
Challenges of Gene Editing Therapies for Genodermatoses

Abstract
:1. Introduction
2. Gene Editing Efficiency
2.1. Editing Efficiencies Already Achieved in Genodermatoses
2.2. Improving Gene Editing Efficiency in the Future
2.3. Improving Efficiency by Selecting for Edited Cells
2.4. How Much Efficiency Is Really Needed?
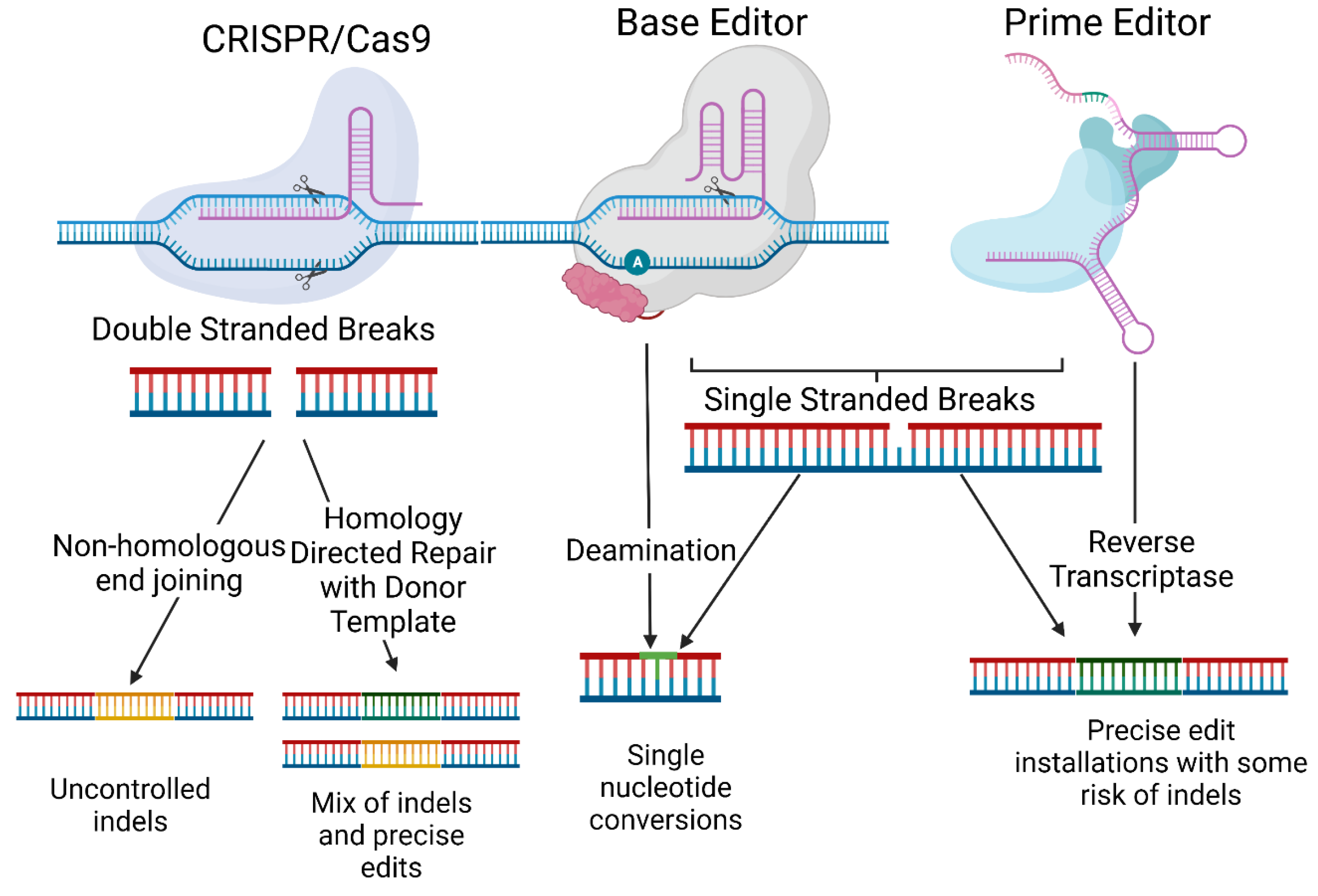
3. Off-Targets
3.1. Interrogating Off-Target Genomic DNA Editing
3.1.1. In Silico Prediction Methods
3.1.2. Experimental Methods
3.1.3. Base and Prime Editors
3.2. Designing Gene Editing Systems to Limit Off-Target Effects
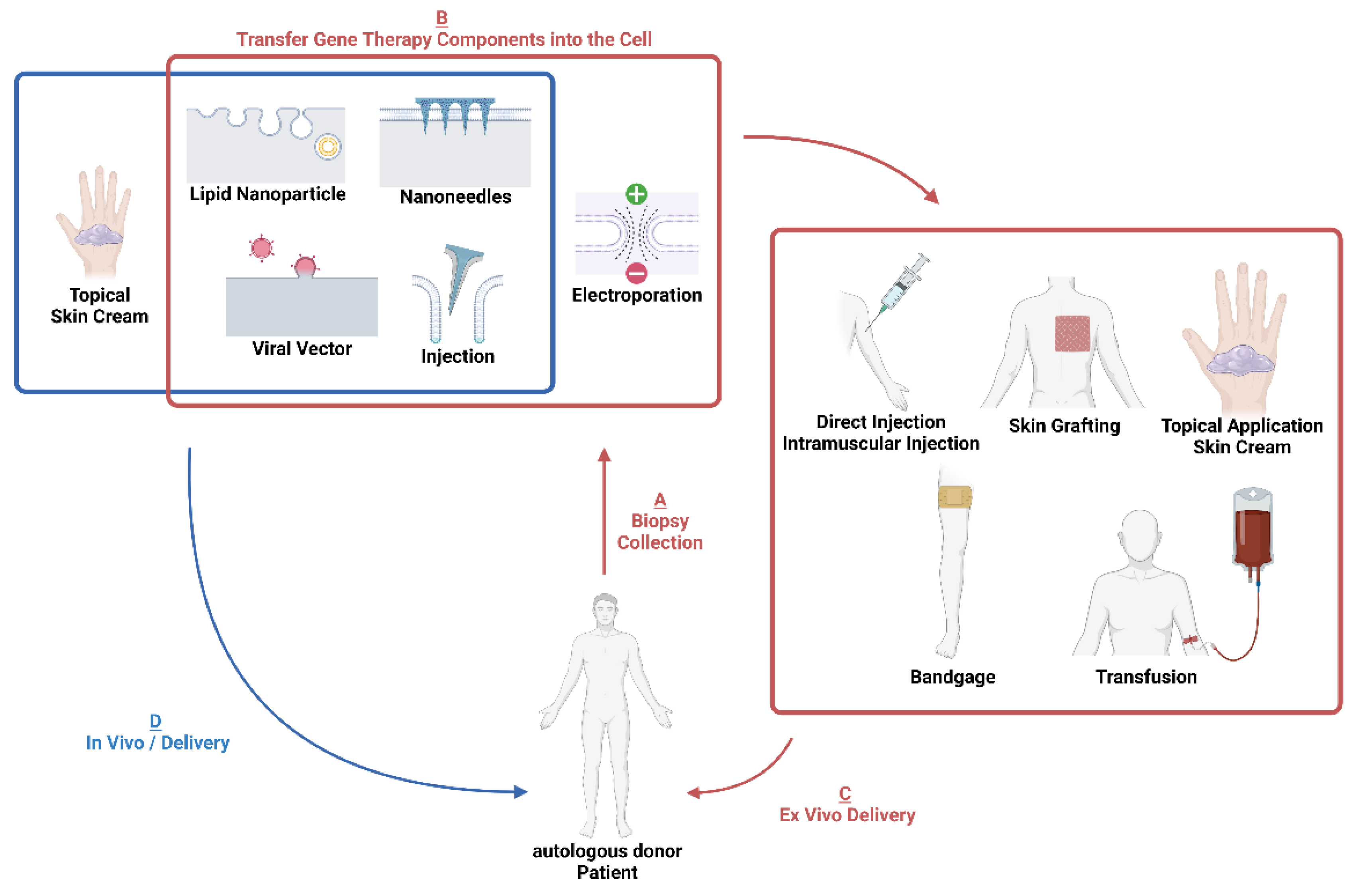
4. Delivery
4.1. Electroporation
4.2. Viral Vectors
4.3. Non-Viral Nanoparticle Vectors
4.4. Micro/Nanoneedles
5. Ex Vivo Therapies
5.1. Injections
5.2. Grafting
6. Animal Models
7. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nyström, A.; Bruckner-Tuderman, L. Gene Therapy for Epidermolysis Bullosa: Sticky Business. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 2035–2036. [Google Scholar] [CrossRef] [Green Version]
- Vahlquist, A. Treatment of Rare Keratinization Disorders: What’s New? Expert Rev. Dermatol. 2011, 6, 211–216. [Google Scholar] [CrossRef]
- Nemudryi, A.A.; Valetdinova, K.R.; Medvedev, S.P.; Zakian, S.M. TALEN and CRISPR/Cas Genome Editing Systems: Tools of Discovery. Acta Nat. 2014, 6, 19–40. [Google Scholar] [CrossRef]
- Dupuy, A.; Valton, J.; Leduc, S.; Armier, J.; Galetto, R.; Gouble, A.; Lebuhotel, C.; Stary, A.; Pâques, F.; Duchateau, P.; et al. Targeted Gene Therapy of Xeroderma Pigmentosum Cells Using Meganuclease and TALENTM. PLoS ONE 2013, 8, e78678. [Google Scholar] [CrossRef]
- Osborn, M.J.; Starker, C.G.; McElroy, A.N.; Webber, B.R.; Riddle, M.J.; Xia, L.; Defeo, A.P.; Gabriel, R.; Schmidt, M.; Von Kalle, C.; et al. TALEN-Based Gene Correction for Epidermolysis Bullosa. Mol. Ther. 2013, 21, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- March, O.P.; Lettner, T.; Klausegger, A.; Ablinger, M.; Kocher, T.; Hainzl, S.; Peking, P.; Lackner, N.; Rajan, N.; Hofbauer, J.P.; et al. Gene Editing–Mediated Disruption of Epidermolytic Ichthyosis–Associated KRT10 Alleles Restores Filament Stability in Keratinocytes. J. Investig. Dermatol. 2019, 139, 1699–1710.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mencía, Á.; Chamorro, C.; Bonafont, J.; Duarte, B.; Holguin, A.; Illera, N.; Llames, S.G.; Escámez, M.J.; Hausser, I.; Del Río, M.; et al. Deletion of a Pathogenic Mutation-Containing Exon of COL7A1 Allows Clonal Gene Editing Correction of RDEB Patient Epidermal Stem Cells. Mol. Ther.-Nucleic Acids 2018, 11, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benati, D.; Miselli, F.; Cocchiarella, F.; Patrizi, C.; Carretero, M.; Baldassarri, S.; Ammendola, V.; Has, C.; Colloca, S.; Del Rio, M.; et al. CRISPR/Cas9-Mediated In Situ Correction of LAMB3 Gene in Keratinocytes Derived from a Junctional Epidermolysis Bullosa Patient. Mol. Ther. 2018, 26, 2592–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacków, J.; Guo, Z.; Hansen, C.; Abaci, H.E.; Doucet, Y.S.; Shin, J.U.; Hayashi, R.; DeLorenzo, D.; Kabata, Y.; Shinkuma, S.; et al. CRISPR/Cas9-Based Targeted Genome Editing for Correction of Recessive Dystrophic Epidermolysis Bullosa Using IPS Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 26846–26852. [Google Scholar] [CrossRef]
- Baker, C.; Hayden, M.S. Gene Editing in Dermatology: Harnessing CRISPR for the Treatment of Cutaneous Disease. F1000Research 2020, 9, 281. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.-J.; Liquori, A.J.; et al. Directed Evolution of Adenine Base Editors with Increased Activity and Therapeutic Application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.K.; Song, B.; Bae, S. Current Status and Challenges of DNA Base Editing Tools; Cell Press: Cambridge, MA, USA, 2020; Volume 28. [Google Scholar]
- Osborn, M.J.; Newby, G.A.; McElroy, A.N.; Knipping, F.; Nielsen, S.C.; Riddle, M.J.; Xia, L.; Chen, W.; Eide, C.R.; Webber, B.R.; et al. Base Editor Correction of COL7A1 in Recessive Dystrophic Epidermolysis Bullosa Patient-Derived Fibroblasts and IPSCs. J. Investig. Dermatol. 2020, 140, 338–347.e5. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Hong, S.-A.; Kim, S.-E.; Lee, A.-Y.; Hwang, G.-H.; Kim, J.H.; Iwata, H.; Kim, S.-C.; Bae, S.; Lee, S.E. Therapeutic Base Editing and Prime Editing of COL7A1 Mutations in Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. 2022, 30, 2664–2679. [Google Scholar] [CrossRef]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained Genome Targeting with Near-PAMless Engineered CRISPR-Cas9 Variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef]
- Kocher, T.; Peking, P.; Klausegger, A.; Murauer, E.M.; Hofbauer, J.P.; Wally, V.; Lettner, T.; Hainzl, S.; Ablinger, M.; Bauer, J.W.; et al. Cut and Paste: Efficient Homology-Directed Repair of a Dominant Negative KRT14 Mutation via CRISPR/Cas9 Nickases. Mol. Ther. 2017, 25, 2585–2598. [Google Scholar] [CrossRef] [Green Version]
- Izmiryan, A.; Ganier, C.; Bovolenta, M.; Schmitt, A.; Mavilio, F.; Hovnanian, A. Ex Vivo COL7A1 Correction for Recessive Dystrophic Epidermolysis Bullosa Using CRISPR/Cas9 and Homology-Directed Repair. Mol. Ther.-Nucleic Acids 2018, 12, 554–567. [Google Scholar] [CrossRef]
- Kuscu, C.; Parlak, M.; Tufan, T.; Yang, J.; Szlachta, K.; Wei, X.; Mammadov, R.; Adli, M. CRISPR-STOP: Gene Silencing through Base-Editing-Induced Nonsense Mutations. Nat. Methods 2017, 14, 710–712. [Google Scholar] [CrossRef]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In Vivo CRISPR Base Editing of PCSK9 Durably Lowers Cholesterol in Primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef]
- García, M.; Bonafont, J.; Martínez-Palacios, J.; Xu, R.; Turchiano, G.; Svensson, S.; Thrasher, A.J.; Larcher, F.; Rio, M.D.; Hernández-Alcoceba, R.; et al. Preclinical Model for Phenotypic Correction of Dystrophic Epidermolysis Bullosa by in Vivo CRISPR-Cas9 Delivery Using Adenoviral Vectors. Mol. Ther.-Methods Clin. Dev. 2022, 27, 96–108. [Google Scholar] [CrossRef]
- Di, W.-L.; Lwin, S.M.; Petrova, A.; Bernadis, C.; Syed, F.; Farzaneh, F.; Moulding, D.; Martinez, A.E.; Sebire, N.J.; Rampling, D.; et al. Generation and Clinical Application of Gene-Modified Autologous Epidermal Sheets in Netherton Syndrome: Lessons Learned from a Phase 1 Trial. Hum. Gene Ther. 2019, 30, 1067–1078. [Google Scholar] [CrossRef]
- De Rosa, L.; Enzo, E.; Zardi, G.; Bodemer, C.; Magnoni, C.; Schneider, H.; De Luca, M. Hologene 5: A Phase II/III Clinical Trial of Combined Cell and Gene Therapy of Junctional Epidermolysis Bullosa. Front. Genet. 2021, 12, 705019. [Google Scholar] [CrossRef]
- Gurevich, I.; Agarwal, P.; Zhang, P.; Dolorito, J.A.; Oliver, S.; Liu, H.; Reitze, N.; Sarma, N.; Bagci, I.S.; Sridhar, K.; et al. In Vivo Topical Gene Therapy for Recessive Dystrophic Epidermolysis Bullosa: A Phase 1 and 2 Trial. Nat. Med. 2022, 28, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Bushman, F.D. Retroviral Insertional Mutagenesis in Humans: Evidence for Four Genetic Mechanisms Promoting Expansion of Cell Clones. Mol. Ther. 2020, 28, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional Oncogenesis in 4 Patients after Retrovirus-Mediated Gene Therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Micklethwaite, K.P.; Gowrishankar, K.; Gloss, B.S.; Li, Z.; Street, J.A.; Moezzi, L.; Mach, M.A.; Sutrave, G.; Clancy, L.E.; Bishop, D.C.; et al. Investigation of Product-Derived Lymphoma Following Infusion of PiggyBac-Modified CD19 Chimeric Antigen Receptor T Cells. Blood 2021, 138, 1391–1405. [Google Scholar] [CrossRef]
- Biasco, L. Integration Site Analysis in Gene Therapy Patients: Expectations and Reality. Hum. Gene Ther. 2017, 28, 1122–1129. [Google Scholar] [CrossRef]
- Drack, A.V.; Bhattarai, S.; Seo, S.; Stone, E.M.; Sheffield, V.; Mullins, R.; Tucker, B.A. Overcoming the Overexpression Toxicity of Gene Replacement Therapy for Bardet Biedl Syndrome Type 1. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4378. [Google Scholar]
- Alhaji, S.Y.; Ngai, S.C.; Abdullah, S. Silencing of Transgene Expression in Mammalian Cells by DNA Methylation and Histone Modifications in Gene Therapy Perspective. Biotechnol. Genet. Eng. Rev. 2019, 35, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, N.; Liou, A.; Patel, P.; Palmer, D.; Grove, N.; Finegold, M.; Piccolo, P.; Donnachie, E.; Rice, K.; Beaudet, A.; et al. Balloon Catheter Delivery of Helper-Dependent Adenoviral Vector Results in Sustained, Therapeutic HFIX Expression in Rhesus Macaques. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 1863–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Institut National de la Santé Et de la Recherche Médicale, France Phase I/II. Ex Vivo Gene Therapy Clinical Trial for RDEB Using Autologous Skin Equivalent Grafts Genetically Corrected With a COL7A1-Encoding SIN Retroviral Vector. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04186650 (accessed on 29 November 2022).
- So, J.Y.; Nazaroff, J.; Iwummadu, C.V.; Harris, N.; Gorell, E.S.; Fulchand, S.; Bailey, I.; McCarthy, D.; Siprashvili, Z.; Marinkovich, M.P.; et al. Long-Term Safety and Efficacy of Gene-Corrected Autologous Keratinocyte Grafts for Recessive Dystrophic Epidermolysis Bullosa. Orphanet J. Rare Dis. 2022, 17, 377. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.C.; Parry, T.J.; Zhang, P.; Majumdar, A.; Krishnan, S.; Regula, L.K.; O’Malley, M.; Coghlan, S.; Yogesha, S.D.; Ramasamy, S.; et al. Preclinical Evaluation of a Modified HSV-1 Vector Encoding Human TGM1 for the Treatment of Autosomal Recessive Congenital Ichthyosis (ARCI). J. Investig. Dermatol. 2020, 141. [Google Scholar] [CrossRef]
- Lwin, S.M.; Syed, F.; Di, W.L.; Kadiyirire, T.; Liu, L.; Guy, A.; Petrova, A.; Abdul-Wahab, A.; Reid, F.; Phillips, R.; et al. Safety and Early Efficacy Outcomes for Lentiviral Fibroblast Gene Therapy in Recessive Dystrophic Epidermolysis Bullosa. JCI Insight 2019, 4, e126243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Shukla, S.; Yang, C.; Zhang, M.; Fatma, Z.; Lingamaneni, M.; Abesteh, S.; Lane, S.T.; Xiong, X.; Wang, Y.; et al. TALEN Outperforms Cas9 in Editing Heterochromatin Target Sites. Nat. Commun. 2021, 12, 606. [Google Scholar] [CrossRef] [PubMed]
- Shinkuma, S.; Guo, Z.; Christiano, A.M. Site-Specific Genome Editing for Correction of Induced Pluripotent Stem Cells Derived from Dominant Dystrophic Epidermolysis Bullosa. Proc. Natl. Acad. Sci. USA 2016, 113, 5676–5681. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Kawagoe, S.; Tamai, K.; Nakagawa, H.; Asahina, A.; Okano, H.J. Footprint-Free Gene Mutation Correction in Induced Pluripotent Stem Cell (IPSC) Derived from Recessive Dystrophic Epidermolysis Bullosa (RDEB) Using the CRISPR/Cas9 and PiggyBac Transposon System. J. Dermatol. Sci. 2020, 98, 163–172. [Google Scholar] [CrossRef]
- Kocher, T.; Wagner, R.N.; Klausegger, A.; Guttmann-Gruber, C.; Hainzl, S.; Bauer, J.W.; Reichelt, J.; Koller, U. Improved Double-Nicking Strategies for COL7A1-Editing by Homologous Recombination. Mol. Ther.-Nucleic Acids 2019, 18, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Bonafont, J.; Mencía, Á.; García, M.; Torres, R.; Rodríguez, S.; Carretero, M.; Chacón-Solano, E.; Modamio-Høybjør, S.; Marinas, L.; León, C.; et al. Clinically Relevant Correction of Recessive Dystrophic Epidermolysis Bullosa by Dual SgRNA CRISPR/Cas9-Mediated Gene Editing. Mol. Ther. 2019, 27, 986–998. [Google Scholar] [CrossRef]
- Richter, M.F.; Zhao, K.T.; Eton, E.; Lapinaite, A.; Newby, G.A.; Thuronyi, B.W.; Wilson, C.; Koblan, L.W.; Zeng, J.; Bauer, D.E.; et al. Phage-Assisted Evolution of an Adenine Base Editor with Improved Cas Domain Compatibility and Activity. Nat. Biotechnol. 2020, 38, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of T to G C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive Guide Selection for CRISPR/Cas9 Genome Editing Experiments and Screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [Green Version]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR Web Toolbox beyond Genome Editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandage, R.; Després, P.C.; Yachie, N.; Landry, C.R. Beditor: A Computational Workflow for Designing Libraries of Guide RNAs for CRISPR-Mediated Base Editing. Genetics 2019, 212, 377–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, G.-H.; Park, J.; Lim, K.; Kim, S.; Yu, J.; Yu, E.; Kim, S.-T.; Eils, R.; Kim, J.-S.; Bae, S. Web-Based Design and Analysis Tools for CRISPR Base Editing. BMC Bioinform. 2018, 19, 542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, J.Y.; Grünewald, J.; Szalay, R.; Shih, J.; Anzalone, A.V.; Lam, K.C.; Shen, M.W.; Petri, K.; Liu, D.R.; Joung, J.K.; et al. PrimeDesign Software for Rapid and Simplified Design of Prime Editing Guide RNAs. Nat. Commun. 2021, 12, 1034. [Google Scholar] [CrossRef] [PubMed]
- Siegner, S.M.; Karasu, M.E.; Schröder, M.S.; Kontarakis, Z.; Corn, J.E. PnB Designer: A Web Application to Design Prime and Base Editor Guide RNAs for Animals and Plants. BMC Bioinform. 2021, 22, 101. [Google Scholar] [CrossRef]
- Park, J.; Yoon, J.; Kwon, D.; Han, M.-J.; Choi, S.; Park, S.; Lee, J.; Lee, K.; Lee, J.; Lee, S.; et al. Enhanced Genome Editing Efficiency of CRISPR PLUS: Cas9 Chimeric Fusion Proteins. Sci. Rep. 2021, 11, 16199. [Google Scholar] [CrossRef]
- Li, G.; Zhang, X.; Zhong, C.; Mo, J.; Quan, R.; Yang, J.; Liu, D.; Li, Z.; Yang, H.; Wu, Z. Small Molecules Enhance CRISPR/Cas9-Mediated Homology-Directed Genome Editing in Primary Cells. Sci. Rep. 2017, 7, 8943. [Google Scholar] [CrossRef] [Green Version]
- Ray, U.; Vartak, S.V.; Raghavan, S.C. NHEJ Inhibitor SCR7 and Its Different Forms: Promising CRISPR Tools for Genome Engineering. Gene 2020, 763, 144997. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Sharma, S.; Ray, U.; Manjunath, M.; Lakshmanan, D.; Vartak, S.V.; Gopinatha, V.K.; Srivastava, M.; Kempegowda, M.; Choudhary, B.; et al. SCR7, an Inhibitor of NHEJ Can Sensitize Tumor Cells to Ionization Radiation. Mol. Carcinog. 2021, 60, 627–643. [Google Scholar] [CrossRef]
- Li, Y.-H.; Wang, X.; Pan, Y.; Lee, D.-H.; Chowdhury, D.; Kimmelman, A.C. Inhibition of Non-Homologous End Joining Repair Impairs Pancreatic Cancer Growth and Enhances Radiation Response. PLoS ONE 2012, 7, e39588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charpentier, M.; Khedher, A.H.Y.; Menoret, S.; Brion, A.; Lamribet, K.; Dardillac, E.; Boix, C.; Perrouault, L.; Tesson, L.; Geny, S.; et al. CtIP Fusion to Cas9 Enhances Transgene Integration by Homology-Dependent Repair. Nat. Commun. 2018, 9, 1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Ruan, J.; Song, J.; Wen, L.; Yang, D.; Zhao, J.; Xia, X.; Chen, Y.E.; Zhang, J.; Xu, J. MiCas9 Increases Large Size Gene Knock-in Rates and Reduces Undesirable on-Target and off-Target Indel Edits. Nat. Commun. 2020, 11, 6082. [Google Scholar] [CrossRef] [PubMed]
- Koblan, L.W.; Doman, J.L.; Wilson, C.; Levy, J.M.; Tay, T.; Newby, G.A.; Maianti, J.P.; Raguram, A.; Liu, D.R. Improving Cytidine and Adenine Base Editors by Expression Optimization and Ancestral Reconstruction. Nat. Biotechnol. 2018, 36, 843–846. [Google Scholar] [CrossRef]
- Li, M.; Zhong, A.; Wu, Y.; Sidharta, M.; Beaury, M.; Zhao, X.; Studer, L.; Zhou, T. Transient Inhibition of P53 Enhances Prime Editing and Cytosine Base-Editing Efficiencies in Human Pluripotent Stem Cells. Nat. Commun. 2022, 13, 6354. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Hussmann, J.A.; Yan, J.; Knipping, F.; Ravisankar, P.; Chen, P.-F.; Chen, C.; Nelson, J.W.; Newby, G.A.; Sahin, M.; et al. Enhanced Prime Editing Systems by Manipulating Cellular Determinants of Editing Outcomes. Cell 2021, 184, 5635–5652.e29. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.W.; Randolph, P.B.; Shen, S.P.; Everette, K.A.; Chen, P.J.; Anzalone, A.V.; An, M.; Newby, G.A.; Chen, J.C.; Hsu, A.; et al. Engineered PegRNAs Improve Prime Editing Efficiency. Nat. Biotechnol. 2022, 40, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Hainzl, S.; Peking, P.; Kocher, T.; Murauer, E.M.; Larcher, F.; Del Rio, M.; Duarte, B.; Steiner, M.; Klausegger, A.; Bauer, J.W.; et al. COL7A1 Editing via CRISPR/Cas9 in Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. 2017, 25, 2573–2584. [Google Scholar] [CrossRef] [Green Version]
- Perret, R.; Olsen, K. Magnetic Selection for Consistent Cellular Starting Material in Autologous Cell Therapy Manufacture. Cell Gene Ther. Insights 2022, 08, 97–112. [Google Scholar] [CrossRef]
- Doss, M.X.; Sachinidis, A. Current Challenges of IPSC-Based Disease Modeling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoepfler, P.S. Key Anticipated Regulatory Issues for Clinical Use of Human Induced Pluripotent Stem Cells. Regen. Med. 2012, 7, 713–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell-Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef]
- Merkle, F.T.; Ghosh, S.; Kamitaki, N.; Mitchell, J.; Avior, Y.; Mello, C.; Kashin, S.; Mekhoubad, S.; Ilic, D.; Charlton, M.; et al. Human Pluripotent Stem Cells Recurrently Acquire and Expand Dominant Negative P53 Mutations. Nature 2017, 545, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Kern, J.S.; Loeckermann, S.; Fritsch, A.; Hausser, I.; Roth, W.; Magin, T.M.; Mack, C.; Müller, M.L.; Paul, O.; Ruther, P.; et al. Mechanisms of Fibroblast Cell Therapy for Dystrophic Epidermolysis Bullosa: High Stability of Collagen VII Favors Long-Term Skin Integrity. Mol. Ther. 2009, 17, 1605–1615. [Google Scholar] [CrossRef]
- Bao, X.R.; Pan, Y.; Lee, C.M.; Davis, T.H.; Bao, G. Tools for Experimental and Computational Analyses of Off-Target Editing by Programmable Nucleases. Nat. Protoc. 2021, 16, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized SgRNA Design to Maximize Activity and Minimize Off-Target Effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheriff, A.; Guri, I.; Zebrowska, P.; Llopis-Hernandez, V.; Brooks, I.R.; Tekkela, S.; Subramaniam, K.; Gebrezgabher, R.; Naso, G.; Petrova, A.; et al. ABE8e Adenine Base Editor Precisely and Efficiently Corrects a Recurrent COL7A1 Nonsense Mutation. Sci. Rep. 2022, 12, 19643. [Google Scholar] [CrossRef] [PubMed]
- Listgarten, J.; Weinstein, M.; Kleinstiver, B.P.; Sousa, A.A.; Joung, J.K.; Crawford, J.; Gao, K.; Hoang, L.; Elibol, M.; Doench, J.G.; et al. Prediction of Off-Target Activities for the End-to-End Design of CRISPR Guide RNAs. Nat. Biomed. Eng. 2018, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Abadi, S.; Yan, W.X.; Amar, D.; Mayrose, I. A Machine Learning Approach for Predicting CRISPR-Cas9 Cleavage Efficiencies and Patterns Underlying Its Mechanism of Action. PLoS Comput. Biol. 2017, 13, e1005807. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Shandilya, H.; D’Alessio, J.M.; O’Connor, K.; Durocher, J.; Gerard, G.F. Mutation Detection Using Surveyor Nuclease. BioTechniques 2004, 36, 702–707. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, H.J.; Kim, H.; Cho, S.W.; Kim, J.-S. Targeted Genome Editing in Human Cells with Zinc Finger Nucleases Constructed via Modular Assembly. Genome Res. 2009, 19, 1279–1288. [Google Scholar] [CrossRef] [Green Version]
- Webber, B.R.; Osborn, M.J.; McElroy, A.N.; Twaroski, K.; Lonetree, C.; DeFeo, A.P.; Xia, L.; Eide, C.; Lees, C.J.; McElmurry, R.T.; et al. CRISPR/Cas9-Based Genetic Correction for Recessive Dystrophic Epidermolysis Bullosa. Npj Regen. Med. 2016, 1, 16014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkins, A.; Chung, C.-H.; Allen, A.G.; Dampier, W.; Gurrola, T.E.; Sariyer, I.K.; Nonnemacher, M.R.; Wigdahl, B. Off-Target Analysis in Gene Editing and Applications for Clinical Translation of CRISPR/Cas9 in HIV-1 Therapy. Front. Genome Ed. 2021, 3, 673022. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Kousholt, A.N.; Harmsen, T.; Leemans, C.; Chen, T.; Jonkers, J.; van Steensel, B. Easy Quantification of Template-Directed CRISPR/Cas9 Editing. Nucleic Acids Res. 2018, 46, e58. [Google Scholar] [CrossRef]
- Conant, D.; Hsiau, T.; Rossi, N.; Oki, J.; Maures, T.; Waite, K.; Yang, J.; Joshi, S.; Kelso, R.; Holden, K.; et al. Inference of CRISPR Edits from Sanger Trace Data. CRISPR J. 2022, 5, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Sentmanat, M.F.; Peters, S.T.; Florian, C.P.; Connelly, J.P.; Pruett-Miller, S.M. A Survey of Validation Strategies for CRISPR-Cas9 Editing. Sci. Rep. 2018, 8, 888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.R. Next-Generation Sequencing in High-Sensitive Detection of Mutations in Tumors: Challenges, Advances, and Applications. J. Mol. Diagn. JMD 2020, 22, 994–1007. [Google Scholar] [CrossRef]
- Martin, F.; Sánchez-Hernández, S.; Gutiérrez-Guerrero, A.; Pinedo-Gomez, J.; Benabdellah, K. Biased and Unbiased Methods for the Detection of Off-Target Cleavage by CRISPR/Cas9: An Overview. Int. J. Mol. Sci. 2016, 17, 1507. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Bae, S.; Park, J.; Kim, E.; Kim, S.; Yu, H.R.; Hwang, J.; Kim, J.-I.; Kim, J.-S. Digenome-Seq: Genome-Wide Profiling of CRISPR-Cas9 off-Target Effects in Human Cells. Nat. Methods 2015, 12, 237–243. [Google Scholar] [CrossRef]
- Kim, D.; Kim, J.-S. DIG-Seq: A Genome-Wide CRISPR off-Target Profiling Method Using Chromatin DNA. Genome Res. 2018, 28, 1894–1900. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-Seq: A Highly Sensitive in Vitro Screen for Genome-Wide CRISPR-Cas9 Nuclease off-Targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-Seq Enables Genome-Wide Profiling of off-Target Cleavage by CRISPR-Cas Nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Lim, K.; Kim, S.-T.; Yoon, S.; Kim, K.; Ryu, S.-M.; Kim, J.-S. Genome-Wide Target Specificities of CRISPR RNA-Guided Programmable Deaminases. Nat. Biotechnol. 2017, 35, 475–480. [Google Scholar] [CrossRef]
- Kim, D.; Kim, D.; Lee, G.; Cho, S.-I.; Kim, J.-S. Genome-Wide Target Specificity of CRISPR RNA-Guided Adenine Base Editors. Nat. Biotechnol. 2019, 37, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Doman, J.L.; Raguram, A.; Newby, G.A.; Liu, D.R. Evaluation and Minimization of Cas9-Independent off-Target DNA Editing by Cytosine Base Editors. Nat. Biotechnol. 2020, 38, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Beal, P.A. Off-Target Editing by CRISPR-Guided DNA Base Editors. Biochemistry 2019, 58, 3727–3734. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Komor, A.C.; Yeh, W.-H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.B.; Liu, D.R. Improving the DNA Specificity and Applicability of Base Editing through Protein Engineering and Protein Delivery. Nat. Commun. 2017, 8, 15790. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Zong, Y.; Gao, Q.; Zhu, Z.; Wang, Y.; Qin, P.; Liang, C.; Wang, D.; Qiu, J.-L.; Zhang, F.; et al. Cytosine, but Not Adenine, Base Editors Induce Genome-Wide off-Target Mutations in Rice. Science 2019, 364, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Zuo, E.; Sun, Y.; Wei, W.; Yuan, T.; Ying, W.; Sun, H.; Yuan, L.; Steinmetz, L.M.; Li, Y.; Yang, H. Cytosine Base Editor Generates Substantial Off-Target Single-Nucleotide Variants in Mouse Embryos. Science 2019, 364, 289–292. [Google Scholar] [CrossRef]
- Liang, P.; Xie, X.; Zhi, S.; Sun, H.; Zhang, X.; Chen, Y.; Chen, Y.; Xiong, Y.; Ma, W.; Liu, D.; et al. Genome-Wide Profiling of Adenine Base Editor Specificity by EndoV-Seq. Nat. Commun. 2019, 10, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath, E.; Shin, H.; Zhang, L.; Phue, J.-N.; Wu, W.W.; Shen, R.-F.; Jang, Y.-Y.; Revollo, J.; Ye, Z. Targeting Specificity of APOBEC-Based Cytosine Base Editor in Human IPSCs Determined by Whole Genome Sequencing. Nat. Commun. 2019, 10, 5353. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Sun, Y.; Yan, R.; Liu, Y.; Zuo, E.; Gu, C.; Han, L.; Wei, Y.; Hu, X.; Zeng, R.; et al. Off-Target RNA Mutation Induced by DNA Base Editing and Its Elimination by Mutagenesis. Nature 2019, 571, 275–278. [Google Scholar] [CrossRef]
- Grünewald, J.; Zhou, R.; Garcia, S.P.; Iyer, S.; Lareau, C.A.; Aryee, M.J.; Joung, J.K. Transcriptome-Wide off-Target RNA Editing Induced by CRISPR-Guided DNA Base Editors. Nature 2019, 569, 433–437. [Google Scholar] [CrossRef]
- Fan, J.; Ding, Y.; Ren, C.; Song, Z.; Yuan, J.; Chen, Q.; Du, C.; Li, C.; Wang, X.; Shu, W. Cytosine and Adenine Deaminase Base-Editors Induce Broad and Nonspecific Changes in Gene Expression and Splicing. Commun. Biol. 2021, 4, 882. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Leete, T.C.; Born, D.A.; Young, L.; Barrera, L.A.; Lee, S.-J.; Rees, H.A.; Ciaramella, G.; Gaudelli, N.M. Cytosine Base Editors with Minimized Unguided DNA and RNA Off-Target Events and High on-Target Activity. Nat. Commun. 2020, 11, 2052. [Google Scholar] [CrossRef]
- Grünewald, J.; Zhou, R.; Iyer, S.; Lareau, C.A.; Garcia, S.P.; Aryee, M.J.; Joung, J.K. CRISPR DNA Base Editors with Reduced RNA Off-Target and Self-Editing Activities. Nat. Biotechnol. 2019, 37, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Moon, S.B.; Ko, J.-H.; Kim, Y.-S.; Kim, D. Unbiased Investigation of Specificities of Prime Editing Systems in Human Cells. Nucleic Acids Res. 2020, 48, 10576–10589. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Fu, Z.-C.; Li, X.; Wang, Y.; Wei, J.; Li, G.; Wang, L.; Wu, J.; Huang, X.; Yang, L.; et al. Genomic and Transcriptomic Analyses of Prime Editing Guide RNA-Independent Off-Target Effects by Prime Editors. CRISPR J. 2022, 5, 276–293. [Google Scholar] [CrossRef]
- Yang, L.; Guell, M.; Byrne, S.; Yang, J.L.; De Los Angeles, A.; Mali, P.; Aach, J.; Kim-Kiselak, C.; Briggs, A.W.; Rios, X.; et al. Optimization of Scarless Human Stem Cell Genome Editing. Nucleic Acids Res. 2013, 41, 9049–9061. [Google Scholar] [CrossRef]
- Uitto, J.; Bruckner-Tuderman, L.; Christiano, A.M.; McGrath, J.A.; Has, C.; South, A.P.; Kopelan, B.; Robinson, E.C. Progress toward Treatment and Cure of Epidermolysis Bullosa: Summary of the DEBRA International Research Symposium EB2015. J. Investig. Dermatol. 2016, 136, 352–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Kriz, A.J.; Sharp, P.A. Target Specificity of the CRISPR-Cas9 System. Quant. Biol. 2014, 2, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Mitsunobu, H.; Yoshioka, S.; Suzuki, T.; Kondo, A.; Nishida, K. Cytosine Base Editing Systems with Minimized Off-Target Effect and Molecular Size. Nat. Commun. 2022, 13, 4531. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Wilson, C.; Doman, J.L.; Liu, D.R. Analysis and Minimization of Cellular RNA Editing by DNA Adenine Base Editors. Sci. Adv. 2019, 5, eaax5717. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Wang, Y.; Liu, Q.; Qiu, Y.; Zhong, Z.; Li, K.; Li, W.; Deng, Z.; Sun, Y. Improving the Precision of Base Editing by Bubble Hairpin Single Guide RNA. mBio 2021, 12, e00342-21. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome Editing with CRISPR–Cas Nucleases, Base Editors, Transposases and Prime Editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Arbab, M.; Shen, M.W.; Mok, B.; Wilson, C.; Matuszek, Ż.; Cassa, C.A.; Liu, D.R. Determinants of Base Editing Outcomes from Target Library Analysis and Machine Learning. Cell 2020, 182, 463–480.e30. [Google Scholar] [CrossRef]
- Stewart, M.P.; Sharei, A.; Ding, X.; Sahay, G.; Langer, R.; Jensen, K.F. In Vitro and Ex Vivo Strategies for Intracellular Delivery. Nature 2016, 538, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balantič, K.; Miklavčič, D.; Križaj, I.; Kramar, P. The Good and the Bad of Cell Membrane Electroporation. Acta Chim. Slov. 2021, 68, 753–764. [Google Scholar] [CrossRef]
- Batista Napotnik, T.; Polajžer, T.; Miklavčič, D. Cell Death Due to Electroporation—A Review. Bioelectrochemistry 2021, 141, 107871. [Google Scholar] [CrossRef]
- Uchida, N.; Drysdale, C.M.; Nassehi, T.; Gamer, J.; Yapundich, M.; DiNicola, J.; Shibata, Y.; Hinds, M.; Gudmundsdottir, B.; Haro-Mora, J.J.; et al. Cas9 Protein Delivery Non-Integrating Lentiviral Vectors for Gene Correction in Sickle Cell Disease. Mol. Ther. Methods Clin. Dev. 2021, 21, 121–132. [Google Scholar] [CrossRef]
- Wu, W.; Lu, Z.; Li, F.; Wang, W.; Qian, N.; Duan, J.; Zhang, Y.; Wang, F.; Chen, T. Efficient in Vivo Gene Editing Using Ribonucleoproteins in Skin Stem Cells of Recessive Dystrophic Epidermolysis Bullosa Mouse Model. Proc. Natl. Acad. Sci. USA 2017, 114, 1660–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picanço-Castro, V.; Pereira, C.G.; Covas, D.T.; Porto, G.S.; Athanassiadou, A.; Figueiredo, M.L. Emerging Patent Landscape for Non-Viral Vectors Used for Gene Therapy. Nat. Biotechnol. 2020, 38, 151–157. [Google Scholar] [CrossRef]
- Flotte, T.R.; Afione, S.A.; Solow, R.; Drumm, M.L.; Markakis, D.; Guggino, W.B.; Zeitlin, P.L.; Carter, B.J. Expression of the Cystic Fibrosis Transmembrane Conductance Regulator from a Novel Adeno-Associated Virus Promoter. J. Biol. Chem. 1993, 268, 3781–3790. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, Y.; He, L.; Pu, W.; Yu, W.; Li, Y.; Wu, Y.-T.; Xu, C.; Wei, Y.; Ding, Q.; et al. In Vivo AAV-CRISPR/Cas9-Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation 2020, 141, 67–79. [Google Scholar] [CrossRef]
- Davis, J.R.; Wang, X.; Witte, I.P.; Huang, T.P.; Levy, J.M.; Raguram, A.; Banskota, S.; Seidah, N.G.; Musunuru, K.; Liu, D.R. Efficient in Vivo Base Editing via Single Adeno-Associated Viruses with Size-Optimized Genomes Encoding Compact Adenine Base Editors. Nat. Biomed. Eng. 2022, 6, 1272–1283. [Google Scholar] [CrossRef]
- Modlich, U.; Baum, C. Preventing and Exploiting the Oncogenic Potential of Integrating Gene Vectors. J. Clin. Investig. 2009, 119, 755–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The Genotoxic Potential of Retroviral Vectors Is Strongly Modulated by Vector Design and Integration Site Selection in a Mouse Model of HSC Gene Therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, M.; Gregori, S.; Hauben, E.; Brown, B.D.; Sergi, L.S.; Naldini, L.; Roncarolo, M.-G. HIV-1-Derived Lentiviral Vectors Directly Activate Plasmacytoid Dendritic Cells, Which in Turn Induce the Maturation of Myeloid Dendritic Cells. Hum. Gene Ther. 2011, 22, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Kenjo, E.; Hozumi, H.; Makita, Y.; Iwabuchi, K.A.; Fujimoto, N.; Matsumoto, S.; Kimura, M.; Amano, Y.; Ifuku, M.; Naoe, Y.; et al. Low Immunogenicity of LNP Allows Repeated Administrations of CRISPR-Cas9 MRNA into Skeletal Muscle in Mice. Nat. Commun. 2021, 12, 7101. [Google Scholar] [CrossRef]
- Ain, Q.U.; Campos, E.V.R.; Huynh, A.; Witzigmann, D.; Hedtrich, S. Gene Delivery to the Skin—How Far Have We Come? Trends Biotechnol. 2021, 39, 474–487. [Google Scholar] [CrossRef]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-Based Analysis of Lipid Nanoparticle-Mediated SiRNA Delivery, Intracellular Trafficking and Endosomal Escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef]
- Paramasivam, P.; Franke, C.; Stöter, M.; Höijer, A.; Bartesaghi, S.; Sabirsh, A.; Lindfors, L.; Arteta, M.Y.; Dahlén, A.; Bak, A.; et al. Endosomal Escape of Delivered MRNA from Endosomal Recycling Tubules Visualized at the Nanoscale. J. Cell Biol. 2022, 221, e202110137. [Google Scholar] [CrossRef]
- Miteva, M.; Kirkbride, K.C.; Kilchrist, K.V.; Werfel, T.A.; Li, H.; Nelson, C.E.; Gupta, M.K.; Giorgio, T.D.; Duvall, C.L. Tuning PEGylation of Mixed Micelles to Overcome Intracellular and Systemic SiRNA Delivery Barriers. Biomaterials 2015, 38, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Lubich, C.; Allacher, P.; de la Rosa, M.; Bauer, A.; Prenninger, T.; Horling, F.M.; Siekmann, J.; Oldenburg, J.; Scheiflinger, F.; Reipert, B.M. The Mystery of Antibodies Against Polyethylene Glycol (PEG)—What Do We Know? Pharm. Res. 2016, 33, 2239–2249. [Google Scholar] [CrossRef]
- Chiappini, C.; De Rosa, E.; Martinez, J.O.; Liu, X.; Steele, J.; Stevens, M.M.; Tasciotti, E. Biodegradable Silicon Nanoneedles Delivering Nucleic Acids Intracellularly Induce Localized in Vivo Neovascularization. Nat. Mater. 2015, 14, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Römgens, A.M.; Bader, D.L.; Bouwstra, J.A.; Baaijens, F.P.T.; Oomens, C.W.J. Monitoring the Penetration Process of Single Microneedles with Varying Tip Diameters. J. Mech. Behav. Biomed. Mater. 2014, 40, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Chiappini, C.; Almeida, C. Silicon Nanoneedles for Drug Delivery. In Semiconducting Silicon Nanowires for Biomedical Applications; Woodhead Publishing: Cambridge, UK, 2014; pp. 144–167. ISBN 978-0-85709-766-8. [Google Scholar]
- Chiappini, C.; Martinez, J.O.; De Rosa, E.; Almeida, C.S.; Tasciotti, E.; Stevens, M.M. Biodegradable Nanoneedles for Localized Delivery of Nanoparticles in Vivo: Exploring the Biointerface. ACS Nano 2015, 9, 5500–5509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Jang, H.; Kim, B.; Kim, M.K.; Wie, D.S.; Lee, H.S.; Kim, D.R.; Lee, C.H. Flexible Elastomer Patch with Vertical Silicon Nanoneedles for Intracellular and Intratissue Nanoinjection of Biomolecules. Sci. Adv. 2018, 4, eaau6972. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Zhang, Z.; Li, L. Micro/Nano Needles for Advanced Drug Delivery. Prog. Nat. Sci. Mater. Int. 2020, 30, 589–596. [Google Scholar] [CrossRef]
- Wan, T.; Pan, Q.; Ping, Y. Microneedle-Assisted Genome Editing: A Transdermal Strategy of Targeting NLRP3 by CRISPR-Cas9 for Synergistic Therapy of Inflammatory Skin Disorders. Sci. Adv. 2021, 7, eabe2888. [Google Scholar] [CrossRef]
- Jayarajan, V.; Kounatidou, E.; Qasim, W.; Di, W.-L. Ex Vivo Gene Modification Therapy for Genetic Skin Diseases-Recent Advances in Gene Modification Technologies and Delivery. Exp. Dermatol. 2021, 30, 887–896. [Google Scholar] [CrossRef]
- Petrof, G.; Martinez-Queipo, M.; Mellerio, J.E.; Kemp, P.; McGrath, J.A. Fibroblast Cell Therapy Enhances Initial Healing in Recessive Dystrophic Epidermolysis Bullosa Wounds: Results of a Randomized, Vehicle-Controlled Trial. Br. J. Dermatol. 2013, 169, 1025–1033. [Google Scholar] [CrossRef]
- Castle Creek Biosciences, LLC. A Phase I/II Study of FCX-007 (Genetically-Modified Autologous Human Dermal Fibroblasts) for Recessive Dystrophic Epidermolysis Bullosa (RDEB). 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT02810951 (accessed on 2 December 2022).
- Ha, D.H.; Kim, H.-K.; Lee, J.; Kwon, H.H.; Park, G.-H.; Yang, S.H.; Jung, J.Y.; Choi, H.; Lee, J.H.; Sung, S.; et al. Mesenchymal Stem/Stromal Cell-Derived Exosomes for Immunomodulatory Therapeutics and Skin Regeneration. Cells 2020, 9, 1157. [Google Scholar] [CrossRef]
- Andrzejewska, A.; Lukomska, B.; Janowski, M. Concise Review: Mesenchymal Stem Cells: From Roots to Boost. Stem Cells Dayt. Ohio 2019, 37, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Sabapathy, V.; Kumar, S. HiPSC-Derived IMSCs: NextGen MSCs as an Advanced Therapeutically Active Cell Resource for Regenerative Medicine. J. Cell. Mol. Med. 2016, 20, 1571–1588. [Google Scholar] [CrossRef] [Green Version]
- Rashidghamat, E.; Kadiyirire, T.; Ayis, S.; Petrof, G.; Liu, L.; Pullabhatla, V.; Ainali, C.; Guy, A.; Aristodemou, S.; McMillan, J.R.; et al. Phase I/II Open-Label Trial of Intravenous Allogeneic Mesenchymal Stromal Cell Therapy in Adults with Recessive Dystrophic Epidermolysis Bullosa. J. Am. Acad. Dermatol. 2020, 83, 447–454. [Google Scholar] [CrossRef]
- Hanson, S.E.; Bentz, M.L.; Hematti, P. Mesenchymal Stem Cell Therapy for Nonhealing Cutaneous Wounds. Plast. Reconstr. Surg. 2010, 125, 510–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; De Rosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the Entire Human Epidermis Using Transgenic Stem Cells. Nature 2017, 551, 327–332. [Google Scholar] [CrossRef]
- Enzo, E.; Secone Seconetti, A.; Forcato, M.; Tenedini, E.; Polito, M.P.; Sala, I.; Carulli, S.; Contin, R.; Peano, C.; Tagliafico, E.; et al. Single-Keratinocyte Transcriptomic Analyses Identify Different Clonal Types and Proliferative Potential Mediated by FOXM1 in Human Epidermal Stem Cells. Nat. Commun. 2021, 12, 2505. [Google Scholar] [CrossRef]
- Herskovitz, I.; Hughes, O.B.; Macquhae, F.; Rakosi, A.; Kirsner, R. Epidermal Skin Grafting. Int. Wound J. 2016, 13 (Suppl. 3), 52–56. [Google Scholar] [CrossRef] [Green Version]
- Hachach-Haram, N.; Bystrzonowski, N.; Kanapathy, M.; Smith, O.; Harding, K.; Mosahebi, A.; Richards, T. A Prospective, Multicentre Study on the Use of Epidermal Grafts to Optimise Outpatient Wound Management. Int. Wound J. 2017, 14, 241–249. [Google Scholar] [CrossRef]
- Heinonen, S.; Männikkö, M.; Klement, J.F.; Whitaker-Menezes, D.; Murphy, G.F.; Uitto, J. Targeted Inactivation of the Type VII Collagen Gene (Col7a1) in Mice Results in Severe Blistering Phenotype: A Model for Recessive Dystrophic Epidermolysis Bullosa. J. Cell Sci. 1999, 112, 3641–3648. [Google Scholar] [CrossRef]
- Fritsch, A.; Loeckermann, S.; Kern, J.S.; Braun, A.; Bösl, M.R.; Bley, T.A.; Schumann, H.; von Elverfeldt, D.; Paul, D.; Erlacher, M.; et al. A Hypomorphic Mouse Model of Dystrophic Epidermolysis Bullosa Reveals Mechanisms of Disease and Response to Fibroblast Therapy. J. Clin. Investig. 2008, 118, 1669–1679. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.M.; Shelke, R.; Nyström, A.; Laver, N.; Sampson, J.F.; Zhiyi, C.; Bhat, N.; Panjwani, N. Collagen VII Deficient Mice Show Morphologic and Histologic Corneal Changes That Phenotypically Mimic Human Dystrophic Epidermolysis Bullosa of the Eye. Exp. Eye Res. 2018, 175, 133–141. [Google Scholar] [CrossRef]
- Webber, B.R.; O’Connor, K.T.; McElmurry, R.T.; Durgin, E.N.; Eide, C.; Lees, C.J.; Riddle, M.J.; Mathews, W.; Frank, N.Y.; Kluth, M.A.; et al. Rapid Generation of Col7a1−/− Mouse Model of Recessive Dystrophic Epidermolysis Bullosa and Partial Rescue via Immunosuppressive Dermal Mesenchymal Stem Cells. Lab. Investig. J. Technol. Methods Pathol. 2017, 97, 1218–1224. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.R.C.; Nyström, A.; Nowell, C.J.; Hausser, I.; Gretzmeier, C.; Robertson, S.J.; Varigos, G.A.; Has, C.; Kern, J.S.; Pang, K.C. Mouse Models for Dominant Dystrophic Epidermolysis Bullosa Carrying Common Human Point Mutations Recapitulate the Human Disease. Dis. Model. Mech. 2021, 14, dmm048082. [Google Scholar] [CrossRef]
- Johnson, A.L.; Peterson, S.M.; Terry, M.M.L.; Ferguson, B.; Colgin, L.M.; Lewis, A.D. Spontaneous KRT5 Gene Mutation in Rhesus Macaques (Macaca Mulatta): A Novel Nonhuman Primate Model of Epidermolysis Bullosa Simplex. Vet. Pathol. 2020, 57, 344–348. [Google Scholar] [CrossRef]
- Kim, S.H.; Choi, H.Y.; So, J.-H.; Kim, C.-H.; Ho, S.-Y.; Frank, M.; Li, Q.; Uitto, J. Zebrafish Type XVII Collagen: Gene Structures, Expression Profiles, and Morpholino “Knock-down” Phenotypes. Matrix Biol. J. Int. Soc. Matrix Biol. 2010, 29, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Mastrodonato, V.; Beznoussenko, G.; Mironov, A.; Ferrari, L.; Deflorian, G.; Vaccari, T. A Genetic Model of CEDNIK Syndrome in Zebrafish Highlights the Role of the SNARE Protein Snap29 in Neuromotor and Epidermal Development. Sci. Rep. 2019, 9, 1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohnekamp, J.; Cryderman, D.E.; Paululat, A.; Baccam, G.C.; Wallrath, L.L.; Magin, T.M. A Drosophila Model of Epidermolysis Bullosa Simplex. J. Investig. Dermatol. 2015, 135, 2031–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Disease | Therapeutic | Gene Therapy Delivery Method | Phase | Outcome | Reference |
|---|---|---|---|---|---|
| Netherton syndrome | Autologous skin sheets containing additional SPINK5 gene | Lentiviral Vector | I | Transient functional correction in 1 Patient | [23] |
| Recessive dystrophic epidermolysis bullosa (RDEB) | Autologous skin sheets containing additional COL7A1 gene | Retroviral Self Inactivating | I/II | In progress | [33] |
| RDEB | Autologous epidermal sheets containing additional COL7A1 gene | Retroviral | I/II | Favourable safety and efficacy outcomes—Phase III in progress | [34] |
| DEB | Topical beremagene geperpavec (carries HSV1-COL7) applied to wounds | Self-inactivating HSV1 | I/II | Durable wound closure with minimal adverse events—Phase III in progress | [25] |
| Autosomal recessive congenital ichthyosis | Topically administered KB105 containing TGM-1 | Self-inactivating HSV-1 | I/II | In progress | [35] |
| junctional epidermolysis bullosa | Epidermal autograft containing LAMB5 | Gamma-retroviral | II/III | In progress | [24] |
| RDEB | Intradermal Injections of COL7A1-modified autologous fibroblasts | Self-inactivating lentivirus | I | Increased C7 observed after 12 months but no mature anchoring Fibrils | [36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brooks, I.R.; Sheriff, A.; Moran, D.; Wang, J.; Jacków, J. Challenges of Gene Editing Therapies for Genodermatoses. Int. J. Mol. Sci. 2023, 24, 2298. https://doi.org/10.3390/ijms24032298
Brooks IR, Sheriff A, Moran D, Wang J, Jacków J. Challenges of Gene Editing Therapies for Genodermatoses. International Journal of Molecular Sciences. 2023; 24(3):2298. https://doi.org/10.3390/ijms24032298
Chicago/Turabian StyleBrooks, Imogen R., Adam Sheriff, Declan Moran, Jingbo Wang, and Joanna Jacków. 2023. "Challenges of Gene Editing Therapies for Genodermatoses" International Journal of Molecular Sciences 24, no. 3: 2298. https://doi.org/10.3390/ijms24032298