural injury or damage is still unclear [343]. Despite this knowledge gap, the presence of activated microglia at the sites of MS lesions seems to be a common feature. Patients with a progressive course of MS exhibit axonal degeneration and chronic active lesions with microglia typically present at the rim of the pathological lesions [274,344] in association with complement factors, antibodies, and immune cells [345,346]. Phagocytic microglia have also been observed in the white matter of the brain tissues obtained from patients with secondary progressive MS [347].
Following activation of NADPH Oxidase 2 (CYBB/NOX2), microglia increase their chemotactic signaling and recruit peripheral immune cells to the brain (Figure 5), which play a role in demyelination and axonal damage [348]. The intersection of MS pathogenesis and autoreactive T and B lymphocytes has been gaining interest in recent years as they appear to play a role as amplifiers and effectors in MS. Specifically, a subset of T-helper cells, the Th17 cells, appear to play a role in the pathogenesis of MS [349] through the secretion of IL12 and related members, interleukin 21 (IL21), and interleukin 23 (IL23). Further, IL21 is known to induce the activation of Th17 cells in an autocrine manner [350]. Other factors involved in the inflammatory pathogenesis of MS include polymorphisms in the T-cell receptor beta locus (TRB), cytotoxic T-lymphocyte associated protein 4 (CTLA4), CD6 molecule (CD6), interleukin 10 (IL10), interleukin 2 receptor subunit beta (IL2R), interleukin 4 receptor (IL4R), interleukin 7 receptor (IL7R), C-C motif chemokine receptor 5 (CCR5), interferon-gamma (IFNG), interferon regulatory factor 8 (IRF8), intercellular adhesion molecule 1 (ICAM1), TNFA, TNF receptor superfamily member 1A (TNFRSF1A/TNFRI, vitamin D receptor (VDR), and estrogen receptors 1 and 2 (ESR1 and ESR2) [351].
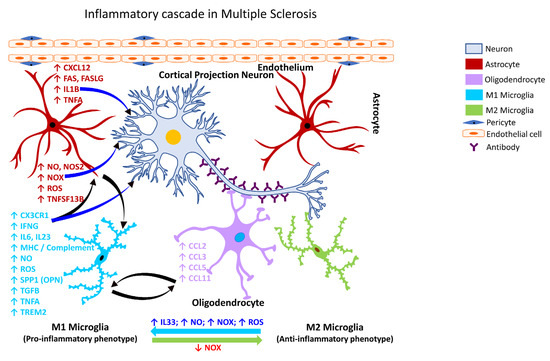
Figure 5.
Inflammatory cascade in Multiple Sclerosis. Schematic representation of the molecular mechanisms associated with glia-mediated functional interactions and systematic perturbations within the CNS to induce neuroinflammation in Multiple Sclerosis. The multifocal inflammatory demyelination of the white matter is driven largely by an inflammatory process besides an autoimmune component that involves innate and adaptive (B and T lymphocytes) immune cells. Various subtypes of myeloid cells are also critical for the pathogenic implications and the blood-derived monocytes represent the highest fraction of infiltrating peripheral cells into the CNS that undergo transformation into monocyte-derived inflammatory phagocytes (macrophages or dendritic cells) leading to neuronal damage. The endothelial cells form the inner lining of the blood vessel with pericytes enveloping the surface of the vasculature forming tight junctions to maintain the BBB integrity. Disruption of the BBB integrity further facilitates the infiltration of autoreactive immune cells into the CNS. Microglia serve as antigen presenting cells and present the myelin antigen to infiltrating B and T-cells, thereby exacerbating the neuroinflammatory cascade. M1 microglia (proinflammatory phenotype, neurotoxic) release various proinflammatory mediators, including free radicals, cytokines, and chemokines that further stimulate other glial cells, thus perpetuating a self-destructive environment that collectively contributes to exacerbating the neuronal injury/damage. M2 microglial cells (anti-inflammatory phenotype, neuroprotective) can polarize to an M1 state and release proinflammatory mediators in the presence of increased levels of NOX, ROS, NO, and IL33 released by M1 microglia and/or astrocytes and thereby augment the neuroinflammatory and neuronal injury process leading to synaptic dysfunction, neuronal injury, and neuronal death. Astrocytes respond by releasing proinflammatory mediators, including free radicals, cytokines, and chemokines, which further contribute to enhancing the endothelial permeability, disrupting BBB integrity, and allowing for infiltration of peripheral immune cells, events that further intensify inflammation and neuronal injury. Feedback regulation of NOX or its inhibition causes M1 microglia to polarize to the M2 state (anti-inflammatory phenotype), which downregulates M1 functions and promotes regulation of neuroinflammation and neurorepair by releasing anti-inflammatory mediators, e.g., cytokines, neurotrophic, and growth factors. Mediators released by specific neural cell types (neuron, astrocyte, microglia or oligodendrocyte) are listed adjacent to each cell type in similar colored text. Curved arrows indicate the direction of signal flow between various neural cells for the inflammation activation process. Red curved arrows show the directional flow of danger/damage signals from neurons to glial cells (astroglia, microglia, oligodendroglia); blue curved arrows show the flow of proinflammatory signals from astrocytes and M1 microglia towards distressed neurons; green curved arrows show the flow of neurotrophic signals from M2 microglia and oligodendroglia towards the distressed neurons as a neuroprotective/neurorescue endeavor; black curved arrows show the directional crosstalk among various glial cells to mount a glial response to neuronal injury/damage. ↑, increase; ↓, decrease; BBB, blood-brain barrier; CCL2, C-C motif chemokine ligand 2 (also referred to as MCP1, monocyte chemoattractant protein 1); CCL3, C-C motif chemokine ligand 3; CCL5, C-C motif chemokine ligand 5; CCL11, C-C motif chemokine ligand 11; CXCL12, C-X-C motif chemokine ligand 12; CX3CR1, C-X3-C motif chemokine receptor 1 (also referred to as fractalkine receptor); FAS, Fas cell surface death receptor; FASLG, Fas ligand; IFNG, interferon gamma; IL1B, interleukin 1 beta; IL6, interleukin 6; IL23, interleukin 23; IL33, interleukin 33; MHC, major histocompatibility complex; NO, nitric oxide; NOS2, nitric oxide synthase 2 (inducible nitric oxide synthase); NOX, NADPH oxidase 1; ROS, reactive oxygen species; SPP1, secreted phosphoprotein 1; TGFB, transforming growth factor beta; TNFA, tumor necrosis factor alpha; TNFSF13B, TNF superfamily member 13b; TREM2, triggering receptor expressed on myeloid cells 2.
4. Signaling Pathways Associated with Reactive Gliosis
4.1. Protein Phosphorylation in Neural Injury and Gliosis
One of the significant interests in astroglial biology is to know how the astrocytes, critical for synapse formation and neuronal maintenance, transform into a pathogenic form. As protein phosphorylation is a principal regulatory mechanism controlling almost all cellular machinery, the generalization is that it may actively modulate astrocyte and microglial communication and response. Despite the significant role of astrocytes in neurodegeneration, the cellular and molecular mechanisms that promote pathogenic astrocyte activity remain unresolved [320].
In the eukaryotic cell, protein phosphorylation refers to the reversible, covalent addition of a phosphate group to the serine, threonine, or tyrosine side chains of acceptor amino acid residues mediated by protein kinases [352,353]. Further, phosphorylation of protein kinases themselves can be associated with the injury process. For example, phosphorylation of the protein kinase R-like endoplasmic reticulum kinase (PERK) generates a distinct reactive state in astrocytes, which alters the astrocytic secretome leading to loss of synaptic function [354]. In addition, phosphorylation of PERK at specific residues has been shown to alter the function of proteins electrostatically by modulating the intermolecular interactions or by allosterically modifying the protein conformation, eventually influencing the protein activity [355].
Proliferation and apoptosis adaptor protein 15 (PEA15), a phosphoprotein enriched in astrocytes, is a cytoplasmic protein that regulates cell proliferation and apoptosis [356]. It has been shown to act as a cytoplasmic anchor for mitogen-activated protein kinase 3 and 1 (MAPK3/1; also known as ERK1/2) and prevents it from translocating to the nucleus, thus interfering with or reducing ERK1/2-mediated gene expression [357]. PEA15 protein is essential for the normal functioning of mature astrocytes [358]. Its levels and phosphorylation state exhibit peak expression in the adult brain [359]. Biogenic neurotransmitters such as noradrenaline, and hormones such as endothelin and vasoactive intestinal peptide are known to modulate the phosphorylation of PEA15 through the activation of kinases, primarily protein kinase C and calcium-calmodulin-dependent protein kinase II, thereby enhancing the intracellular levels of Ca2+ in the astrocytes [358]. Increased Ca2+ is known to elicit reactive gliosis and increase GFAP immunoreactivity, which is thought to be mediated by the activation of calpain I [360].
4.2. Activation of NF-κB Pathway in Neural Injury and Gliosis
NF-κB is a family of pleiotropic inducible transcription factors discovered in 1986 that are well known to regulate both innate and adaptive immune functions, besides serving as a pivotal signaling pathway (Figure 6) for inflammatory stimuli [361,362]. NF-κB signaling has been categorized into two major pathways, the canonical and non-canonical pathways, both central to the regulation of the immune and inflammatory response despite their distinct signaling mechanisms [362,363].
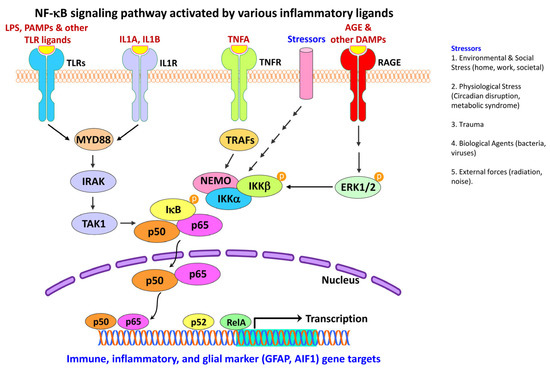
Figure 6.
NF-κB signaling in neuroinflammatory response. Schematic representation depicts activation and regulation of NF-κB signaling pathways by various inflammatory ligands. NF-κB activation is initiated when ligands stimulate various cell-surface receptors, e.g., TLR2, IL1R, TNFR or RAGE. Upon stimulation, TLRs and IL1R assembles the cytoplasmic MYD88 complex, which recruits IRAK and TAK1. Stimulation of TNFR recruits TRAFs. TAK1 and TRAFs activate the downstream kinase IKK, which in turn phosphorylate the NF-κB inhibitor IκBα, leading to ubiquitin-dependent IκBα degradation and NF-κB activation. Once activated, TRAFs function as an E3 ubiquitin ligase that catalyzes the synthesis of K63-linked polyubiquitin chains conjugated to itself, NEMO. A complex signal transduction process then starts as soon as TNFRs are activated. IKK is ultimately triggered and leads to the phosphorylation of IκB, which results in IκB ubiquitination and degradation. When IκB is degraded, the remaining NF-κB dimer (e.g., p65/p50 or p50/p50 subunit or their putative combination) translocates to the nucleus, which further binds to a DNA consensus sequence of target immune, inflammatory or glial marker (e.g., GFAP, AIF1) genes. Similarly, the binding of AGE and other DAMP ligands to RAGE receptor, causes ERK1/2 phosphorylation that further leads to activation and phosphorylation of the NEMO-IKK complex, NF-κB activation, and subsequent translocation of the NF-κB dimer (e.g., p65/p50 or p50/p50 subunit or their putative combination) to the nucleus to regulate transcription of immune and inflammatory genes including glial markers, e.g., GFAP, AIF1. Over-expression of RAGE produces vicious cycles that perpetuate oxidative stress and contribute to neuroinflammation by nuclear factor-kB (NF-kB) up-regulation. Black arrows indicate the activation process and multiple arrows indicate several unknown intermediates in the signaling cascade. AGE, advanced glycation end-products; AIF1, allograft inflammatory factor 1 (also known as IBA1, ionized calcium-binding adapter molecule 1); DAMP, damage-associated molecular patterns; ERK1/2, extracellular signal-regulated kinase 1 and 2; GFAP, glial fibrillary acidic protein; IKKα, inhibitor of nuclear factor kappa-B kinase subunit alpha (IKKA; also known as CHUK, component of inhibitor of nuclear factor kappa B kinase complex); IKKβ, inhibitor of nuclear factor kappa-B kinase subunit beta (IKKB; also known as IKBKB); IL1A, interleukin 1 alpha; IL1B, interleukin 1 beta; IL1R, interleukin 1 receptor; IRAK, interleukin 1 receptor associated kinase 1; IRAK, interleukin 1 receptor-associated kinase; IκB, inhibitor of nuclear factor kappa B; LPS, lipopolysaccharide; MYD88, MYD88 innate immune signal transduction adaptor (also known as myeloid differentiation primary response gene 88; NEMO, NF-kappaB essential modulator (also known as IKBKG, inhibitor of nuclear factor kappa-B kinase regulatory subunit gamma); NF-κB, nuclear factor kappa B; P, phosphate group; p50, protein p50 (the functional subunit of the 105 kD precursor protein coded by the gene NFKB1, nuclear factor kappa B subunit 1); p52, protein p52 (the functional subunit of the 100 kD precursor protein coded by the gene NFKB2, nuclear factor kappa B subunit 2); p65, protein p65 (also known as RELA, RELA proto-oncogene, NF-KB Subunit or V-Rel avian reticuloendotheliosis viral oncogene homolog A); PAMPs, pathogen-associated molecular patterns; RAGE, receptor for advanced glycation end-products; TAK1, transforming growth factor beta activated kinase 1; TLR, toll-like receptor; TNFA, tumor necrosis factor alpha; TNFR, tumor necrosis factor receptor; TRAF, tumor necrosis actor receptor-associated factor.
The NF-κB family consists of five related structural members: nuclear factor kappa B subunit 1 (NFKB1; p50), nuclear factor kappa B subunit 2 (NFKB2; p52), REL proto-oncogene, NF-KB subunit (REL; also known as V-Rel avian reticuloendotheliosis viral oncogene homolog; c-Rel), RELA proto-oncogene, NF-KB subunit (RELA/NFKB3; also known as V-Rel avian reticuloendotheliosis viral oncogene homolog A; p65), and RELB proto-oncogene, NF-KB subunit (RELB; also known as V-Rel avian reticuloendotheliosis viral oncogene homolog B). These five members mediate the transcription of target genes by binding to a specific DNA element, the κB enhancer, through a variety of homo- or hetero-dimer formations [4,362]. Deregulation of NF-κB is a hallmark of chronic inflammatory diseases [362].
Within the nucleus, active NF-κB promotes the transcription of NF-κB-dependent genes, such as the NLR family pyrin domain containing 3 (NLRP3), interleukin 1 beta (IL1B), and interleukin 18 (IL18), key components of the inflammasome [362]. NF-κB also has a role in regulating the activation of inflammasomes [364].
The activated NF-κB pathway is evident in the neurons of the hippocampus and entorhinal cortex of post-mortem brain samples from patients with AD [365,366], in the dopaminergic neurons within the SNPc of post-mortem brain samples from patients with PD [367], and in peripheral immune cells of patients with HD [368]. Increased accumulation of NF-κB in astrocytes is also reported in the R6/2 animal model of HD [369]. Inducible NF-κB has been detected at presynaptic sites in the cortex, hippocampus, and cerebellum and is known to be responsive to the agents such as glutamic acid (glutamate). Such NF-κB amounts are highly variable in the neuronal nucleus and are referred to as active NF-κB, as they are induced by neuronal synaptic activity [370].
NF-κB is also a core transcription factor for various inflammatory and immune mediators [371]. Following spinal cord injury, downregulation of SKI proto-oncogene (SKI) has been shown to inhibit glial inflammation by inhibiting the activity of the NF-κB, thus highlighting the role of NF-κB in the activation of astrocytes and the release of inflammatory mediators [372]. Other studies have shown that systemic inflammation or aging modulates hypothalamic function through NF-κB activation and microglia-neuron cross-talk [373,374]. Thus, NF-κB is a family of transcription factors that are strongly implicated in the physiological and pathological processes involving inflammation, immunity, cell proliferation, cell differentiation, and cell survival.
Inhibition of NF-κB in microglia was shown to reduce propagation and neurotoxicity of the human microtubule-associated protein tau, as well as restore learning and memory function in aged P301S transgenic mice (PS19 mice), which express the P301S mutant form [375]. NF-κB activation has also been shown to occur in the microglia of the SOD1G93A mouse model of ALS, especially at the later stages of the disease [376]. Further, the reduction of the inhibitor of nuclear factor-kappa B kinase subunit beta (IKKB) levels and thus of NF-κB activity in the SOD1G93A transgenic mice extended their median survival rate [274,376].
NF-κB mediates the synergistic activation of the positive-feedback loop for IL6-signaling (IL6 amplifier), which is further activated following IL6 and/or IL17 stimulation of non-immune cells [131]. IL6 amplifier is also activated by simultaneous stimulation of NF-κB and STAT3 to locally induce chemokines, causing chronic inflammation [131]. This provides evidence of crosstalk between the NF-κB and the JAK/STAT signaling pathways.
4.3. Activation of JAK/STAT Pathway in Neural Injury and Gliosis
The JAK/STAT signaling cascade is one of the critical communication pathways pivots in orchestrating several cellular processes, including the immune function. The JAK/STAT pathway (Figure 7) constitutes a rapid transfer of signals from cell-membrane receptors to the nucleus and induces the expression of various essential mediators of inflammation [377,378], as well as of innate and adaptive immunity [379]. Thus, the JAK/STAT pathway serves as a vital downstream mediator for a myriad of cytokines, hormones, and growth factors [4,37,316]; hematopoiesis, adipogenesis, inflammation, and apoptosis [378]. Extracellular factors, e.g., leptin and growth hormone also affect JAK/STAT signaling and control gene expression [4].
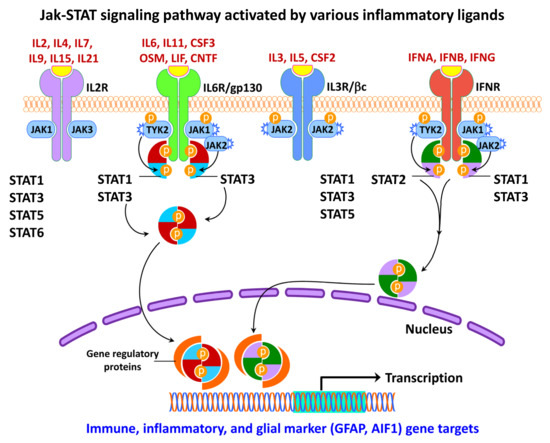
Figure 7.
JAK-STAT signaling in neuroinflammatory response. Schematic representation depicting activation and regulation of JAK/STAT signaling pathways by various inflammatory ligands. The JAK family has four main members: JAK1, JAK2, JAK3, and TYK2. Black arrows indicate the activation process. Inflammatory mediators and growth factors bind to their corresponding receptors leading to receptor dimerization and recruitment of related JAKs. These mediators include, but are not limited to, interferons, such as interferon alpha, interferon beta, and interferon gamma; interleukin 2, interleukin 3, interleukin 4, interleukin 5, interleukin 6, interleukin 7, interleukin 9, interleukin 11, interleukin 15, interleukin 21; colony stimulating factor 2, colony stimulating factor 3; oncostatin M; leukemia inhibitory factor; and ciliary neurotrophic factor. JAK activation leads to tyrosine phosphorylation of the receptors and the formation of docking sites for various STATs. The activated JAKs phosphorylate (P) and activate STATs. Some JAKs are associated with many different cytokine receptors, but JAK3 associates with only IL2, predominantly to its gamma chain subunit. The STAT family consists of six members: STAT1, STAT2, STAT3, STAT4, STAT5 and STAT6, which upon phosphorylation dissociate from the receptor to form homodimers or heterodimers and translocate to the nucleus and bind to transcription factor binding sites on the target DNA to regulate transcription of immune and inflammatory genes including glial markers, e.g., GFAP, AIF1. AIF1, allograft inflammatory factor 1 (also known as IBA1, ionized calcium-binding adapter molecule 1); CNTF, ciliary neurotrophic factor; CSF2, colony stimulating factor 2; CSF3, colony stimulating factor 3; GFAP, glial fibrillary acidic protein; gp130, glycoprotein 130; IFNA, interferon alpha; IFNB, interferon beta; IFNG, interferon gamma; IFNR, interferon receptor; IL11, interleukin 11; IL15, interleukin 15; IL2, interleukin 2; IL21, interleukin 21; IL2R, interleukin 2 receptor; IL3, interleukin 3; IL3R/βC, interleukin 3 receptor beta chain subunit; IL4, interleukin 4; IL5, interleukin 5; IL6, interleukin 6; IL6R, interleukin 6 receptor; IL7, interleukin 7; IL9, interleukin 9; JAK1, janus kinase 1; JAK2, janus kinase 2; JAK3, janus kinase 3; JAK-STAT, janus kinase-signal transducer and activator of transcription; LIF, leukemia inhibitory factor; OSM, oncostatin M; P, phosphate group; STAT1, signal transducer and activator of transcription 1; STAT2, signal transducer and activator of transcription 2; STAT3, signal transducer and activator of transcription 3; STAT4, signal transducer and activator of transcription 4; STAT5, signal transducer and activator of transcription 5; STAT6, signal transducer and activator of transcription 6; TYK2, tyrosine kinase 2.
Ligands activate receptors linked to JAK/STAT and initiate the process of phosphorylation. Phosphorylated JAKs proceed towards activating several substrates and provide docking sites for STATs, which in turn become phosphorylated and exhibit the full spectrum of STAT activity [380]. During this signaling process, phosphorylated STATs are released from the receptor complex, undergo dimerization through -SH2 domain interactions, and translocate into the nucleus. Therefore, activating the receptors associated with JAKs is critical to initiate the JAK trans-phosphorylation and subsequent recruitment of one or more STATs to be phosphorylated [380].
In general, STATs often regulate transcription through the following sequential mechanisms: (a) first, STAT is interlinked with the N-terminal coiled-coil DNA binding site linker Src-homology 2 (SH2) and C-terminal to facilitate transcription activation, (b) second, transcriptional complexes are formed between STAT and other non-STAT transcription factors to initiate transcription, (c) third, STAT binds to non-STAT DNA binding domain to augment STAT-mediated transcription, and (d) fourth, STAT and non-STAT transcription factors bind to discrete DNA binding sites and synergistically coordinate transcription [378].
The cytoplasmic JAK family comprises four isoforms, JAK1, JAK2, JAK3, and TYK2 [37,380], whereas STAT has been classified into seven isoforms including STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B and STAT6 [381]. Overall, dysregulation of JAK/STAT activity is linked with inflammatory, autoimmune, cancer, and metabolic diseases [4].
It has been well accepted that activation of STAT3 is regulated by post-translational modification, particularly by phosphorylation at the serine and tyrosine residues. Selectively blocking the JAK/STAT and NF-κB pathways have been shown to reduce microgliosis cell death [382]. The JAK/STAT pathway has been shown to be linked to astrocyte differentiation, glial activation, and glial gene responses upon neuronal injury [37,316]. These early studies showed the target genes of STAT3 constitute cytokines and growth factors (IL6; interleukin 10/IL10; interleukin 6 cytokine family signal transducer/IL6ST or gp130; oncostatin M/OSM), apoptosis-related genes (BCL2 apoptosis regulator/BCL2; BCL2-Like 1/BCL2L1 or BCL-XL), acute phase response factors (C-reactive protein/CRP; alpha2-macroglobulin/A2M), transcription factors (Fos proto-oncogene, AP-1 transcription factor subunit/FOS or C-FOS; Jun proto-oncogene, AP-1 transcription factor subunit/JUN or C-JUN), and cell cycle-related genes (cyclin-dependent kinase inhibitor 1A/CDKN1A or p21; cyclin D1/CCND1; cyclin E1/CCNE1).
A crucial function for STAT3 in regulating GFAP expression has been identified during astrogenesis and astrogliosis, in which signaling molecules like cytokines act via the IL6/gp130-receptor to activate the JAK/STAT and MAPK pathways [316,383]. Following an injury to the striatal dopaminergic nerve terminals mediated by the dopaminergic neurotoxicant, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), the JAK2/STAT3 pathway is specifically activated to upregulate GFAP expression in striatal astrocytes [316].
Altered JAK2/STAT3 signaling is involved in several neurodegenerative diseases and appears to be independent of an inflammatory event [384]. Inhibiting the JAK/STAT3 pathway has been shown to increase the frequency of Huntingtin protein aggregates in reactive astrocytes, a pathological hallmark of HD [385]. In addition, STAT3, STAT4, and TYK2 genetic variants have been associated with MS susceptibility [386]. Taken together, the JAK/STAT pathway is critical for cytokine-mediated signal transduction associated with a multitude of functions such as cellular proliferation, differentiation, and immune homeostasis.
4.4. Activation of MAPK Pathway in Neural Injury and Gliosis
The MAPK pathway is a major signaling cascade, which regulates a diverse feature of cellular processes such as initiation, propagation, proliferation, differentiation, and apoptosis. The MAPK family (Figure 8) are serine/threonine kinases that direct cellular responses arising from various stimuli including growth factors, extracellular matrix proteins, inflammatory mediators and stressors [4,387].
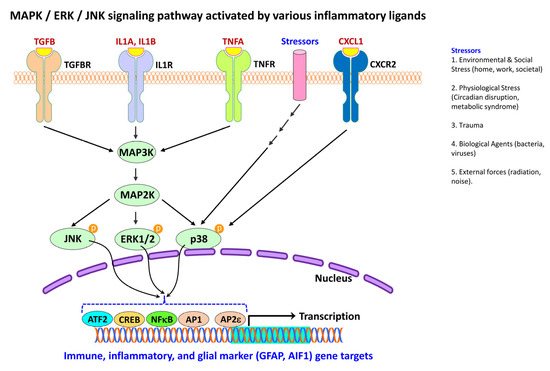
Figure 8.
MAPK/ERK/JNK signaling in neuroinflammatory response. Schematic representation depicts MAP kinase activation by growth factors, stressor and cytokines which triggers a phosphorylation cascade. Agonist binding to TGFBR, IL1R, TNFR, stress receptors or CXCR2 leads to activation of the MAP3K, which has potential to activate various other MAP kinases by phosphorylating and activating the respective MAP2K. MAP2K has a restricted specificity for MAP kinase substrates. Upon activation, JNK, ERK1/2, and p38 further regulate gene transcription through activation and recruitment of transcription factors such as ATF2, CREB, NFkB, AP1 and AP2ε to the nucleus to regulate transcription of immune and inflammatory genes including glial markers, e.g., GFAP and AIF1. Arrows indicate the activation process in the signaling cascade and multiple arrows indicate several intermediates in the signaling cascade. AIF1, allograft inflammatory factor 1 (also known as IBA1, ionized calcium-binding adapter molecule 1); AP1, activator protein 1 (also known as JUN, Jun protooncogene AP1 transcription factor subunit or V-Jun avian sarcoma virus 17 oncogene homolog); AP2ε, transcription factor AP 2 epsilon (also known as TFAP2E); ATF2, activating transcription factor 2; CREB, cyclic AMP response element binding protein; CXCL1, C-X-C motif chemokine ligand 1; CXCR2, C-X-C motif chemokine receptor 2; ERK 1/2, extracellular signal-regulated kinase 1 and 2; GFAP, glial fibrillary acidic protein; IL1A, interleukin 1alpha; IL1B, interleukin 1 beta; IL1R, interleukin 1 receptor; JNK, c-Jun N-terminal kinase; MAP2K, mitogen activated protein kinase kinase; MAP3K, mitogen activated protein kinase kinase kinase; NFkB, nuclear factor kappa-B; P, phosphate group; p38, p38 mitogen activated protein kinase (p38 MAPK); TGFB, transforming growth factor beta; TGFBR, transforming growth factor beta receptor; TNFA, tumor necrosis factor alpha; TNFR, tumor necrosis factor receptor.
In eukaryotic cells, at least three distinct types of MAPK cascades are known. These include mitogen-activated protein kinases 1 and 2 (MAPK3/ERK1 and MAPK1/ERK2; also referred to as p42/44 MAPK or p42/44 ERK), mitogen-activated protein kinase 8 (MAPK8; also referred to as c-Jun N-terminal kinase/JNK or stress-activated protein kinase/SAPK), and mitogen-activated protein kinase 14 (MAPK14; also referred to as p38 MAPK) signal transduction pathways [10]. ERK has been shown to be activated by mitogens and differentiation signals [4], and the ERK/MAPK signaling pathway forms the core signaling network for regulating cellular development, growth, and division. The ribosomal protein S6 kinase A1 (RSK) is a downstream effector of the ERK/MAPK signaling cascade [388].
JNK signaling cascade is widely involved in multiple physiological processes and mediates its actions through three alternate splicing isoforms, JNK α, β, and γ [389]. JNK activation is also associated with oncogene transformation growth factor-mediated signaling. Because inflammatory stimuli and stress activate JNK [4], it has been implicated in apoptosis, survival signaling, cell differentiation, and cytochrome c release from mitochondria.
Mitogen-activated protein kinase 14 (MAPK14; also known as p38 MAPK) is activated by toxicants, UV irradiation, heat shock, osmotic stress, proinflammatory cytokines, lipopolysaccharides, protein synthesis inhibitors, and certain mitogens [389]. It has been shown that the specific isoform p38δ is critical for interferon signaling, where it directs the phosphorylation and activation of phospholipase A2 in the cytoplasm. IFNA or IFNG-mediated activation of p38 MAPK also produces phosphorylation of the transcription factor STAT1 on Ser727 [389]. Collectively, activation of MAPKs, including ERK1/2, JNK, and p38 MAPK, leads to phosphorylation and activation of p38 transcriptions factors known to be abundant in cytoplasm or nucleus, that trigger the inflammatory responses in response to a variety of insults such as physical, chemical, biological, psychological, infectious, or microbial factors [4].
In post-mortem brain samples obtained from patients with AD, phosphorylated p38 is seen both in neurons and glia in the vicinity of the amyloid plaques [390]. However, in mouse models of AD, p38 MAPK is seen only within microglial cells [391]. Reactive astrocytes in the brains of patients with ALS have been shown to contain the active form of ERK, JNK, and p38 MAPK [385,392]. In mouse models of ALS, accumulation and activation of p38 MAPK is an essential feature to form hypertrophic astrocytes and reactive microglial state that accounts for the initiation and progression of motor neuron pathology [393].
5. GFAP Gene Expression as an Index of Astroglial Activation
The glial reaction to nervous system damage often termed gliosis, represents a hallmark of all types of neural injury [37,38]. Microglia and astrocytes are differentially involved in engulfing synapses at the sites of neuronal injury and reactive gliosis [394]. While the molecular events related to microglial activation following neuronal injury are limited, experimental studies have shown that like astroglia, activated microglia also serve as microsensors of brain injury, including that engendered by disease or toxicants [37,38]. Reactive astrogliosis is also seen in neurological disease states including neurodegeneration [385,395], epilepsy [396], ischemic stroke [397], demyelination [398,399], and traumatic brain injury [400,401].
When astrocytes react to injury, they undergo hypertrophy and upregulate the expression of GFAP. The basal levels of GFAP in astrocytes of different brain regions is variable [402,403]. For example, the hippocampal astrocytes display higher GFAP expression than the cortical or striatal astrocyte populations [404,405,406].
GFAP is a type-III intermediate filament and is the signature molecule among several intermediate filaments in astrocytes [407]. It is a relatively non-soluble acidic cytoskeletal protein. GFAP intermediate filament has a diameter of 10 nm, with the alpha-helical rod domain flanked by a flexible N-terminal tail domain [408]. The human GFAP consists of 432 amino acids with head and tail domains flanking a central α-helical rod domain encoded by a gene on chromosome 17q21 [409,410]. The gene consists of eight introns and nine exons, with four alternative exons and two alternative introns (3 kb, mRNA). A recent study demonstrated that mutations in the intron regions of the GFAP gene result in gene splicing and expression of various GFAP isoforms, aggregation, and accumulation of which leads to astrocyte dysfunction [411].
To date, six isoforms of GFAP have been identified [410,412] and classified as GFAP alpha (α), beta (β), gamma (γ), delta (δ), epsilon (ε), and kappa (κ). GFAPα is the major component of the astrocytic cytoskeleton and scaffolding required for the maintenance of its shape, structure, and plasticity. In humans, the promoter region of the GFAP gene extends from -2162 to +47, containing binding sites for several transcription factors [413]. The inhibition of the histone deacetylase activity causes a GFAPα to GFAPδ isoform shift mediated by serine and arginine-rich splicing factor 6 (SRSF6; also known as SR splicing factor 6/SR6) [414] resulting in downregulation of GFAP expression, which is suggestive of a regulatory function. The GFAP protein undergoes post-translational modifications via citrullination, glycosylation, lipoxidation, and phosphorylation, which influence its properties and function, including cytoskeletal assembly [412]. Experimental studies using rodent models and human cell lines have revealed that microRNAs (miRNA) regulate GFAP. However, direct regulation of the human GFAP gene by miRNA remains to be elucidated [413].
6. Concluding Remarks
Occupational brain injury and toxicant-induced neuronal damage are increasingly recognized to dysregulate the homeostatic signaling cascade in the healthy brain, causing gliosis and neuroinflammation. With the advent of new knowledge in the morphometric and cytokine field, the critical role of toxicant-induced acute or chronic neuroinflammation through gliosis is emerging, providing a foundation for understanding the pathophysiology of various neurological disorders. The polarization of microglia, release of inflammatory cytokines, microglial activation, reactive astrocytes, and high expression of GFAP provides robust evidence to the molecular underpinnings of neural injury and a benchmark for understanding various injury- or disease- models. Consistent with these findings, this review identifies and synthesizes the evidence about how a wide variety of ligands insult the homeostatic gene signature that could eventually trigger the pathological signaling cascades, including protein phosphorylation, NF-κB, JAK/STAT, and MAPK pathway, which can potentially sustain even after the cessation of the toxicant exposure or occupational injury.
Nevertheless, transient changes in the phenotype expression can be anticipated in the early phase of head injury, concussion, and TBI; however, the long-term phenotypic changes require a significant number of resident cells to change and secrete signaling molecules that sequentially activate various intermediate steps culminating in signal transduction and gene expression. In neurodegenerative diseases, understanding the specificity of the toxicant’s effect and their cytokine signature, along with any identifiable intermediate steps in the signaling cascades, can provide avenues for strategic therapeutic intervention.
Neurotoxicological studies aim to link the mechanistic processes within the cells and tissues to elucidate the physiological substrates of impaired functions associated with a toxicant. These substrates form a broad spectrum that reflects the biological organization hierarchy, bringing neuroinflammation into the context. At one end of the spectrum are the brain cells, including glia, and at the other is an organism exhibiting neurological disorders originating from the aftermath of gliosis. Cellular interactions in the brain compartment are becoming essential components of tissue functions. Microglia and astroglia exhibit an intimate association at the physical, molecular, and biochemical levels. They exist in proximity with neurons and endothelial cells of the BBB to maintain cellular interaction and transport of small molecules, which are critical for neuron-glia omnidirectional signaling primarily at the tripartite synapses.
This review has provided a comprehensive vision of recent advancements in the field to identify the possible targets as the result of occupational injury or the toxicant’s effect, bringing the neuroinflammation perspective into place for the implication of gliosis and subsequent progression of the neurodegenerative events in diseases including AD, PD, CJD, ALS, and MS. New research directions should include experimental models in which heterogeneous cell interactions are preserved during toxicant exposure in neuroinflammation arising from an occupational injury or toxicant-induced neuronal damage. In addition to in vivo assessment, a system of co-cultures, brain slices, and primary cultures prepared from the brains of toxicant-exposed animals should make it possible to design the strategy from the cell biology endpoint of the spectrum towards the tissue level, and from there, perhaps our view of the other end of the spectrum will be insightful.
Author Contributions
K.S. and D.P. conceived and designed the review article. D.P. conducted the literature search and drafted the manuscript. K.S. prepared the tables, illustrations, and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The National Institute for Occupational Safety and Health provided funding through the NORA (9390BN0 to K.S.) and NTRC (number 9390KK9 to K.S.) projects. The APC charges were waived.
Institutional Review Board Statement
The study did not require ethical approval as this is a review article.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this review article as no datasets were generated or analyzed.
Conflicts of Interest
The authors declare no conflict of interest.
Disclaimer
The findings and conclusions in this report are those of the author(s) and do not necessarily represent the official position of the National Institute for Occupational Safety and Health, Centers for Disease Control and Prevention.
References
- Kotas, M.E.; Medzhitov, R. Homeostasis, inflammation, and disease susceptibility. Cell 2015, 160, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831, Erratum in Nat. Immunol. 2021, 22, 254. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef]
- Postolache, T.T.; Wadhawan, A.; Can, A.; Lowry, C.A.; Woodbury, M.; Makkar, H.; Hoisington, A.J.; Scott, A.J.; Potocki, E.; Benros, M.E.; et al. Inflammation in Traumatic Brain Injury. J. Alzheimer’s Dis. 2020, 74, 1–28. [Google Scholar] [CrossRef]
- Wojtulewicz, K.; Krawczyńska, A.; Tomaszewska-Zaremba, D.; Wójcik, M.; Herman, A.P. Effect of Acute and Prolonged Inflammation on the Gene Expression of Proinflammatory Cytokines and Their Receptors in the Anterior Pituitary Gland of Ewes. Int. J. Mol. Sci. 2020, 21, 6939. [Google Scholar] [CrossRef]
- Rahimi, R.A.; Luster, A.D. Chemokines: Critical Regulators of Memory T Cell Development, Maintenance, and Function. Adv. Immunol. 2018, 138, 71–98. [Google Scholar] [CrossRef]
- Schirmer, L.; Velmeshev, D.; Holmqvist, S.; Kaufmann, M.; Werneburg, S.; Jung, D.; Vistnes, S.; Stockley, J.H.; Young, A.; Steindel, M.; et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019, 573, 75–82. [Google Scholar] [CrossRef]
- Yu, L.; Su, X.; Li, S.; Zhao, F.; Mu, D.; Qu, Y. Microglia and Their Promising Role in Ischemic Brain Injuries: An Update. Front. Cell Neurosci. 2020, 14, 211. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Ekdahl, C.T.; Claasen, J.H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wei, Y.Z.; Wang, G.Q.; Li, D.D.; Shi, J.S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Microglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell Neurosci. 2019, 12, 531. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.L.; Schafer, D.P. Microglia: Architects of the Developing Nervous System. Trends Cell Biol. 2016, 26, 587–597. [Google Scholar] [CrossRef]
- Li, T.; Pang, S.; Yu, Y.; Wu, X.; Guo, J.; Zhang, S. Proliferation of parenchymal microglia is the main source of microgliosis after ischaemic stroke. Brain 2013, 136 Pt 12, 3578–3588. [Google Scholar] [CrossRef]
- Konishi, H.; Okamoto, T.; Hara, Y.; Komine, O.; Tamada, H.; Maeda, M.; Osako, F.; Kobayashi, M.; Nishiyama, A.; Kataoka, Y.; et al. Astrocytic phagocytosis is a compensatory mechanism for microglial dysfunction. EMBO J. 2020, 39, e104464. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kadluczka, J.; Kuter, K.Z. Beyond the GFAP-Astrocyte Protein Markers in the Brain. Biomolecules 2021, 11, 1361. [Google Scholar] [CrossRef]
- Ogbodo, J.O.; Arazu, A.V.; Iguh, T.C.; Onwodi, N.J.; Ezike, T.C. Volatile organic compounds: A proinflammatory activator in autoimmune diseases. Front. Immunol. 2022, 13, 928379. [Google Scholar] [CrossRef]
- Zatta, P.; Drago, D.; Bolognin, S.; Sensi, S.L. Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol. Sci. 2009, 30, 346–355. [Google Scholar] [CrossRef]
- Varacallo, M.; Knoblauch, D.K. Occupational Injuries and Workers’ Compensation Management Strategies; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Mayo Clinic. Understanding Brain Injury, A Guide for Employers. MC1298rev0111, 2011, mc1298-2. Available online: https://biaia.org/ (accessed on 1 December 2022).
- Available online: https://injuryfacts.nsc.org/all-injuries/deaths-by-demographics/top-10-preventable-injuries/ (accessed on 1 December 2022).
- Pek, S.; Turner, N.; Tucker, S.; Kelloway, E.K.; Morrish, J. Injunctive safety norms, young worker risk-taking behaviors, and workplace injuries. Accid. Anal. Prev. 2017, 106, 202–210. [Google Scholar] [CrossRef] [PubMed]
- NIOSH. Slip, Trip, and Fall Prevention for Health Care Workers. DHHS (NIOSH) Publication No. 2011-123. 2010. Available online: http://www.cdc.gov/niosh/docs/2011-123 (accessed on 1 December 2022).
- NIOSH. Preventing Slips, Trips, and Falls in Wholesale and Retail Trade Establishments DHHS (NIOSH) Publication No. 2013−100, 2013-100.pdf. 2012. Available online: https://www.cdc.gov/ (accessed on 1 December 2022).
- Anderson, V.P.; Mulhern, B. Focus on risk management. Don’t let costly slip and fall injuries trip you up. Chain Storage Age 2010, 86, 42–43. [Google Scholar]
- Terry, D.P.; Iverson, G.L.; Panenka, W.; Colantonio, A.; Silverberg, N.D. Workplace and non-workplace mild traumatic brain injuries in an outpatient clinic sample: A case-control study. PLoS ONE 2018, 13, e0198128. [Google Scholar] [CrossRef] [PubMed]
- Tiesman, H.; Konda, S.; Bell, J. The Epidemiology of Fatal Occupational Traumatic Brain Injury in the United States. Am. J. Prev. Med. 2011, 41, 61–67. [Google Scholar] [CrossRef]
- Konda, S.; Tiesman, H.; Reichard, A. Nonfatal occupational traumatic brain injuries treated in U.S. hospital emergency departments: 1998–2007. Inj. Prev. 2015, 21, 115–120. [Google Scholar] [CrossRef]
- Ruiz-Hernandez, A.; Kuo, C.C.; Rentero-Garrido, P.; Tang, W.Y.; Redon, J.; Ordovas, J.M.; Navas-Acien, A.; Tellez-Plaza, M. Environmental chemicals and DNA methylation in adults: A systematic review of the epidemiologic evidence. Clin. Epigenetics 2015, 7, 55. [Google Scholar] [CrossRef]
- Zhang, Q.; Niu, Q.; Li, L.Y.; Yang, L.; Guo, X.L.; Huang, J.X.; Wang, L.P.; Liang, Y.X. Establishment of a poisoned animal model of toxic encephalopathy induced by 1,2-dichloroethane. Int. J. Immunopathol. Pharmacol. 2011, 24 (Suppl. S1), 79S–83S. [Google Scholar]
- Chen, S.; Zhang, Z.; Lin, H.; Chen, Z.; Wang, Z.; Wang, W. 1,2-Dichloroethane-induced toxic encephalopathy: A case series with morphological investigations. J. Neurol. Sci. 2015, 351, 36–40. [Google Scholar] [CrossRef]
- Yang, J.; Wang, T.; Jin, X.; Wang, G.; Zhao, F.; Jin, Y. Roles of Crosstalk between Astrocytes and Microglia in Triggering Neuroinflammation and Brain Edema Formation in 1,2-Dichloroethane-Intoxicated Mice. Cells 2021, 10, 2647. [Google Scholar] [CrossRef]
- Wang, T.; Sun, Q.; Yang, J.; Wang, G.; Zhao, F.; Chen, Y.; Jin, Y. Reactive astrocytes induced by 2-chloroethanol modulate microglia polarization through IL-1β, TNF-α, and iNOS upregulation. Food Chem. Toxicol. 2021, 157, 112550. [Google Scholar] [CrossRef]
- Zhong, Y.; Liang, B.; Meng, H.; Ye, R.; Li, Z.; Du, J.; Wang, B.; Zhang, B.; Huang, Y.; Lin, X.; et al. 1,2-Dichloroethane induces cortex demyelination by depressing myelin basic protein via inhibiting aquaporin 4 in mice. Ecotoxicol. Environ. Saf. 2022, 231, 113180. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; O’Callaghan, J.P. Signaling Mechanisms Underlying Toxicant-Induced Gliosis. In The Role of Glia in Neurotoxicity, 2nd ed.; Aschner, M., Costa, L.G., Eds.; CRC Press: Boca Raton, FL, USA, 2005; pp. 141–171. [Google Scholar]
- O’Callaghan, J.P.; Sriram, K. Glial fibrillary acidic protein and related glial proteins as biomarkers of neurotoxicity. Expert Opin. Drug Saf. 2005, 4, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Tjalkens, R.B.; Popichak, K.A.; Kirkley, K.A. Inflammatory Activation of Microglia and Astrocytes in Manganese Neurotoxicity. Adv. Neurobiol. 2017, 18, 159–181. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Rosengren, L.E.; Aurell, A.; Kjellstrand, P.; Haglid, K.G. Astrogliosis in the cerebral cortex of gerbils after long-term exposure to 1,1,1-trichloroethane. Scand. J. Work Environ. Health 1985, 11, 447–455. [Google Scholar] [CrossRef]
- Shi, J.; Ma, Y.; Zheng, M.; Ruan, Z.; Liu, J.; Tian, S.; Zhang, D.; He, X.; Li, G. Effect of sub-acute exposure to acrylamide on GABAergic neurons and astrocytes in weaning rat cerebellum. Toxicol. Ind. Health 2012, 28, 10–20. [Google Scholar] [CrossRef]
- Garcia, S.J.; Seidler, F.J.; Qiao, D.; Slotkin, T.A. Chlorpyrifos targets developing glia: Effects on glial fibrillary acidic protein. Brain Res. Dev. Brain Res. 2002, 133, 151–161. [Google Scholar] [CrossRef]
- Campos-Ordonez, T.; Zarate-Lopez, D.; Galvez-Contreras, A.Y.; Moy-Lopez, N.; Guzman-Muniz, J.; Gonzalez-Perez, O. Cyclohexane produces behavioral deficits associated with astrogliosis and microglial reactivity in the adult hippocampus mouse brain. Cell Mol. Neurobiol. 2015, 35, 503–512. [Google Scholar] [CrossRef]
- Rosengren, L.E.; Kjellstrand, P.; Aurell, A.; Haglid, K.G. Irreversible effects of dichloromethane on the brain after long term exposure: A quantitative study of DNA and the glial cell marker proteins S-100 and GFA. Br. J. Ind. Med. 1986, 43, 291–299. [Google Scholar] [CrossRef]
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; McKinney, W.; Jackson, M.C.; Cumpston, A.; Cumpston, J.L.; Cumpston, J.B.; Leonard, H.D.; Kashon, M.; et al. Biological effects of inhaled hydraulic fracturing sand dust VII. Neuroinflammation and altered synaptic protein expression. Toxicol. Appl. Pharmacol. 2020, 409, 115300. [Google Scholar] [CrossRef]
- Olanow, C.W.; Good, P.F.; Shinotoh, H.; Hewitt, K.A.; Vingerhoets, F.; Snow, B.J.; Beal, M.F.; Calne, D.B.; Perl, D.P. Manganese intoxication in the rhesus monkey: A clinical, imaging, pathologic, and biochemical study. Neurology 1996, 46, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Vezér, T.; Kurunczi, A.; Náray, M.; Papp, A.; Nagymajtényi, L. Behavioral effects of subchronic inorganic manganese exposure in rats. Am. J. Ind. Med. 2007, 50, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; Goldsmith, W.T.; Jackson, M.; McKinney, W.; Frazer, D.G.; Robinson, V.A.; Castranova, V. Neurotoxicity following acute inhalation exposure to the oil dispersant COREXIT EC9500A. J. Toxicol. Environ. Health A 2011, 74, 1405–1418. [Google Scholar] [CrossRef]
- Opris, R.V.; Toma, V.; Baciu, A.M.; Moldovan, R.; Dume, B.; Berghian-Sevastre, A.; Moldovan, B.; Clichici, S.; David, L.; Filip, G.A.; et al. Neurobehavioral and Ultrastructural Changes Induced by Phytosynthesized Silver-Nanoparticle Toxicity in an In Vivo Rat Model. Nanomaterials 2021, 12, 58. [Google Scholar] [CrossRef]
- Fukui, K.; Utsumi, H.; Tamada, Y.; Nakajima, T.; Ibata, Y. Selective increase in astrocytic elements in the rat dentate gyrus after chronic toluene exposure studied by GFAP immunocytochemistry and electron microscopy. Neurosci. Lett. 1996, 203, 85–88. [Google Scholar] [CrossRef]
- Gotohda, T.; Kuwada, A.; Morita, K.; Kubo, S.; Tokunaga, I. Elevation of steroid 5 alpha-reductase mRNA levels in rat cerebellum by toluene inhalation: Possible relation to GFAP expression. J. Toxicol. Sci. 2000, 25, 223–231. [Google Scholar] [CrossRef][Green Version]
- Svenson, D.W.; Davidson, C.J.; Thakur, C.; Bowen, S.E. Acute exposure to abuse-like concentrations of toluene induces inflammation in mouse lungs and brain. J. Appl. Toxicol. 2022, 42, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, A.C.; Guilarte, T.R. Cellular and subcellular localization of peripheral benzodiazepine receptors after trimethyltin neurotoxicity. J. Neurochem. 2000, 74, 1694–1704. [Google Scholar] [CrossRef]
- Fiedorowicz, A.; Figiel, I.; Kamińska, B.; Zaremba, M.; Wilk, S.; Oderfeld-Nowak, B. Dentate granule neuron apoptosis and glia activation in murine hippocampus induced by trimethyltin exposure. Brain Res. 2001, 912, 116–127. [Google Scholar] [CrossRef]
- Antonini, J.M.; Sriram, K.; Benkovic, S.A.; Roberts, J.R.; Stone, S.; Chen, B.T.; Schwegler-Berry, D.; Jefferson, A.M.; Billig, B.K.; Felton, C.M.; et al. Mild steel welding fume causes manganese accumulation and subtle neuroinflammatory changes but not overt neuronal damage in discrete brain regions of rats after short-term inhalation exposure. Neurotoxicology 2009, 30, 915–925. [Google Scholar] [CrossRef]
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; Roberts, J.R.; Chapman, R.S.; Chen, B.T.; Soukup, J.M.; Ghio, A.J.; Antonini, J.M. Dopaminergic neurotoxicity following pulmonary exposure to manganese-containing welding fumes. Arch. Toxicol. 2010, 84, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Ajtai, B.M.; Kálmán, M. Glial fibrillary acidic protein expression but no glial demarcation follows the lesion in the molecular layer of cerebellum. Brain Res. 1998, 802, 285–288. [Google Scholar] [CrossRef]
- Krum, J.M.; Phillips, T.M.; Rosenstein, J.M. Changes in astroglial GLT-1 expression after neural transplantation or stab wounds. Exp. Neurol. 2002, 174, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Isono, M.; Goda, M.; Kobayashi, H.; Wu, J.L. TGF-alpha overexpression induces astrocytic hypertrophy after cortical stab wound injury. Neurol. Res. 2003, 25, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Laskawi, R.; Rohlmann, A.; Landgrebe, M.; Wolff, J.R. Rapid astroglial reactions in the motor cortex of adult rats following peripheral facial nerve lesions. Eur. Arch. Otorhinolaryngol. 1997, 254, 81–85. [Google Scholar] [CrossRef]
- Carbonell, W.S.; Mandell, J.W. Transient neuronal but persistent astroglial activation of ERK/MAP kinase after focal brain injury in mice. J. Neurotrauma 2003, 20, 327–336. [Google Scholar] [CrossRef]
- Zhu, W.; Umegaki, H.; Shinkai, T.; Kurotani, S.; Suzuki, Y.; Endo, H.; Iguchi, A. Different glial reactions to hippocampal stab wounds in young adult and aged rats. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 117–122. [Google Scholar] [CrossRef]
- Cheung, W.M.; Wang, C.K.; Kuo, J.S.; Lin, T.N. Changes in the level of glial fibrillary acidic protein (GFAP) after mild and severe focal cerebral ischemia. Chin. J. Physiol. 1999, 42, 227–235. [Google Scholar]
- Endoh, M.; Maiese, K.; Wagner, J. Expression of the inducible form of nitric oxide synthase by reactive astrocytes after transient global ischemia. Brain Res. 1994, 651, 92–100. [Google Scholar] [CrossRef]
- Soltys, Z.; Janeczko, K.; Orzyłowska-Sliwińska, O.; Zaremba, M.; Januszewski, S.; Oderfeld-Nowak, B. Morphological transformations of cells immunopositive for GFAP, TrkA or p75 in the CA1 hippocampal area following transient global ischemia in the rat. A quantitative study. Brain Res. 2003, 987, 186–193. [Google Scholar] [CrossRef]
- Butler, T.L.; Kassed, C.A.; Sanberg, P.R.; Willing, A.E.; Pennypacker, K.R. Neurodegeneration in the rat hippocampus and striatum after middle cerebral artery occlusion. Brain Res. 2002, 929, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y.P.; Cai, J.; Shields, L.B.; Tuchek, C.A.; Shi, R.; Li, J.; Shields, C.B.; Xu, X.M. A Compact Blast-Induced Traumatic Brain Injury Model in Mice. J. Neuropathol. Exp. Neurol. 2016, 75, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Witcher, K.G.; Bray, C.E.; Chunchai, T.; Zhao, F.; O’Neil, S.M.; Gordillo, A.J.; Campbell, W.A.; McKim, D.B.; Liu, X.; Dziabis, J.E.; et al. Traumatic Brain Injury Causes Chronic Cortical Inflammation and Neuronal Dysfunction Mediated by Microglia. J. Neurosci. 2021, 41, 1597–1616. [Google Scholar] [CrossRef] [PubMed]
- Ravula, A.R.; Rodriguez, J.; Younger, D.; Perumal, V.; Shao, N.; Rama Rao, K.V.; Pfister, B.; Chandra, N. Animal model of repeated low-level blast traumatic brain injury displays acute and chronic neurobehavioral and neuropathological changes. Exp. Neurol. 2022, 349, 113938. [Google Scholar] [CrossRef]
- Panter, S.S.; McSwigan, J.D.; Sheppard, J.R.; Emory, C.R.; Frey, W.H., 2nd. Glial fibrillary acidic protein and Alzheimer’s disease. Neurochem. Res. 1985, 10, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Morimatsu, M.; Hirai, S.; Takahashi, K. Alzheimer’s neurofibrillary tangles are penetrated by astroglial processes and appear eosinophilic in their final stages. Acta Neuropathol. 1987, 72, 214–217. [Google Scholar] [CrossRef]
- Delacourte, A. General and dramatic glial reaction in Alzheimer brains. Neurology 1990, 40, 33–37. [Google Scholar] [CrossRef]
- Vijayan, V.K.; Geddes, J.W.; Anderson, K.J.; Chang-Chui, H.; Ellis, W.G.; Cotman, C.W. Astrocyte hypertrophy in the Alzheimer’s disease hippocampal formation. Exp. Neurol. 1991, 112, 72–78. [Google Scholar] [CrossRef]
- Cullen, K.M. Perivascular astrocytes within Alzheimer’s disease plaques. Neuroreport 1997, 8, 1961–1966. [Google Scholar] [CrossRef]
- Kumar, A.; Koistinen, N.A.; Malarte, M.L.; Nennesmo, I.; Ingelsson, M.; Ghetti, B.; Lemoine, L.; Nordberg, A. Astroglial tracer BU99008 detects multiple binding sites in Alzheimer’s disease brain. Mol. Psychiatry 2021, 26, 5833–5847. [Google Scholar] [CrossRef]
- Astillero-Lopez, V.; Gonzalez-Rodriguez, M.; Villar-Conde, S.; Flores-Cuadrado, A.; Martinez-Marcos, A.; Ubeda-Banon, I.; Saiz-Sanchez, D. Neurodegeneration and astrogliosis in the entorhinal cortex in Alzheimer’s disease: Stereological layer-specific assessment and proteomic analysis. Alzheimers Dement. 2022, 18, 2468–2480. [Google Scholar] [CrossRef] [PubMed]
- Harpin, M.L.; Delaère, P.; Javoy-Agid, F.; Bock, E.; Jacque, C.; Delpech, B.; Villarroya, H.; Duyckaerts, C.; Hauw, J.J.; Baumann, N. Glial fibrillary acidic protein and beta A4 protein deposits in temporal lobe of aging brain and senile dementia of the Alzheimer type: Relation with the cognitive state and with quantitative studies of senile plaques and neurofibrillary tangles. J. Neurosci. Res. 1990, 27, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Troost, D.; Sillevis Smitt, P.A.; de Jong, J.M.; Swaab, D.F. Neurofilament and glial alterations in the cerebral cortex in amyotrophic lateral sclerosis. Acta Neuropathol. 1992, 84, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Kobayashi, K.; Isaki, K. Microglial and astrocytic change in brains of Creutzfeldt-Jakob disease: An immunocytochemical and quantitative study. Clin. Neuropathol. 1999, 18, 51–60. [Google Scholar] [PubMed]
- Zanusso, G.; Colaizzo, E.; Poleggi, A.; Masullo, C.; Romeo, R.; Ferrari, S.; Bongianni, M.; Fiorini, M.; Tiple, D.; Vaianella, L.; et al. Biochemical and Neuropathological Findings in a Creutzfeldt-Jakob Disease Patient with the Rare Val180Ile-129Val Haplotype in the Prion Protein Gene. Int. J. Mol. Sci. 2022, 23, 10210. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Eikelenboom, M.J.; Gveric, D.; Keir, G.; Chapman, M.; Lazeron, R.H.; Cuzner, M.L.; Polman, C.H.; Uitdehaag, B.M.; Thompson, E.J.; et al. Markers for different glial cell responses in multiple sclerosis: Clinical and pathological correlations. Brain 2002, 125 Pt 7, 1462–1473. [Google Scholar] [CrossRef]
- Nowacki, P.; Koziarska, D.; Masztalewicz, M. Microglia and astroglia proliferation within the normal appearing white matter in histologically active and inactive multiple sclerosis. Folia Neuropathol. 2019, 57, 249–257. [Google Scholar] [CrossRef]
- Hishikawa, N.; Hashizume, Y.; Yoshida, M.; Sobue, G. Widespread occurrence of argyrophilic glial inclusions in Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2001, 27, 362–372. [Google Scholar] [CrossRef]
- Flores-Cuadrado, A.; Saiz-Sanchez, D.; Mohedano-Moriano, A.; Lamas-Cenjor, E.; Leon-Olmo, V.; Martinez-Marcos, A.; Ubeda-Bañon, I. Astrogliosis and sexually dimorphic neurodegeneration and microgliosis in the olfactory bulb in Parkinson’s disease. NPJ Parkinson’s Dis. 2021, 7, 11. [Google Scholar] [CrossRef]
- Arnold, S.E.; Franz, B.R.; Trojanowski, J.Q.; Moberg, P.J.; Gur, R.E. Glial fibrillary acidic protein-immunoreactive astrocytosis in elderly patients with schizophrenia and dementia. Acta Neuropathol. 1996, 91, 269–277. [Google Scholar] [CrossRef]
- Zhang, C.; Hou, L.; Yang, J.; Che, Y.; Sun, F.; Li, H.; Wang, Q. 2,5-Hexanedione induces dopaminergic neurodegeneration through integrin αMβ2/NADPH oxidase axis-mediated microglial activation. Cell Death Dis. 2018, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Zhang, X.; Jiao, Y.; Duan, L.; Dai, L.; Yan, H. Chronic acrylamide exposure resulted in dopaminergic neuron loss, neuroinflammation and motor impairment in rats. Toxicol. Appl. Pharmacol. 2022, 451, 116190. [Google Scholar] [CrossRef]
- Platt, B.; Fiddler, G.; Riedel, G.; Henderson, Z. Aluminium toxicity in the rat brain: Histochemical and immunocytochemical evidence. Brain Res. Bull. 2001, 55, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Tiwari, M.N.; Dixit, A.; Upadhyay, G.; Patel, D.K.; Singh, D.; Prakash, O.; Singh, M.P. Nigrostriatal proteomics of cypermethrin-induced dopaminergic neurodegeneration: Microglial activation-dependent and -independent regulations. Toxicol. Sci. 2011, 122, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Hubbs, A.F.; Fluharty, K.L.; Edwards, R.J.; Barnabei, J.L.; Grantham, J.T.; Palmer, S.M.; Kelly, F.; Sargent, L.M.; Reynolds, S.H.; Mercer, R.R.; et al. Accumulation of Ubiquitin and Sequestosome-1 Implicate Protein Damage in Diacetyl-Induced Cytotoxicity. Am. J. Pathol. 2016, 186, 2887–2908. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Binukumar, B.K.; Bal, A.; Gill, K.D. Chronic dichlorvos exposure: Microglial activation, proinflammatory cytokines and damage to nigrostriatal dopaminergic system. NeuroMolecular Med. 2011, 13, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Levesque, S.; Taetzsch, T.; Lull, M.E.; Kodavanti, U.; Stadler, K.; Wagner, A.; Johnson, J.A.; Duke, L.; Kodavanti, P.; Surace, M.J.; et al. Diesel exhaust activates and primes microglia: Air pollution, neuroinflammation, and regulation of dopaminergic neurotoxicity. Environ. Health Perspect. 2011, 119, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Zucconi, G.G.; Laurenzi, M.A.; Semprevivo, M.; Torni, F.; Lindgren, J.A.; Marinucci, E. Microglia activation and cell death in response to diethyl-dithiocarbamate acute administration. J. Comp. Neurol. 2002, 446, 135–150. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, B.; Zhu, M.T.; Li, M.; Wang, H.J.; Wang, M.; Ouyang, H.; Chai, Z.F.; Feng, W.Y.; Zhao, Y.L. Microglial activation, recruitment and phagocytosis as linked phenomena in ferric oxide nanoparticle exposure. Toxicol. Lett. 2011, 205, 26–37. [Google Scholar] [CrossRef]
- Zhao, F.; Cai, T.; Liu, M.; Zheng, G.; Luo, W.; Chen, J. Manganese induces dopaminergic neurodegeneration via microglial activation in a rat model of manganism. Toxicol. Sci. 2009, 107, 156–164. [Google Scholar] [CrossRef]
- Gonzalez-Cuyar, L.F.; Nelson, G.; Criswell, S.R.; Ho, P.; Lonzanida, J.A.; Checkoway, H.; Seixas, N.; Gelman, B.B.; Evanoff, B.A.; Murray, J.; et al. Quantitative neuropathology associated with chronic manganese exposure in South African mine workers. Neurotoxicology 2014, 45, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Zhang, C.; Wang, K.; Liu, X.; Wang, H.; Che, Y.; Sun, F.; Zhou, X.; Zhao, X.; Wang, Q. Paraquat and maneb co-exposure induces noradrenergic locus coeruleus neurodegeneration through NADPH oxidase-mediated microglial activation. Toxicology 2017, 380, 1–10. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Kim, J.H.; Greenamyre, J.T. Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci. Lett. 2003, 341, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, S.; Hou, L.; Jing, L.; Ruan, Z.; Peng, B.; Zhang, X.; Hong, J.S.; Zhao, J.; Wang, Q. Microglial activation contributes to cognitive impairments in rotenone-induced mouse Parkinson’s disease model. J. Neuroinflamm. 2021, 18, 4. [Google Scholar] [CrossRef]
- Patchin, E.S.; Anderson, D.S.; Silva, R.M.; Uyeminami, D.L.; Scott, G.M.; Guo, T.; Van Winkle, L.S.; Pinkerton, K.E. Size-Dependent Deposition, Translocation, and Microglial Activation of Inhaled Silver Nanoparticles in the Rodent Nose and Brain. Environ. Health Perspect. 2016, 124, 1870–1875. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Upadhyay, P.; Lopez, C.H.; Miyashiro, M.K.; Van Winkle, L.S.; Thomasy, S.M.; Pinkerton, K.E. Inhalation of Silver Silicate Nanoparticles Leads to Transient and Differential Microglial Activation in the Rodent Olfactory Bulb. Toxicol. Pathol. 2022, 50, 763–775. [Google Scholar] [CrossRef]
- Koczyk, D.; Oderfeld-Nowak, B. Long-term microglial and astroglial activation in the hippocampus of trimethyltin-intoxicated rat: Stimulation of NGF and TrkA immunoreactivities in astroglia but not in microglia. Int. J. Dev. Neurosci. 2000, 18, 591–606. [Google Scholar] [CrossRef]
- Sriram, K.; Jefferson, A.M.; Lin, G.X.; Afshari, A.; Zeidler-Erdely, P.C.; Meighan, T.G.; McKinney, W.; Jackson, M.; Cumpston, A.; Cumpston, J.L.; et al. Neurotoxicity following acute inhalation of aerosols generated during resistance spot weld-bonding of carbon steel. Inhal. Toxicol. 2014, 26, 720–732. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; Stone, S.; Afshari, A.; Keane, M.J.; McKinney, W.; Jackson, M.; Chen, B.T.; Schwegler-Berry, D.; et al. Modifying welding process parameters can reduce the neurotoxic potential of manganese-containing welding fumes. Toxicology 2015, 328, 168–178. [Google Scholar] [CrossRef]
- Katchanov, J.; Waeber, C.; Gertz, K.; Gietz, A.; Winter, B.; Brück, W.; Dirnagl, U.; Veh, R.W.; Endres, M. Selective neuronal vulnerability following mild focal brain ischemia in the mouse. Brain Pathol. 2003, 13, 452–464. [Google Scholar] [CrossRef]
- Huh, Y.; Jung, J.W.; Park, C.; Ryu, J.R.; Shin, C.Y.; Kim, W.K.; Ryu, J.H. Microglial activation and tyrosine hydroxylase immunoreactivity in the substantia nigral region following transient focal ischemia in rats. Neurosci. Lett. 2003, 349, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Ginsberg, M.D.; Busto, R.; Dietrich, W.D. Sequential analysis of subacute and chronic neuronal, astrocytic and microglial alterations after transient global ischemia in rats. Acta Neuropathol. 1998, 95, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Dihné, M.; Block, F. Focal ischemia induces transient expression of IL-6 in the substantia nigra pars reticulata. Brain Res. 2001, 889, 165–173. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Styren, S.D.; O’Malley, M.E.; Goss, J.R.; Kochanek, P.; Marion, D.; Evans, C.H.; Robbins, P.D. Interleukin-1 receptor antagonist suppresses neurotrophin response in injured rat brain. Ann. Neurol. 1996, 39, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.; Zetterberg, H.; Jolly, A.; Cole, J.H.; De Simoni, S.; Jenkins, P.O.; Feeney, C.; Owen, D.R.; Lingford-Hughes, A.; Howes, O.; et al. Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain 2018, 141, 459–471. [Google Scholar] [CrossRef]
- Wierzba-Bobrowicz, T.; Gwiazda, E.; Kosno-Kruszewska, E.; Lewandowska, E.; Lechowicz, W.; Bertrand, E.; Szpak, G.M.; Schmidt-Sidor, B. Morphological analysis of active microglia--rod and ramified microglia in human brains affected by some neurological diseases (SSPE, Alzheimer’s disease and Wilson’s disease). Folia Neuropathol. 2002, 40, 125–131. [Google Scholar]
- Versijpt, J.J.; Dumont, F.; Van Laere, K.J.; Decoo, D.; Santens, P.; Audenaert, K.; Achten, E.; Slegers, G.; Dierckx, R.A.; Korf, J. Assessment of neuroinflammation and microglial activation in Alzheimer’s disease with radiolabelled PK11195 and single photon emission computed tomography. A pilot study. Eur. Neurol. 2003, 50, 39–47. [Google Scholar] [CrossRef]
- Walker, D.G.; Tang, T.M.; Lue, L.F. Studies on Colony Stimulating Factor Receptor-1 and Ligands Colony Stimulating Factor-1 and Interleukin-34 in Alzheimer’s Disease Brains and Human Microglia. Front. Aging Neurosci. 2017, 9, 244. [Google Scholar] [CrossRef]
- Singh-Bains, M.K.; Linke, V.; Austria, M.D.R.; Tan, A.Y.S.; Scotter, E.L.; Mehrabi, N.F.; Faull, R.L.M.; Dragunow, M. Altered microglia and neurovasculature in the Alzheimer’s disease cerebellum. Neurobiol. Dis. 2019, 132, 104589. [Google Scholar] [CrossRef]
- Fixemer, S.; Ameli, C.; Hammer, G.; Salamanca, L.; Uriarte Huarte, O.; Schwartz, C.; Gérardy, J.J.; Mechawar, N.; Skupin, A.; Mittelbronn, M.; et al. Microglia phenotypes are associated with subregional patterns of concomitant tau, amyloid-β and α-synuclein pathologies in the hippocampus of patients with Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. Commun. 2022, 10, 36. [Google Scholar] [CrossRef]
- Fracassi, A.; Marcatti, M.; Tumurbaatar, B.; Woltjer, R.; Moreno, S.; Taglialatela, G. TREM2-induced activation of microglia contributes to synaptic integrity in cognitively intact aged individuals with Alzheimer’s neuropathology. Brain Pathol. 2022, 33, e13108. [Google Scholar] [CrossRef] [PubMed]
- Cagnin, A.; Brooks, D.J.; Kennedy, A.M.; Gunn, R.N.; Myers, R.; Turkheimer, F.E.; Jones, T.; Banati, R.B. In-vivo measurement of activated microglia in dementia. Lancet 2001, 358, 461–467, Erratum in Lancet 2001, 358, 766. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.M.; Grace, G.M.; Munoz, D.G.; He, B.P.; Strong, M.J. Cognitive impairment in sporadic ALS: A pathologic continuum underlying a multisystem disorder. Neurology 2001, 57, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Libon, D.J.; Toledo, J.B.; Xie, S.X.; McCluskey, L.; Elman, L.; Geser, F.; Lee, V.M.; Grossman, M.; Trojanowski, J.Q. Microglial activation and TDP-43 pathology correlate with executive dysfunction in amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 123, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Takao, M.; Hatsuta, H.; Funabe, S.; Ito, S.; Obi, T.; Tanaka, F.; Kuroiwa, Y.; Murayama, S. Increased number of astrocytes and macrophages/microglial cells in the corpus callosum in amyotrophic lateral sclerosis. Neuropathology 2013, 33, 591–599. [Google Scholar] [CrossRef]
- Gray, F.; Chrétien, F.; Adle-Biassette, H.; Dorandeu, A.; Ereau, T.; Delisle, M.B.; Kopp, N.; Ironside, J.W.; Vital, C. Neuronal apoptosis in Creutzfeldt-Jakob disease. J. Neuropathol. Exp. Neurol. 1999, 58, 321–328. [Google Scholar] [CrossRef]
- Schwarz, S.C.; Seufferlein, T.; Liptay, S.; Schmid, R.M.; Kasischke, K.; Foster, O.J.; Daniel, S.; Schwarz, J. Microglial activation in multiple system atrophy: A potential role for NF-kappaB/rel proteins. Neuroreport 1998, 9, 3029–3032. [Google Scholar] [CrossRef]
- Banati, R.B.; Daniel, S.E.; Blunt, S.B. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov. Disord. 1998, 13, 221–227. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Breidert, T.; Rousselet, E.; Hunot, S.; Hartmann, A.; Michel, P.P. The role of glial reaction and inflammation in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2003, 991, 214–228. [Google Scholar] [CrossRef]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef]
- Harry, G.J.; Schmitt, T.J.; Gong, Z.; Brown, H.; Zawia, N.; Evans, H.L. Lead-induced alterations of glial fibrillary acidic protein (GFAP) in the developing rat brain. Toxicol. Appl. Pharmacol. 1996, 139, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.R.; Liu, J.C.; Bao, J.S.; Bai, Q.Q.; Wang, G.Q. Interaction of Microglia and Astrocytes in the Neurovascular Unit. Front. Immunol. 2020, 11, 1024. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275 Pt 3, 305–315. [Google Scholar] [CrossRef]
- Murakami, M.; Hirano, T. The molecular mechanisms of chronic inflammation development. Front. Immunol. 2012, 3, 323. [Google Scholar] [CrossRef]
- Yeh, H.; Ikezu, T. Transcriptional and Epigenetic Regulation of Microglia in Health and Disease. Trends Mol. Med. 2019, 25, 96–111. [Google Scholar] [CrossRef]
- Cramer, T.; Gill, R.; Thirouin, Z.S.; Vaas, M.; Sampath, S.; Martineau, F.; Noya, S.B.; Panzanelli, P.; Sudharshan, T.J.J.; Colameo, D.; et al. Cross-talk between GABAergic postsynapse and microglia regulate synapse loss after brain ischemia. Sci. Adv. 2022, 8, eabj0112. [Google Scholar] [CrossRef]
- Van Everbroeck, B.; Dobbeleir, I.; De Waele, M.; De Leenheir, E.; Lübke, U.; Martin, J.J.; Cras, P. Extracellular protein deposition correlates with glial activation and oxidative stress in Creutzfeldt-Jakob and Alzheimer’s disease. Acta Neuropathol. 2004, 108, 194–200. [Google Scholar] [CrossRef]
- Neidert, N.; von Ehr, A.; Zöller, T.; Spittau, B. Microglia-Specific Expression of Olfml3 Is Directly Regulated by Transforming Growth Factor β1-Induced Smad2 Signaling. Front. Immunol. 2018, 9, 1728. [Google Scholar] [CrossRef]
- Sears, J.M.; Graves, J.M.; Blanar, L.; Bowman, S.M. Case identification of work-related traumatic brain injury using the occupational injury and illness classification system. J. Occup. Environ. Med. 2013, 55, 507–513. [Google Scholar] [CrossRef]
- Dorsett, C.R.; McGuire, J.L.; DePasquale, E.A.; Gardner, A.E.; Floyd, C.L.; McCullumsmith, R.E. Glutamate Neurotransmission in Rodent Models of Traumatic Brain Injury. J. Neurotrauma 2017, 34, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Hou, J.; Lu, J.; Zhu, Z.; Yang, Y.; Peng, W.; Pi, R. A novel simple traumatic brain injury mouse model. Chin. Neurosurg. J. 2022, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [PubMed]
- Faul, M.; Wald, M.M.; Xu, L.; Coronado, V.G. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations, and Deaths 2002–2006; US Department of Health and Human Services, CDC: Atlanta, GA, USA, 2010. [Google Scholar]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 572, Erratum in Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef]
- Peterson, A.B.; Zhou, H.; Thomas, K.E. Disparities in traumatic brain injury-related deaths-United States, 2020. J. Saf. Res. 2022, 83, 419–426. [Google Scholar] [CrossRef] [PubMed]
- De Crescenzo, L.A.; Gabella, B.A.; Johnson, J. Interrupted time series design to evaluate ICD-9-CM to ICD-10-CM coding changes on trends in Colorado emergency department visits related to traumatic brain injury. Inj. Epidemiol. 2021, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.; Heitger, M.; Macleod, A.D. Concussion and mild head injury. Pract. Neurol. 2006, 6, 342–357. [Google Scholar] [CrossRef]
- Available online: https://www.cdc.gov/headsup/basics/concussion_whatis.html (accessed on 1 December 2022).
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Kenny, E.M.; Bayır, H. Therapies targeting lipid peroxidation in traumatic brain injury. Brain Res. 2016, 1640, 57–76. [Google Scholar] [CrossRef]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef]
- Tyburski, A.L.; Cheng, L.; Assari, S.; Darvish, K.; Elliott, M.B. Frequent mild head injury promotes trigeminal sensitivity concomitant with microglial proliferation, astrocytosis, and increased neuropeptide levels in the trigeminal pain system. J. Headache Pain 2017, 18, 16. [Google Scholar] [CrossRef] [PubMed]
- Verboon, L.N.; Patel, H.C.; Greenhalgh, A.D. The Immune System’s Role in the Consequences of Mild Traumatic Brain Injury (Concussion). Front. Immunol. 2021, 12, 620698. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Tripodis, Y.; Alvarez, V.E.; Huber, B.; Kiernan, P.T.; Daneshvar, D.H.; Mez, J.; Montenigro, P.H.; Solomon, T.M.; Alosco, M.L.; et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol. Commun. 2016, 4, 112. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Nayak, D.; Atanasijevic, T.; Koretsky, A.P.; Latour, L.L.; McGavern, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014, 505, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Madathil, S.K.; Wilfred, B.S.; Urankar, S.E.; Yang, W.; Leung, L.Y.; Gilsdorf, J.S.; Shear, D.A. Early Microglial Activation Following Closed-Head Concussive Injury Is Dominated by Pro-Inflammatory M-1 Type. Front. Neurol. 2018, 9, 964. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist 2014, 20, 160–172. [Google Scholar] [CrossRef]
- Witcher, K.G.; Eiferman, D.S.; Godbout, J.P. Priming the inflammatory pump of the CNS after traumatic brain injury. Trends Neurosci. 2015, 38, 609–620. [Google Scholar] [CrossRef]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 Adrenergic-Neurotrophin Feedforward Loop Promotes Pancreatic Cancer. Cancer Cell. 2018, 34, 863–867, Erratum in Cancer Cell. 2018, 33, 75–90.e7. [Google Scholar] [CrossRef]
- Available online: https://ntp.niehs.nih.gov/whatwestudy/tox21/history/index.html (accessed on 1 December 2022).
- Thomas, R.S.; Paules, R.S.; Simeonov, A.; Fitzpatrick, S.C.; Crofton, K.M.; Casey, W.M.; Mendrick, D.L. The US Federal Tox21 Program: A strategic and operational plan for continued leadership. ALTEX 2018, 35, 163–168. [Google Scholar] [CrossRef]
- Richard, A.M.; Huang, R.; Waidyanatha, S.; Shinn, P.; Collins, B.J.; Thillainadarajah, I.; Grulke, C.M.; Williams, A.J.; Lougee, R.R.; Judson, R.S.; et al. The Tox21 10K Compound Library: Collaborative Chemistry Advancing Toxicology. Chem. Res. Toxicol. 2021, 34, 189–216. [Google Scholar] [CrossRef]
- Pezzoli, G.; Strada, O.; Silani, V.; Zecchinelli, A.; Perbellini, L.; Javoy-Agid, F.; Ghidoni, P.; Motti, E.D.; Masini, T.; Scarlato, G.; et al. Clinical and pathological features in hydrocarbon-induced parkinsonism. Ann. Neurol. 1996, 40, 922–925. [Google Scholar] [CrossRef]
- Pezzoli, G.; Canesi, M.; Antonini, A.; Righini, A.; Perbellini, L.; Barichella, M.; Mariani, C.B.; Tenconi, F.; Tesei, S.; Zecchinelli, A.; et al. Hydrocarbon exposure and Parkinson’s disease. Neurology 2000, 55, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.C.; Chang, L.H.; Huang, S.S.; Huang, Y.J.; Chih, C.L.; Kuo, H.C.; Lee, Y.H.; Lee, I.H. Aryl hydrocarbon receptor modulates stroke-induced astrogliosis and neurogenesis in the adult mouse brain. J. Neuroinflamm. 2019, 16, 187. [Google Scholar] [CrossRef] [PubMed]
- Ramos-García, N.A.; Orozco-Ibarra, M.; Estudillo, E.; Elizondo, G.; Gómez Apo, E.; Chávez Macías, L.G.; Sosa-Ortiz, A.L.; Torres-Ramos, M.A. Aryl Hydrocarbon Receptor in Post-Mortem Hippocampus and in Serum from Young, Elder, and Alzheimer’s Patients. Int. J. Mol. Sci. 2020, 21, 1983. [Google Scholar] [CrossRef]
- Cho, J.; Sohn, J.; Noh, J.; Jang, H.; Kim, W.; Cho, S.K.; Seo, H.; Seo, G.; Lee, S.K.; Noh, Y.; et al. Association between exposure to polycyclic aromatic hydrocarbons and brain cortical thinning: The Environmental Pollution-Induced Neurological EFfects (EPINEF) study. Sci. Total Environ. 2020, 737, 140097. [Google Scholar] [CrossRef]
- Kim, Y.T.; Kim, W.; Bae, M.J.; Choi, J.E.; Kim, M.J.; Oh, S.S.; Park, K.S.; Park, S.; Lee, S.K.; Koh, S.B.; et al. The effect of polycyclic aromatic hydrocarbons on changes in the brain structure of firefighters: An analysis using data from the Firefighters Research on Enhancement of Safety & Health study. Sci. Total Environ. 2022, 816, 151655. [Google Scholar] [CrossRef]
- Macchi, Z.A.; Carlisle, T.C.; Filley, C.M. Prognosis in substance abuse-related acute toxic leukoencephalopathy: A scoping review. J. Neurol. Sci. 2022, 442, 120420. [Google Scholar] [CrossRef]
- Naskali, L.; Oksanen, H.; Tähti, H. Astrocytes as targets for CNS effects of organic solvents in vitro. Neurotoxicology 1994, 15, 609–612. [Google Scholar]
- Yamaguchi, H.; Kidachi, Y.; Ryoyama, K. Toluene at environmentally relevant low levels disrupts differentiation of astrocyte precursor cells. Arch. Environ. Health 2002, 57, 232–238. [Google Scholar] [CrossRef]
- Riegel, A.C.; Zapata, A.; Shippenberg, T.S.; French, E.D. The abused inhalant toluene increases dopamine release in the nucleus accumbens by directly stimulating ventral tegmental area neurons. Neuropsychopharmacology 2007, 32, 1558–1569. [Google Scholar] [CrossRef]
- Beckley, J.T.; Woodward, J.J. The abused inhalant toluene differentially modulates excitatory and inhibitory synaptic transmission in deep-layer neurons of the medial prefrontal cortex. Neuropsychopharmacology 2011, 36, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.H.; Tang, Y.C.; Chien, T.H.; Chen, H.H. Toluene exposure during the brain growth spurt reduced behavioral responses to nicotine in young adult rats: A potential role for nicotinic acetylcholine receptors in fetal solvent syndrome. Toxicol. Sci. 2008, 101, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Gotohda, T.; Tokunaga, I.; Kubo, S.; Kitamura, O.; Ishigami, A. Toluene inhalation induces glial cell line-derived neurotrophic factor, transforming growth factor and tumor necrosis factor in rat cerebellum. Leg. Med. 2002, 4, 21–28. [Google Scholar] [CrossRef]
- Montes, S.; Yee-Rios, Y.; Páez-Martínez, N. Environmental enrichment restores oxidative balance in animals chronically exposed to toluene: Comparison with melatonin. Brain Res. Bull. 2019, 144, 58–67. [Google Scholar] [CrossRef]
- Zheng, W.; Monnot, A.D. Regulation of brain iron and copper homeostasis by brain barrier systems: Implication in neurodegenerative diseases. Pharmacol. Ther. 2012, 133, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef]
- Li, W.J.; Jiang, H.; Song, N.; Xie, J.X. Dose- and time-dependent alpha-synuclein aggregation induced by ferric iron in SK-N-SH cells. Neurosci. Bull. 2010, 26, 205–210. [Google Scholar] [CrossRef]
- Kenkhuis, B.; Somarakis, A.; de Haan, L.; Dzyubachyk, O.; IJsselsteijn, M.E.; de Miranda, N.F.C.C.; Lelieveldt, B.P.F.; Dijkstra, J.; van Roon-Mom, W.M.C.; Höllt, T.; et al. Iron loading is a prominent feature of activated microglia in Alzheimer’s disease patients. Acta Neuropathol. Commun. 2021, 9, 27. [Google Scholar] [CrossRef]
- Laabbar, W.; Abbaoui, A.; Elgot, A.; Mokni, M.; Amri, M.; Masmoudi-Kouki, O.; Gamrani, H. Aluminum induced oxidative stress, astrogliosis and cell death in rat astrocytes, is prevented by curcumin. J. Chem. Neuroanat. 2021, 112, 101915. [Google Scholar] [CrossRef] [PubMed]
- Walton, J.R.; Wang, M.X. APP expression, distribution and accumulation are altered by aluminum in a rodent model for Alzheimer’s disease. J. Inorg. Biochem. 2009, 103, 1548–1554. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Zhao, Y.; Hill, J.M.; Percy, M.E.; Lukiw, W.J. Aluminum and its potential contribution to Alzheimer’s disease (AD). Front. Aging Neurosci. 2014, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bhattacharjee, S.; Jones, B.M.; Hill, J.; Dua, P.; Lukiw, W.J. Regulation of neurotropic signaling by the inducible, NF-kB-sensitive miRNA-125b in Alzheimer’s disease (AD) and in primary human neuronal-glial (HNG) cells. Mol. Neurobiol. 2014, 50, 97–106. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell Neurosci. 2015, 9, 124. [Google Scholar] [CrossRef]
- Rodier, J. Manganese poisoning in Moroccan miners. Br. J. Ind. Med. 1955, 12, 21–35. [Google Scholar] [CrossRef]
- Donaldson, J. The physiopathologic significance of manganese in brain: Its relation to schizophrenia and neurodegenerative disorders. Neurotoxicology. 1987, 8, 451–462. [Google Scholar] [PubMed]
- Roels, H.; Lauwerys, R.; Buchet, J.P.; Genet, P.; Sarhan, M.J.; Hanotiau, I.; de Fays, M.; Bernard, A.; Stanescu, D. Epidemiological survey among workers exposed to manganese: Effects on lung, central nervous system, and some biological indices. Am. J. Ind. Med. 1987, 11, 307–327, Erratum in Am. J. Ind. Med. 1987, 12, 119–120. [Google Scholar] [CrossRef]
- Mergler, D.; Huel, G.; Bowler, R.; Iregren, A.; Bélanger, S.; Baldwin, M.; Tardif, R.; Smargiassi, A.; Martin, L. Nervous system dysfunction among workers with long-term exposure to manganese. Environ. Res. 1994, 64, 151–180. [Google Scholar] [CrossRef] [PubMed]
- Mergler, D.; Baldwin, M. Early manifestations of manganese neurotoxicity in humans: An update. Environ. Res. 1997, 73, 92–100. [Google Scholar] [CrossRef]
- Bader, M.; Dietz, M.C.; Ihrig, A.; Triebig, G. Biomonitoring of manganese in blood, urine and axillary hair following low-dose exposure during the manufacture of dry cell batteries. Int. Arch. Occup. Environ. Health 1999, 72, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Lucchini, R.G.; Albini, E.; Benedetti, L.; Borghesi, S.; Coccaglio, R.; Malara, E.C.; Parrinello, G.; Garattini, S.; Resola, S.; Alessio, L. High prevalence of Parkinsonian disorders associated to manganese exposure in the vicinities of ferroalloy industries. Am. J. Ind. Med. 2007, 50, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Bowler, R.M.; Nakagawa, S.; Drezgic, M.; Roels, H.A.; Park, R.M.; Diamond, E.; Mergler, D.; Bouchard, M.; Bowler, R.P.; Koller, W. Sequelae of fume exposure in confined space welding: A neurological and neuropsychological case series. Neurotoxicology 2007, 28, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Bowler, R.M.; Roels, H.A.; Nakagawa, S.; Drezgic, M.; Diamond, E.; Park, R.; Koller, W.; Bowler, R.P.; Mergler, D.; Bouchard, M.; et al. Dose-effect relationships between manganese exposure and neurological, neuropsychological and pulmonary function in confined space bridge welders. Occup. Environ. Med. 2007, 64, 167–177. [Google Scholar] [CrossRef]
- Guilarte, T.R. Manganese neurotoxicity: New perspectives from behavioral, neuroimaging, and neuropathological studies in humans and non-human primates. Front. Aging Neurosci. 2013, 5, 23. [Google Scholar] [CrossRef]
- Park, R.M. Neurobehavioral deficits and parkinsonism in occupations with manganese exposure: A review of methodological issues in the epidemiological literature. Saf. Health Work 2013, 4, 123–135. [Google Scholar] [CrossRef]
- Verina, T.; Schneider, J.S.; Guilarte, T.R. Manganese exposure induces α-synuclein aggregation in the frontal cortex of non-human primates. Toxicol. Lett. 2013, 217, 177–183. [Google Scholar] [CrossRef]
- Aschner, M.; Aschner, J.L. Manganese transport across the blood-brain barrier: Relationship to iron homeostasis. Brain Res. Bull. 1990, 24, 857–860. [Google Scholar] [CrossRef]
- Aschner, M.; Gannon, M. Manganese (Mn) transport across the rat blood-brain barrier: Saturable and transferrin-dependent transport mechanisms. Brain Res. Bull. 1994, 33, 345–349. [Google Scholar] [CrossRef]
- Crossgrove, J.S.; Allen, D.D.; Bukaveckas, B.L.; Rhineheimer, S.S.; Yokel, R.A. Manganese distribution across the blood-brain barrier. I. Evidence for carrier-mediated influx of managanese citrate as well as manganese and manganese transferrin. Neurotoxicology 2003, 24, 3–13. [Google Scholar] [CrossRef]
- Fitsanakis, V.A.; Piccola, G.; Marreilha dos Santos, A.P.; Aschner, J.L.; Aschner, M. Putative proteins involved in manganese transport across the blood-brain barrier. Hum. Exp. Toxicol. 2007, 26, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.E.; Umbreit, J.N.; Moore, E.G.; Hainsworth, L.N.; Porubcin, M.; Simovich, M.J.; Nakada, M.T.; Dolan, K.; Garrick, M.D. Separate pathways for cellular uptake of ferric and ferrous iron. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G767–G774. [Google Scholar] [CrossRef] [PubMed]
- Crossgrove, J.S.; Yokel, R.A. Manganese distribution across the blood-brain barrier. IV. Evidence for brain influx through store-operated calcium channels. Neurotoxicology 2005, 26, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Finney, L.A.; O’Halloran, T.V. Transition metal speciation in the cell: Insights from the chemistry of metal ion receptors. Science 2003, 300, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.H.; Orvig, C. Boon and bane of metal ions in medicine. Science 2003, 300, 936–939. [Google Scholar] [CrossRef]
- Thomassen, Y.; Ellingsen, D.G.; Hetland, S.; Sand, G. Chemical speciation and sequential extraction of Mn in workroom aerosols: Analytical methodology and results from a field study in Mn alloy plants. J. Environ. Monit. 2001, 3, 555–559. [Google Scholar] [CrossRef]
- Reaney, S.H.; Bench, G.; Smith, D.R. Brain accumulation and toxicity of Mn(II) and Mn(III) exposures. Toxicol. Sci. 2006, 93, 114–124. [Google Scholar] [CrossRef]
- Hunter, D.D.; Undem, B.J. Identification and substance P content of vagal afferent neurons innervating the epithelium of the guinea pig trachea. Am. J. Respir. Crit. Care Med. 1999, 159, 1943–1948. [Google Scholar] [CrossRef]
- Oberdörster, G.; Sharp, Z.; Atudorei, V.; Elder, A.; Gelein, R.; Lunts, A.; Kreyling, W.; Cox, C. Extrapulmonary translocation of ultrafine carbon particles following whole-body inhalation exposure of rats. J. Toxicol. Environ. Health A 2002, 65, 1531–1543. [Google Scholar] [CrossRef]
- Oberdörster, G.; Sharp, Z.; Atudorei, V.; Elder, A.; Gelein, R.; Kreyling, W.; Cox, C. Translocation of inhaled ultrafine particles to the brain. Inhal. Toxicol. 2004, 16, 437–445. [Google Scholar] [CrossRef]
- Elder, A.; Gelein, R.; Silva, V.; Feikert, T.; Opanashuk, L.; Carter, J.; Potter, R.; Maynard, A.; Ito, Y.; Finkelstein, J.; et al. Translocation of inhaled ultrafine manganese oxide particles to the central nervous system. Environ Health Perspect. 2006, 114, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.V.; Shukla, G.S.; Srivastava, R.S.; Singh, H.; Gupta, V.P. An exploratory study of manganese exposure to welders. Clin. Toxicol. 1981, 18, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Sjogren, U.; Hagglund, B.; Sundqvist, G.; Wing, K. Factors affecting the long-term results of endodontic treatment. J. Endod. 1990, 16, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Racette, B.A.; McGee-Minnich, L.; Moerlein, S.M.; Mink, J.W.; Videen, T.O.; Perlmutter, J.S. Welding-related parkinsonism: Clinical features, treatment, and pathophysiology. Neurology 2001, 56, 8–13. [Google Scholar] [CrossRef]
- Bowler, R.M.; Gysens, S.; Diamond, E.; Booty, A.; Hartney, C.; Roels, H.A. Neuropsychological sequelae of exposure to welding fumes in a group of occupationally exposed men. Int. J. Hyg. Environ. Health 2003, 206, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Racette, B.A.; Tabbal, S.D.; Jennings, D.; Good, L.; Perlmutter, J.S.; Evanoff, B. Prevalence of parkinsonism and relationship to exposure in a large sample of Alabama welders. Neurology 2005, 64, 230–235. [Google Scholar] [CrossRef]
- Josephs, K.A.; Ahlskog, J.E.; Klos, K.J.; Kumar, N.; Fealey, R.D.; Trenerry, M.R.; Cowl, C.T. Neurologic manifestations in welders with pallidal MRI T1 hyperintensity. Neurology 2005, 64, 2033–2039. [Google Scholar] [CrossRef]
- Park, R.M.; Schulte, P.A.; Bowman, J.D.; Walker, J.T.; Bondy, S.C.; Yost, M.G.; Touchstone, J.A.; Dosemeci, M. Potential occupational risks for neurodegenerative diseases. Am. J. Ind. Med. 2005, 48, 63–77. [Google Scholar] [CrossRef]
- Park, R.M.; Bowler, R.M.; Eggerth, D.E.; Diamond, E.; Spencer, K.J.; Smith, D.; Gwiazda, R. Issues in neurological risk assessment for occupational exposures: The Bay Bridge welders. Neurotoxicology 2006, 27, 373–384. [Google Scholar] [CrossRef]
- Racette, B.A.; Searles Nielsen, S.; Criswell, S.R.; Sheppard, L.; Seixas, N.; Warden, M.N.; Checkoway, H. Dose-dependent progression of parkinsonism in manganese-exposed welders. Neurology 2017, 88, 344–351. [Google Scholar] [CrossRef]
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; Roberts, J.R.; Wirth, O.; Hayashi, Y.; Krajnak, K.M.; Soukup, J.M.; Ghio, A.J.; Reynolds, S.H.; et al. Mitochondrial dysfunction and loss of Parkinson’s disease-linked proteins contribute to neurotoxicity of manganese-containing welding fumes. FASEB J. 2010, 24, 4989–5002. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Lin, G.X.; Jefferson, A.M.; Roberts, J.R.; Andrews, R.N.; Kashon, M.L.; Antonini, J.M. Manganese accumulation in nail clippings as a biomarker of welding fume exposure and neurotoxicity. Toxicology 2012, 291, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Ze, Y.; Sheng, L.; Zhao, X.; Hong, J.; Ze, X.; Yu, X.; Pan, X.; Lin, A.; Zhao, Y.; Zhang, C.; et al. TiO2 nanoparticles induced hippocampal neuroinflammation in mice. PLoS ONE 2014, 9, e92230. [Google Scholar] [CrossRef] [PubMed]
- Disdier, C.; Chalansonnet, M.; Gagnaire, F.; Gaté, L.; Cosnier, F.; Devoy, J.; Saba, W.; Lund, A.K.; Brun, E.; Mabondzo, A. Brain Inflammation, Blood Brain Barrier dysfunction and Neuronal Synaptophysin Decrease after Inhalation Exposure to Titanium Dioxide Nano-aerosol in Aging Rats. Sci. Rep. 2017, 7, 12196. [Google Scholar] [CrossRef] [PubMed]
- Samiei, F.; Shirazi, F.H.; Naserzadeh, P.; Dousti, F.; Seydi, E.; Pourahmad, J. Toxicity of multi-wall carbon nanotubes inhalation on the brain of rats. Environ. Sci. Pollut. Res. Int. 2020, 27, 12096–12111, Erratum in Environ. Sci. Pollut. Res. Int. 2020, 27, 29699. [Google Scholar] [CrossRef]
- Hubbs, A.F.; Porter, D.W.; Mercer, R.R.; Castranova, V.; Sargent, L.M.; Sriram, K. Nanoparticulates. In Haschek and Rousseaux’s Handbook of Toxicologic Pathology, 4th ed.; Haschek, W., Rousseaux, C., Wallig, M., Eds.; Elsevier Press: Amsterdam, The Netherlands, 2023; Volume 3, Chapter 30; ISBN 9780443161537. [Google Scholar]
- Klein, A.L.; Nugent, G.; Cavendish, J.; Geldenhuys, W.J.; Sriram, K.; Porter, D.; Fladeland, R.; Lockman, P.R.; Sherman, J.H. Nanoparticles as a Tool in Neuro-Oncology Theranostics. Pharmaceutics 2021, 13, 948. [Google Scholar] [CrossRef]
- Thiruvengadam, M.; Rajakumar, G.; Swetha, V.; Ansari, M.A.; Alghamdi, S.; Almehmadi, M.; Halawi, M.; Kungumadevi, L.; Raja, V.; Sabura Sarbudeen, S.; et al. Recent Insights and Multifactorial Applications of Carbon Nanotubes. Micromachines 2021, 12, 1502. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, X.; Sun, L.; Wei, Y.; Wei, X. Cellular Toxicity and Immunological Effects of Carbon-based Nanomaterials. Part Fibre Toxicol. 2019, 16, 18. [Google Scholar] [CrossRef]
- Donaldson, K.; Aitken, R.; Tran, L.; Stone, V.; Duffin, R.; Forrest, G.; Alexander, A. Carbon nanotubes: A review of their properties in relation to pulmonary toxicology and workplace safety. Toxicol Sci. 2006, 92, 5–22. [Google Scholar] [CrossRef]
- Mercer, R.R.; Scabilloni, J.F.; Hubbs, A.F.; Wang, L.; Battelli, L.A.; McKinney, W.; Castranova, V.; Porter, D.W. Extrapulmonary transport of MWCNT following inhalation exposure. Part Fibre Toxicol. 2013, 10, 38. [Google Scholar] [CrossRef]
- Albini, A.; Pagani, A.; Pulze, L.; Bruno, A.; Principi, E.; Congiu, T.; Gini, E.; Grimaldi, A.; Bassani, B.; De Flora, S.; et al. Environmental impact of multi-wall carbon nanotubes in a novel model of exposure: Systemic distribution, macrophage accumulation, and amyloid deposition. Int. J. Nanomed. 2015, 10, 6133–6145. [Google Scholar] [CrossRef]
- Huang, C.L.; Hsiao, I.L.; Lin, H.C.; Wang, C.F.; Huang, Y.J.; Chuang, C.Y. Silver nanoparticles affect on gene expression of inflammatory and neurodegenerative responses in mouse brain neural cells. Environ. Res. 2015, 136, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sun, W.; An, L. Nano-CuO impairs spatial cognition associated with inhibiting hippocampal long-term potentiation via affecting glutamatergic neurotransmission in rats. Toxicol. Ind. Health 2018, 34, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Varlamova, E.G.; Turovsky, E.A.; Babenko, V.A.; Plotnikov, E.Y. The Mechanisms Underlying the Protective Action of Selenium Nanoparticles against Ischemia/Reoxygenation Are Mediated by the Activation of the Ca2+ Signaling System of Astrocytes and Reactive Astrogliosis. Int. J. Mol. Sci. 2021, 22, 12825. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Obst, J.; Simon, E.; Mancuso, R.; Gomez-Nicola, D. The Role of Microglia in Prion Diseases: A Paradigm of Functional Diversity. Front. Aging Neurosci. 2017, 9, 207. [Google Scholar] [CrossRef]
- Hristovska, I.; Robert, M.; Combet, K.; Honnorat, J.; Comte, J.C.; Pascual, O. Sleep decreases neuronal activity control of microglial dynamics in mice. Nat. Commun. 2022, 13, 6273. [Google Scholar] [CrossRef]
- Osborn, L.M.; Kamphuis, W.; Wadman, W.J.; Hol, E.M. Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 121–141. [Google Scholar] [CrossRef]
- Cicognola, C.; Janelidze, S.; Hertze, J.; Zetterberg, H.; Blennow, K.; Mattsson-Carlgren, N.; Hansson, O. Plasma glial fibrillary acidic protein detects Alzheimer pathology and predicts future conversion to Alzheimer dementia in patients with mild cognitive impairment. Alzheimer’s Res. Ther. 2021, 13, 68. [Google Scholar] [CrossRef]
- World Health Organization. Global action plan on the public health response to dementia 2017–2025. Geneva: World Health Organization; 2017. Licence: CC BY-NC-SA 3.0 IGO. Available online: https://apps.who.int/iris/bitstream/handle/10665/259615/9789241513487-eng.pdf?sequence=1 (accessed on 1 December 2022).
- Fong, T.G.; Inouye, S.K. The inter-relationship between delirium and dementia: The importance of delirium prevention. Nat. Rev. Neurol. 2022, 18, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Dejanovic, B.; Wu, T.; Tsai, M.C.; Graykowski, D.; Gandham, V.D.; Rose, C.M.; Bakalarski, C.E.; Ngu, H.; Wang, Y.; Pandey, S.; et al. Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nat. Aging 2022, 2, 837–850. [Google Scholar] [CrossRef]
- Rossi, F.; Bianchini, E. Synergistic induction of nitric oxide by beta-amyloid and cytokines in astrocytes. Biochem. Biophys. Res. Commun. 1996, 225, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Blasko, I.; Veerhuis, R.; Stampfer-Kountchev, M.; Saurwein-Teissl, M.; Eikelenboom, P.; Grubeck-Loebenstein, B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol. Dis. 2000, 7, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.K.; Karlo, J.C.; Kao, S.C.; Landreth, G.E. Beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001, 21, 1179–1188. [Google Scholar] [CrossRef]
- Liao, Y.F.; Wang, B.J.; Cheng, H.T.; Kuo, L.H.; Wolfe, M.S. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J. Biol. Chem. 2004, 279, 49523–49532. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef]
- Park, K.M.; Bowers, W.J. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell. Signal. 2010, 22, 977–983. [Google Scholar] [CrossRef]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef]
- Goldgaber, D.; Harris, H.W.; Hla, T.; Maciag, T.; Donnelly, R.J.; Jacobsen, J.S.; Vitek, M.P.; Gajdusek, D.C. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc. Natl. Acad. Sci. USA 1989, 86, 7606–7610. [Google Scholar] [CrossRef]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; Macleod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.G.; Zhu, S.G.; Jones, R.A.; Griffin, W.S.; Mrak, R.E. Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Exp. Neurol. 2000, 163, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Hoozemans, J.J.; Veerhuis, R.; Janssen, I.; Rozemuller, A.J.; Eikelenboom, P. Interleukin-1beta induced cyclooxygenase 2 expression and prostaglandin E2 secretion by human neuroblastoma cells: Implications for Alzheimer’s disease. Exp. Gerontol. 2001, 36, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef]
- Griffin, W.S.; Liu, L.; Li, Y.; Mrak, R.E.; Barger, S.W. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflamm. 2006, 3, 5. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Tachida, Y.; Nakagawa, K.; Saito, T.; Saido, T.C.; Honda, T.; Saito, Y.; Murayama, S.; Endo, T.; Sakaguchi, G.; Kato, A.; et al. Interleukin-1 beta up-regulates TACE to enhance alpha-cleavage of APP in neurons: Resulting decrease in Abeta production. J. Neurochem. 2008, 104, 1387–1393. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef]
- Ringheim, G.E.; Szczepanik, A.M.; Petko, W.; Burgher, K.L.; Zhu, S.Z.; Chao, C.C. Enhancement of beta-amyloid precursor protein transcription and expression by the soluble interleukin-6 receptor/interleukin-6 complex. Brain Res. Mol. Brain Res. 1998, 55, 35–44. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Orellana, D.I.; Gonzalez-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Dugan, L.L.; Ali, S.S.; Shekhtman, G.; Roberts, A.J.; Lucero, J.; Quick, K.L.; Behrens, M.M. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS ONE 2009, 4, e5518. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, P.; Ceballos-Diaz, C.; Beccard, A.; Janus, C.; Dickson, D.; Golde, T.E.; Das, P. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J. Immunol. 2010, 184, 5333–5343. [Google Scholar] [CrossRef] [PubMed]
- Spooren, A.; Kolmus, K.; Laureys, G.; Clinckers, R.; De Keyser, J.; Haegeman, G.; Gerlo, S. Interleukin-6, a mental cytokine. Brain Res. Rev. 2011, 67, 157–183. [Google Scholar] [CrossRef] [PubMed]
- Curran, B.; O’Connor, J.J. The pro-inflammatory cytokine interleukin-18 impairs long-term potentiation and NMDA receptor-mediated transmission in the rat hippocampus in vitro. Neuroscience 2001, 108, 83–90. [Google Scholar] [CrossRef]
- Ojala, J.O.; Sutinen, E.M.; Salminen, A.; Pirttila, T. Interleukin-18 increases expression of kinases involved in tau phosphorylation in SH-SY5Y neuroblastoma cells. J. Neuroimmunol. 2008, 205, 86–93. [Google Scholar] [CrossRef]
- Sutinen, E.M.; Pirttilä, T.; Anderson, G.; Salminen, A.; Ojala, J.O. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-β production in human neuron-like cells. J. Neuroinflammation 2012, 9, 199. [Google Scholar] [CrossRef]
- Yermakova, A.V.; Rollins, J.; Callahan, L.M.; Rogers, J.; O’Banion, M.K. Cyclooxygenase-1 in human Alzheimer and control brain: Quantitative analysis of expression by microglia and CA3 hippocampal neurons. J. Neuropathol. Exp. Neurol. 1999, 58, 1135–1146. [Google Scholar] [CrossRef]
- Liu, B.; Gao, H.M.; Wang, J.Y.; Jeohn, G.H.; Cooper, C.L.; Hong, J.S. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann. N. Y. Acad. Sci. 2002, 962, 318–331. [Google Scholar] [CrossRef]
- Bishop, A.; Anderson, J.E. No signaling in the CNS: From the physiological to the pathological. Toxicology 2005, 208, 193–205. [Google Scholar] [CrossRef]
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef]
- Muzio, L.; Viotti, A.; Martino, G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front. Neurosci. 2021, 15, 742065. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123.e6. [Google Scholar] [CrossRef]
- Brea, D.; Veiga-Fernandes, H. Inflammation in the gut is encoded by neurons in the brain. Nature 2022, 602, 217–218. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? Acc. Chem. Res. 2019, 52, 2026–2035. [Google Scholar] [CrossRef]
- Cheng, Y.; Song, Y.; Chen, H.; Li, Q.; Gao, Y.; Lu, G.; Luo, C. Ferroptosis Mediated by Lipid Reactive Oxygen Species: A Possible Causal Link of Neuroinflammation to Neurological Disorders. Oxidative Med. Cell. Longev. 2021, 5005136. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.H.; Hartley, D.M.; Koh, J.Y.; Choi, D.W. AMPA receptor activation potentiates zinc neurotoxicity. Neuron 1993, 10, 43–49. [Google Scholar] [CrossRef]
- Narayanan, S.E.; Rehuman, N.A.; Harilal, S.; Vincent, A.; Rajamma, R.G.; Behl, T.; Uddin, M.S.; Ashraf, G.M.; Mathew, B. Molecular mechanism of zinc neurotoxicity in Alzheimer’s disease. Environ. Sci. Pollut. Res. Int. 2020, 27, 43542–43552. [Google Scholar] [CrossRef]
- Ghosh, A.; Comerota, M.M.; Wan, D.; Chen, F.; Propson, N.E.; Hwang, S.H.; Hammock, B.D.; Zheng, H. An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 2020, 12, eabb1206. [Google Scholar] [CrossRef] [PubMed]
- DeMaagd, G.; Philip, A. Parkinson’s Disease and Its Management: Part 1: Disease Entity, Risk Factors, Pathophysiology, Clinical Presentation, and Diagnosis. Pharm. Ther. 2015, 40, 504–532. [Google Scholar]
- Mastrangelo, L. The Genetics of Parkinson Disease. Adv. Genet. 2017, 98, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; Chapter 5. [Google Scholar]
- Wilkaniec, A.; Lenkiewicz, A.M.; Babiec, L.; Murawska, E.; Jęśko, H.M.; Cieślik, M.; Culmsee, C.; Adamczyk, A. Exogenous Alpha-Synuclein Evoked Parkin Downregulation Promotes Mitochondrial Dysfunction in Neuronal Cells. Implications for Parkinson’s Disease Pathology. Front. Aging Neurosci. 2021, 13, 591475. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Pathoanatomy of Parkinson’s disease. J. Neurol. 2000, 247 (Suppl. S2), II3–II10. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Vaccari, C.; El Dib, R.; Gomaa, H.; Lopes, L.C.; de Camargo, J.L. Paraquat and Parkinson’s disease: A systematic review and meta-analysis of observational studies. J. Toxicol. Environ. Health B Crit. Rev. 2019, 22, 172–202. [Google Scholar] [CrossRef]
- Selmaj, K.W.; Farooq, M.; Norton, W.T.; Raine, C.S.; Brosnan, C.F. Proliferation of astrocytes in vitro in response to cytokines. A primary role for tumor necrosis factor. J. Immunol. 1990, 144, 129–135. [Google Scholar] [CrossRef]
- Merrill, J.E. Effects of interleukin-1 and tumor necrosis factor-alpha on astrocytes, microglia, oligodendrocytes, and glial precursors in vitro. Dev. Neurosci. 1991, 13, 130–137. [Google Scholar] [CrossRef]
- Sedgwick, J.D.; Riminton, D.S.; Cyster, J.G.; Korner, H. Tumor necrosis factor: A master-regulator of leukocyte movement. Immunol. Today 2000, 21, 110–113. [Google Scholar] [CrossRef]
- Leon, L.R. Invited review: Cytokine regulation of fever: Studies using gene knockout mice. J. Appl. Physiol. 2002, 92, 2648–2655. [Google Scholar] [CrossRef]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Pickering, M.; Cumiskey, D.; O’Connor, J.J. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. Exp. Physiol. 2005, 90, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential Regulation of AMPA Receptor and GABA Receptor Trafficking by Tumor Necrosis Factor-{alpha}. J. Neurosci. 2005, 25, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-[alpha]. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef]
- Doble, A. The role of excitotoxicity in neurodegenerative disease: Implications for therapy. Pharmacol. Ther. 1999, 81, 163–221. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef]
- Lu, K.T.; Wang, Y.W.; Wo, Y.Y.; Yang, Y.L. Extracellular signal-regulated kinase-mediated IL-1-induced cortical neuron damage during traumatic brain injury. Neurosci. Lett. 2005, 386, 40–45. [Google Scholar] [CrossRef]
- Huang, Y.; Smith, D.E.; Ibanez-Sandoval, O.; Sims, J.E.; Friedman, W.J. Neuron-specific effects of interleukin-1beta are mediated by a novel isoform of the IL-1 receptor accessory protein. J. Neurosci. 2011, 31, 18048–18059. [Google Scholar] [CrossRef]
- Kaur, C.; Sivakumar, V.; Zou, Z.; Ling, E.A. Microglia-derived proinflammatory cytokines tumor necrosis factor-alpha and interleukin-1beta induce Purkinje neuronal apoptosis via their receptors in hypoxic neonatal rat brain. Brain Struct. Funct. 2014, 219, 151–170. [Google Scholar] [CrossRef] [PubMed]
- Vawter, M.P.; Dillon-Carter, O.; Tourtellotte, W.W.; Carvey, P.; Freed, W.J. TGFbeta1 and TGFbeta2 concentrations are elevated in Parkinson’s disease in ventricular cerebrospinal fluid. Exp. Neurol. 1996, 142, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Goris, A.; Williams-Gray, C.H.; Foltynie, T.; Brown, J.; Maranian, M.; Walton, A.; Compston, D.A.; Barker, R.A.; Sawcer, S.J. Investigation of TGFB2 as a candidate gene in multiple sclerosis and Parkinson’s disease. J. Neurol. 2007, 254, 846–848. [Google Scholar] [CrossRef] [PubMed]
- Hegarty, S.V.; Sullivan, A.M.; O’Keeffe, G.W. Roles for the TGFbeta superfamily in the development and survival of midbrain dopaminergic neurons. Mol. Neurobiol. 2014, 50, 559–573. [Google Scholar] [CrossRef]
- Tesseur, I.; Nguyen, A.; Chang, B.; Li, L.; Woodling, N.S.; Wyss-Coray, T.; Luo, J. Deficiency in neuronal TGF-beta signaling leads to nigrostriatal degeneration and activation of TGF-beta signaling protects against MPTP neurotoxicity in mice. J. Neurosci. 2017, 37, 4584–4592. [Google Scholar] [CrossRef]
- Ebadi, M.; Sharma, S.K. Peroxynitrite and mitochondrial dysfunction in the pathogenesis of Parkinson’s disease. Antioxid. Redox Signal 2003, 5, 319–335. [Google Scholar] [CrossRef]
- Zhang, L.; Dawson, V.L.; Dawson, T.M. Role of nitric oxide in Parkinson’s disease. Pharmacol. Ther. 2006, 109, 33–41. [Google Scholar] [CrossRef]
- Tsang, A.H.; Chung, K.K. Oxidative and nitrosative stress in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 643–650. [Google Scholar] [CrossRef]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; Herrero, M.T.; García-Martín, E.; Agúndez, J.A. An Update on the Role of Nitric Oxide in the Neurodegenerative Processes of Parkinson’s Disease. Curr. Med. Chem. 2016, 23, 2666–2679. [Google Scholar] [CrossRef]
- Sircar, E.; Rai, S.R.; Wilson, M.A.; Schlossmacher, M.G.; Sengupta, R. Neurodegeneration: Impact of S-nitrosylated Parkin, DJ-1 and PINK1 on the pathogenesis of Parkinson’s disease. Arch. Biochem. Biophys. 2021, 704, 108869. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Langston, J.W.; Forno, L.S.; Tetrud, J.; Reeves, A.G.; Kaplan, J.A.; Karluk, D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999, 46, 598–605. [Google Scholar] [CrossRef]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Mice deficient in TNF receptors are protected against dopaminergic neurotoxicity: Implications for Parkinson’s disease. FASEB J. 2002, 16, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Benkovic, S.A.; Hebert, M.A.; Miller, D.B.; O’Callaghan, J.P. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: Key signaling pathway for astrogliosis in vivo? J. Biol. Chem. 2004, 279, 19936–19947. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Matheson, J.M.; Benkovic, S.A.; Miller, D.B.; Luster, M.I.; O’Callaghan, J.P. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: Role of TNF-alpha. FASEB J. 2006, 20, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Miller, D.B.; O’Callaghan, J.P. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: Role of tumor necrosis factor-alpha. J. Neurochem. 2006, 96, 706–718. [Google Scholar] [CrossRef]
- Mastroeni, D.; Nolz, J.; Sekar, S.; Delvaux, E.; Serrano, G.; Cuyugan, L.; Liang, W.S.; Beach, T.G.; Rogers, J.; Coleman, P.D. Laser-captured microglia in the Alzheimer’s and Parkinson’s brain reveal unique regional expression profiles and suggest a potential role for hepatitis B in the Alzheimer’s brain. Neurobiol. Aging 2018, 63, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.W.; Chang, N.P.; Krishnagiri, M.; Patel, A.P.; Lindman, M.; Angel, J.P.; Kung, P.L.; Atkins, C.; Daniels, B.P. Fibrillar α-synuclein induces neurotoxic astrocyte activation via RIP kinase signaling and NF-κB. Cell Death Dis. 2021, 12, 756. [Google Scholar] [CrossRef]
- Haïk, S.; Marcon, G.; Mallet, A.; Tettamanti, M.; Welaratne, A.; Giaccone, G.; Azimi, S.; Pietrini, V.; Fabreguettes, J.R.; Imperiale, D.; et al. Doxycycline in Creutzfeldt-Jakob disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014, 13, 150–158. [Google Scholar] [CrossRef]
- Watson, N.; Kirby, J.; Kurudzhu, H.; Leitch, M.; MacKenzie, J.; Smith-Bathgate, B.; Smith, C.; Summers, D.; Green, A.J.E.; Pal, S. Impact of the COVID-19 pandemic on Creutzfeldt-Jakob disease surveillance and patient care in the United Kingdom. Eur. J. Neurol. 2022, 29, 1222–1226. [Google Scholar] [CrossRef]
- Aguzzi, A.; Calella, A.M. Prions: Protein aggregation and infectious diseases. Physiol. Rev. 2009, 89, 1105–1152. [Google Scholar] [CrossRef] [PubMed]
- Zerr, I. Laboratory Diagnosis of Creutzfeldt-Jakob Disease. N. Engl. J. Med. 2022, 386, 1345–1350. [Google Scholar] [CrossRef]
- Safar, J.G. Molecular pathogenesis of sporadic prion diseases in man. Prion 2012, 6, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Uttley, L.; Carroll, C.; Wong, R.; Hilton, D.A.; Stevenson, M. Creutzfeldt-Jakob disease: A systematic review of global incidence, prevalence, infectivity, and incubation. Lancet Infect. Dis. 2020, 20, e2–e10. [Google Scholar] [CrossRef] [PubMed]
- Stoeck, K.; Bodemer, M.; Zerr, I. Pro- and anti-inflammatory cytokines in the CSF of patients with Creutzfeldt-Jakob disease. J Neuroimmunol. 2006, 172, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Nicola, D.; Fransen, N.L.; Suzzi, S.; Perry, V.H. Regulation of microglial proliferation during chronic neurodegeneration. J. Neurosci. 2013, 33, 2481–2493. [Google Scholar] [CrossRef]
- Deininger, M.H.; Bekure-Nemariam, K.; Trautmann, K.; Morgalla, M.; Meyermann, R.; Schluesener, H.J. Cyclooxygenase-1 and -2 in brains of patients who died with sporadic Creutzfeldt-Jakob disease. J. Mol. Neurosci. 2003, 20, 25–30. [Google Scholar] [CrossRef]
- Llorens, F.; López-González, I.; Thüne, K.; Carmona, M.; Zafar, S.; Andréoletti, O.; Zerr, I.; Ferrer, I. Subtype and regional-specific neuroinflammation in sporadic creutzfeldt-jakob disease. Front. Aging Neurosci. 2014, 6, 198. [Google Scholar] [CrossRef]
- Guentchev, M.; Siedlak, S.L.; Jarius, C.; Tagliavini, F.; Castellani, R.J.; Perry, G.; Smith, M.A.; Budka, H. Oxidative damage to nucleic acids in human prion disease. Neurobiol. Dis. 2002, 9, 275–281. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Szpak, G.M.; Lewandowska, E.; Lechowicz, W.; Wierzba-Bobrowicz, T.; Kulczycki, J.; Bertrand, E.; Pasennik, E.; Dymecki, J. The brain immune response in human prion diseases. Microglial activation and microglial disease. I. Sporadic Creutzfeldt-Jakob disease. Folia Neuropathol. 2006, 44, 202–213. [Google Scholar] [PubMed]
- McGeer, P.L.; McGeer, E.G. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; NYGC ALS Consortium; Fagegaltier, D.; Harris, B.T.; et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29, 1164–1177.e5. [Google Scholar] [CrossRef] [PubMed]
- Lall, D.; Lorenzini, I.; Mota, T.A.; Bell, S.; Mahan, T.E.; Ulrich, J.D.; Davtyan, H.; Rexach, J.E.; Muhammad, A.K.M.G.; Shelest, O.; et al. C9orf72 deficiency promotes microglial-mediated synaptic loss in aging and amyloid accumulation. Neuron 2021, 109, 2275–2291.e8. [Google Scholar] [CrossRef] [PubMed]
- Apolloni, S.; Amadio, S.; Parisi, C.; Matteucci, A.; Potenza, R.L.; Armida, M.; Popoli, P.; D’Ambrosi, N.; Volonté, C. Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis. Models Mech. 2014, 7, 1101–1109. [Google Scholar] [CrossRef]
- Maniatis, S.; Äijö, T.; Vickovic, S.; Braine, C.; Kang, K.; Mollbrink, A.; Fagegaltier, D.; Andrusivová, Ž.; Saarenpää, S.; Saiz-Castro, G.; et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 2019, 364, 89–93. [Google Scholar] [CrossRef]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- D’Erchia, A.M.; Gallo, A.; Manzari, C.; Raho, S.; Horner, D.S.; Chiara, M.; Valletti, A.; Aiello, I.; Mastropasqua, F.; Ciaccia, L.; et al. Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS. Sci. Rep. 2017, 7, 10046. [Google Scholar] [CrossRef]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef]
- Dols-Icardo, O.; Montal, V.; Sirisi, S.; López-Pernas, G.; Cervera-Carles, L.; Querol-Vilaseca, M.; Muñoz, L.; Belbin, O.; Alcolea, D.; Molina-Porcel, L.; et al. Motor cortex transcriptome reveals microglial key events in amyotrophic lateral sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e829. [Google Scholar] [CrossRef]
- Konjevic Sabolek, M.; Held, K.; Beltrán, E.; Niedl, A.G.; Meinl, E.; Hohlfeld, R.; Lassmann, H.; Dornmair, K. Communication of CD8+ T cells with mononuclear phagocytes in multiple sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132 Pt 5, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis—the plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, C.; van der Poel, M.; Fernández-Zapata, C.; Schlickeiser, S.; Leman, J.K.H.; Hsiao, C.C.; Mizee, M.R.; Adelia Vincenten, M.C.J.; Kunkel, D.; Huitinga, I.; et al. Single-cell mass cytometry reveals complex myeloid cell composition in active lesions of progressive multiple sclerosis. Acta Neuropathol. Commun. 2020, 8, 136. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.F.; Wu, S.P.; Lin, G.J.; Shieh, C.C.; Hsu, C.S.; Chen, J.W.; Chen, S.H.; Hong, J.S.; Chen, S.J. Microglial Nox2 Plays a Key Role in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2021, 12, 638381. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Fugger, L.; Friese, M.A.; Bell, J.I. From genes to function: The next challenge to understanding multiple sclerosis. Nat. Rev. Immunol. 2009, 9, 408–417. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Sriram, K. Focused microwave irradiation of the brain preserves in vivo protein phosphorylation: Comparison with other methods of sacrifice and analysis of multiple phosphoproteins. J. Neurosci. Methods 2004, 135, 159–168. [Google Scholar] [CrossRef]
- Seok, S.H. Structural Insights into Protein Regulation by Phosphorylation and Substrate Recognition of Protein Kinases/Phosphatases. Life 2021, 11, 957. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schätzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J.; et al. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 2020, 105, 855–866.e5. [Google Scholar] [CrossRef] [PubMed]
- Beltrao, P.; Bork, P.; Krogan, N.J.; van Noort, V. Evolution and functional cross-talk of protein post-translational modifications. Mol. Syst. Biol. 2013, 9, 714. [Google Scholar] [CrossRef] [PubMed]
- Greig, F.H.; Nixon, G.F. Phosphoprotein enriched in astrocytes (PEA)-15: A potential therapeutic target in multiple disease states. Pharmacol. Ther. 2014, 143, 265–274. [Google Scholar] [CrossRef]
- Greig, F.H.; Kennedy, S.; Gibson, G.; Ramos, J.W.; Nixon, G.F. PEA-15 (Phosphoprotein Enriched in Astrocytes 15) Is a Protective Mediator in the Vasculature and Is Regulated During Neointimal Hyperplasia. J. Am. Heart Assoc. 2017, 6, e006936. [Google Scholar] [CrossRef] [PubMed]
- Estellés, A.; Yokoyama, M.; Nothias, F.; Vincent, J.D.; Glowinski, J.; Vernier, P.; Chneiweiss, H. The major astrocytic phosphoprotein PEA-15 is encoded by two mRNAs conserved on their full length in mouse and human. J. Biol. Chem. 1996, 271, 14800–14806. [Google Scholar] [CrossRef]
- Danziger, N.; Yokoyama, M.; Jay, T.; Cordier, J.; Glowinski, J.; Chneiweiss, H. Cellular expression, developmental regulation, and phylogenic conservation of PEA-15, the astrocytic major phosphoprotein and protein kinase C substrate. J. Neurochem. 1995, 64, 1016–1025. [Google Scholar] [CrossRef]
- Du, S.; Rubin, A.; Klepper, S.; Barrett, C.; Kim, Y.C.; Rhim, H.W.; Lee, E.B.; Park, C.W.; Markelonis, G.J.; Oh, T.H. Calcium influx and activation of calpain I mediate acute reactive gliosis in injured spinal cord. Exp. Neurol. 1999, 157, 96–105. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Terai, K.; Matsuo, A.; McGeer, P.L. Enhancement of immunoreactivity for NF-kappa B in the hippocampal formation and cerebral cortex of Alzheimer’s disease. Brain Res. 1996, 735, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Martí, E.; López, E.; Tortosa, A. NF-kB immunoreactivity is observed in association with beta A4 diffuse plaques in patients with Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 1998, 24, 271–277. [Google Scholar] [CrossRef]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef]
- Träger, U.; Andre, R.; Magnusson-Lind, A.; Miller, J.R.; Connolly, C.; Weiss, A.; Grueninger, S.; Silajdžić, E.; Smith, D.L.; Leavitt, B.R.; et al. Characterisation of immune cell function in fragment and full-length Huntington’s disease mouse models. Neurobiol. Dis. 2015, 73, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.Y.; Chen, Y.C.; Chen, H.M.; Tu, P.H.; Chern, Y. A critical role of astrocyte-mediated nuclear factor-κB-dependent inflammation in Huntington’s disease. Hum. Mol. Genet. 2013, 22, 1826–1842. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kaltschmidt, C. NF-kappa B: A crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997, 20, 252–258. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.Y.; Ran, R.; Da, C.M.; Wang, Z.Q.; Zhou, K.S.; Zhang, H.H. Ski Regulates the Inflammatory Response of Reactive Astrocytes Induced by Oxygen Glucose Deprivation/Reoxygenation (OGD/R) Through the NF-κB Pathway. Neuroscience 2022, 490, 250–263. [Google Scholar] [CrossRef]
- Li, J.; Tang, Y.; Cai, D. IKKβ/NF-κB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat. Cell Biol. 2012, 14, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, J.; Purkayastha, S.; Tang, Y.; Zhang, H.; Yin, Y.; Li, B.; Liu, G.; Cai, D. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 2013, 497, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Fan, L.; Khawaja, R.R.; Liu, B.; Zhan, L.; Kodama, L.; Chin, M.; Li, Y.; Le, D.; Zhou, Y.; et al. Microglial NF-κB drives tau spreading and toxicity in a mouse model of tauopathy. Nat. Commun. 2022, 13, 1969. [Google Scholar] [CrossRef] [PubMed]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Yan, Z.; Gibson, S.A.; Buckley, J.A.; Qin, H.; Benveniste, E.N. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clin. Immunol. 2018, 189, 4–13. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546, Erratum in Drugs 2017, 77, 939. [Google Scholar] [CrossRef]
- Kacimi, R.; Giffard, R.G.; Yenari, M.A. Endotoxin-activated microglia injure brain derived endothelial cells via NF-κB, JAK-STAT and JNK stress kinase pathways. J. Inflamm. 2011, 8, 7. [Google Scholar] [CrossRef]
- Takizawa, T.; Nakashima, K.; Namihira, M.; Ochiai, W.; Uemura, A.; Yanagisawa, M.; Fujita, N.; Nakao, M.; Taga, T. DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev. Cell. 2001, 1, 749–758. [Google Scholar] [CrossRef]
- Jain, M.; Singh, M.K.; Shyam, H.; Mishra, A.; Kumar, S.; Kumar, A.; Kushwaha, J. Role of JAK/STAT in the Neuroinflammation and its Association with Neurological Disorders. Ann. Neurosci. 2021, 28, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Carrillo-de Sauvage, M.A.; Ceyzériat, K.; Escartin, C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front. Cell Neurosci. 2015, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gibson, S.A.; Benveniste, E.N.; Qin, H. Opportunities for Translation from the Bench: Therapeutic Intervention of the JAK/STAT Pathway in Neuroinflammatory Diseases. Crit. Rev. Immunol. 2015, 35, 505–527. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, K.L.; Camps, M.; Rommel, C.; Mackay, C.R. Targeting dual-specificity phosphatases: Manipulating MAP kinase signalling and immune responses. Nat. Rev. Drug Discov. 2007, 6, 391–403. [Google Scholar] [CrossRef]
- Anjum, R.; Blenis, J. The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747–758. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Hensley, K.; Floyd, R.A.; Zheng, N.Y.; Nael, R.; Robinson, K.A.; Nguyen, X.; Pye, Q.N.; Stewart, C.A.; Geddes, J.; Markesbery, W.R.; et al. p38 kinase is activated in the Alzheimer’s disease brain. J. Neurochem. 1999, 72, 2053–2058. [Google Scholar] [CrossRef]
- Koistinaho, M.; Kettunen, M.I.; Goldsteins, G.; Keinänen, R.; Salminen, A.; Ort, M.; Bures, J.; Liu, D.; Kauppinen, R.A.; Higgins, L.S.; et al. Beta-amyloid precursor protein transgenic mice that harbor diffuse A beta deposits but do not form plaques show increased ischemic vulnerability: Role of inflammation. Proc. Natl. Acad. Sci. USA 2002, 99, 1610–1615. [Google Scholar] [CrossRef]
- Bendotti, C.; Atzori, C.; Piva, R.; Tortarolo, M.; Strong, M.J.; DeBiasi, S.; Migheli, A. Activated p38MAPK is a novel component of the intracellular inclusions found in human amyotrophic lateral sclerosis and mutant SOD1 transgenic mice. J. Neuropathol. Exp. Neurol. 2004, 63, 113–119. [Google Scholar] [CrossRef]
- Tortarolo, M.; Veglianese, P.; Calvaresi, N.; Botturi, A.; Rossi, C.; Giorgini, A.; Migheli, A.; Bendotti, C. Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol. Cell Neurosci. 2003, 23, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Luo, L.; Wang, J.; Shen, H.; Li, Y.; Mamtilahun, M.; Liu, C.; Shi, R.; Lee, J.H.; Tian, H.; et al. Stroke subtype-dependent synapse elimination by reactive gliosis in mice. Nat. Commun. 2021, 12, 6943, Erratum in Nat. Commun. 2022, 13, 1183. [Google Scholar] [CrossRef] [PubMed]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef] [PubMed]
- Steinhäuser, C.; Grunnet, M.; Carmignoto, G. Crucial role of astrocytes in temporal lobe epilepsy. Neuroscience 2016, 323, 157–169. [Google Scholar] [CrossRef]
- Rakers, C.; Schleif, M.; Blank, N.; Matušková, H.; Ulas, T.; Händler, K.; Torres, S.V.; Schumacher, T.; Tai, K.; Schultze, J.L.; et al. Stroke target identification guided by astrocyte transcriptome analysis. Glia 2019, 67, 619–633. [Google Scholar] [CrossRef]
- Tassoni, A.; Farkhondeh, V.; Itoh, Y.; Itoh, N.; Sofroniew, M.V.; Voskuhl, R.R. The astrocyte transcriptome in EAE optic neuritis shows complement activation and reveals a sex difference in astrocytic C3 expression. Sci. Rep. 2019, 9, 10010. [Google Scholar] [CrossRef]
- Rawji, K.S.; Gonzalez Martinez, G.A.; Sharma, A.; Franklin, R.J.M. The Role of Astrocytes in Remyelination. Trends Neurosci. 2020, 43, 596–607. [Google Scholar] [CrossRef]
- Filous, A.R.; Silver, J. Targeting astrocytes in CNS injury and disease: A translational research approach. Prog. Neurobiol. 2016, 144, 173–187. [Google Scholar] [CrossRef]
- Boghdadi, A.G.; Teo, L.; Bourne, J.A. The Neuroprotective Role of Reactive Astrocytes after Central Nervous System Injury. J. Neurotrauma 2020, 37, 681–691. [Google Scholar] [CrossRef]
- Griemsmann, S.; Höft, S.P.; Bedner, P.; Zhang, J.; von Staden, E.; Beinhauer, A.; Degen, J.; Dublin, P.; Cope, D.W.; Richter, N.; et al. Characterization of Panglial Gap Junction Networks in the Thalamus, Neocortex, and Hippocampus Reveals a Unique Population of Glial Cells. Cereb. Cortex 2015, 25, 3420–3433. [Google Scholar] [CrossRef]
- Ben Haim, L.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 2017, 18, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Brock, T.O.; O’Callaghan, J.P. Quantitative changes in the synaptic vesicle proteins synapsin I and p38 and the astrocyte-specific protein glial fibrillary acidic protein are associated with chemical-induced injury to the rat central nervous system. J. Neurosci. 1987, 7, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.M.; O’Callaghan, J.P. A direct comparison of GFAP immunocytochemistry and GFAP concentration in various regions of ethanol-fixed rat and mouse brain. J. Neurosci. Methods 1995, 58, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Diaz-Castro, B.; Shigetomi, E.; Monte, E.; Octeau, J.C.; Yu, X.; Cohn, W.; Rajendran, P.S.; Vondriska, T.M.; Whitelegge, J.P.; et al. Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron 2017, 95, 531–549.e9. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef]
- Van Asperen, J.V.; Robe, P.A.J.T.; Hol, E.M. GFAP Alternative Splicing and the Relevance for Disease-A Focus on Diffuse Gliomas. ASN Neuro 2022, 14, 17590914221102065. [Google Scholar] [CrossRef]
- Bongcam-Rudloff, E.; Nistér, M.; Betsholtz, C.; Wang, J.L.; Stenman, G.; Huebner, K.; Croce, C.M.; Westermark, B. Human glial fibrillary acidic protein: Complementary DNA cloning, chromosome localization, and messenger RNA expression in human glioma cell lines of various phenotypes. Cancer Res. 1991, 51, 1553–1560. [Google Scholar]
- Radu, R.; Petrescu, G.E.D.; Gorgan, R.M.; Brehar, F.M. GFAPδ: A Promising Biomarker and Therapeutic Target in Glioblastoma. Front. Oncol. 2022, 12, 859247. [Google Scholar] [CrossRef]
- Lin, N.H.; Yang, A.W.; Chang, C.H.; Perng, M.D. Elevated GFAP isoform expression promotes protein aggregation and compromises astrocyte function. FASEB J. 2021, 35, e21614. [Google Scholar] [CrossRef]
- Messing, A.; Brenner, M. GFAP at 50. ASN Neuro. 2020, 12, 1759091420949680. [Google Scholar] [CrossRef]
- Brenner, M.; Messing, A. Regulation of GFAP Expression. ASN Neuro 2021, 13, 1759091420981206. [Google Scholar] [CrossRef] [PubMed]
- Kanski, R.; van Strien, M.E.; van Tijn, P.; Hol, E.M. A star is born: New insights into the mechanism of astrogenesis. Cell Mol. Life Sci. 2014, 71, 433–447. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).