
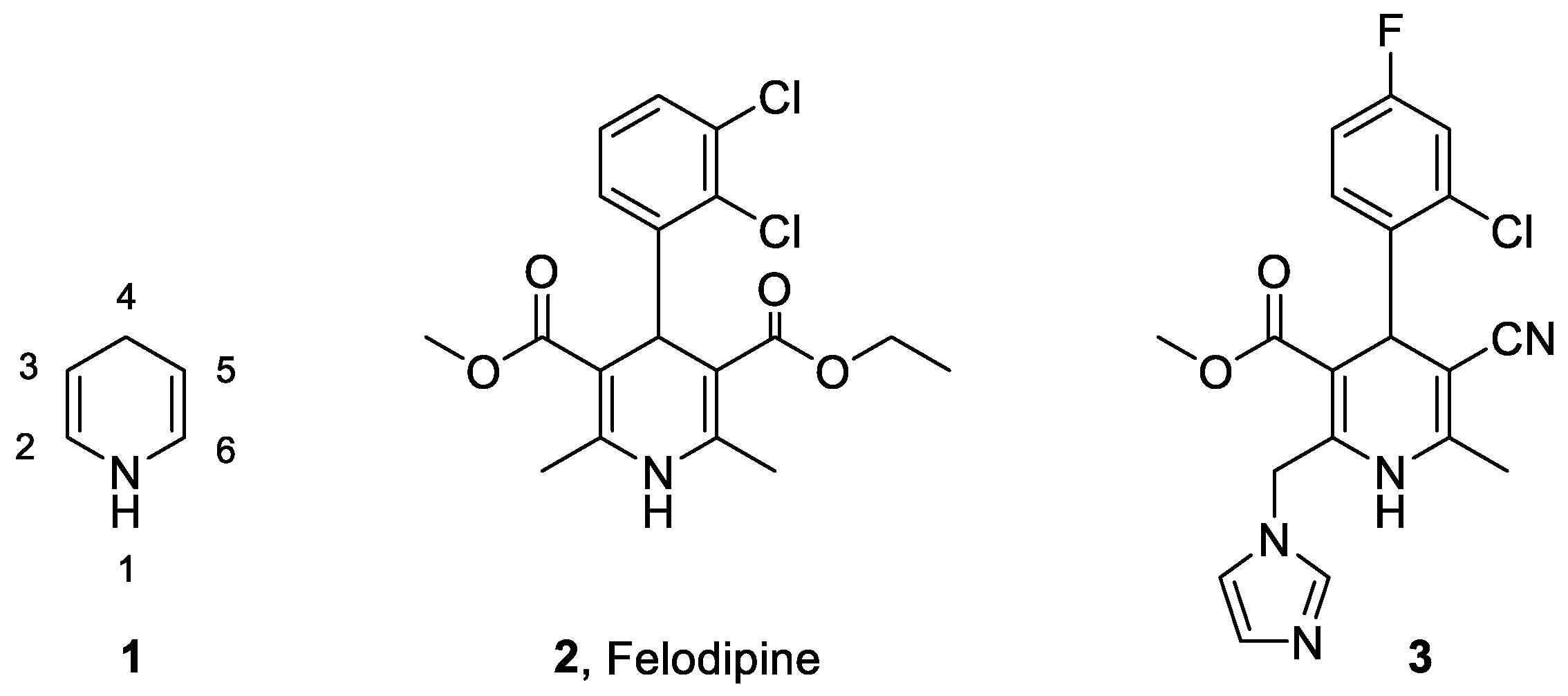
Novel 1,4-Dihydropyridine Derivatives as Mineralocorticoid Receptor Antagonists

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Selection of New 1,4-DHPs by Molecular Docking Studies on Virtual Focused Libraries
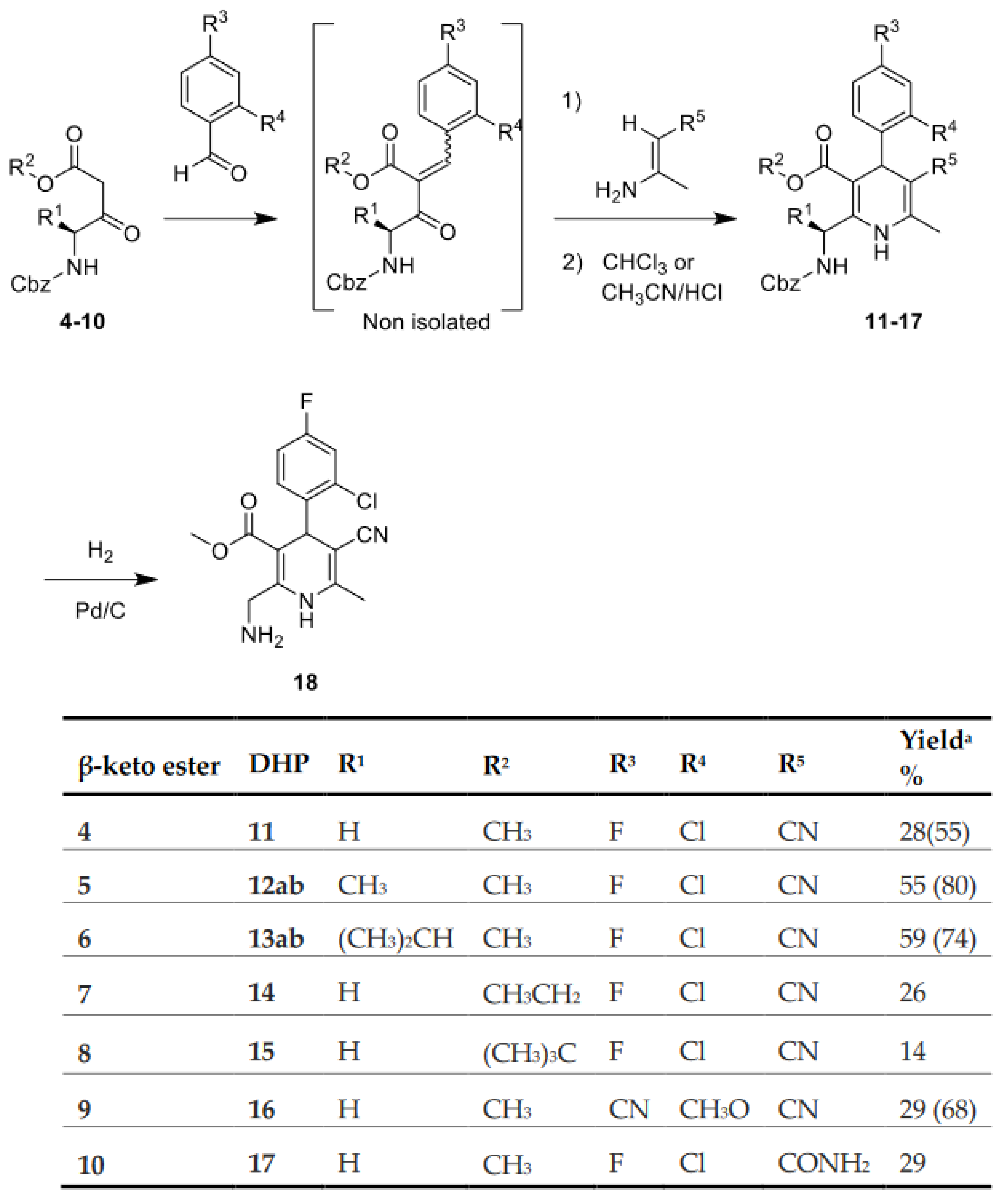
2.2. Synthesis of 1,4-DHPs
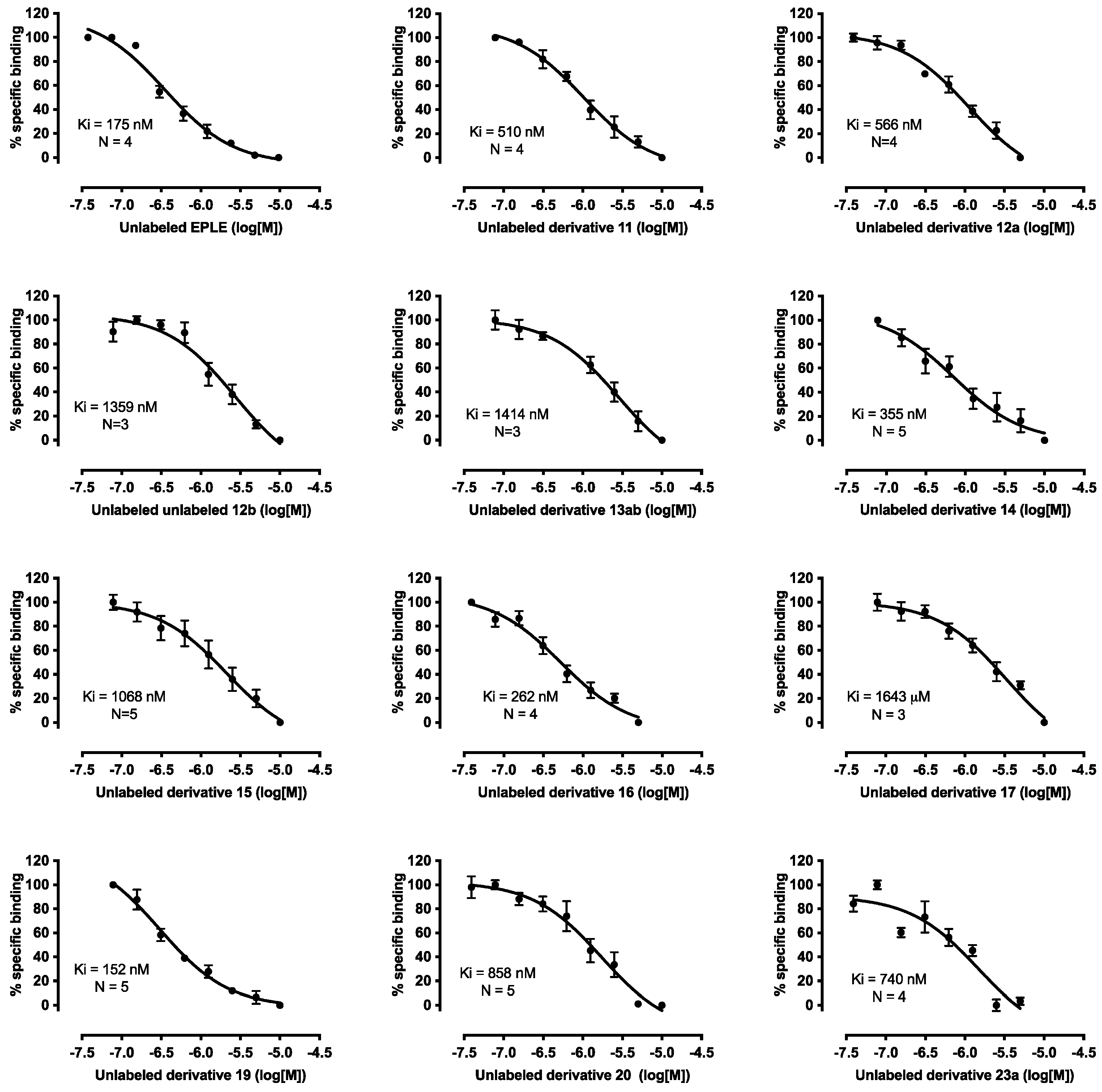
2.3. Biological Evaluation
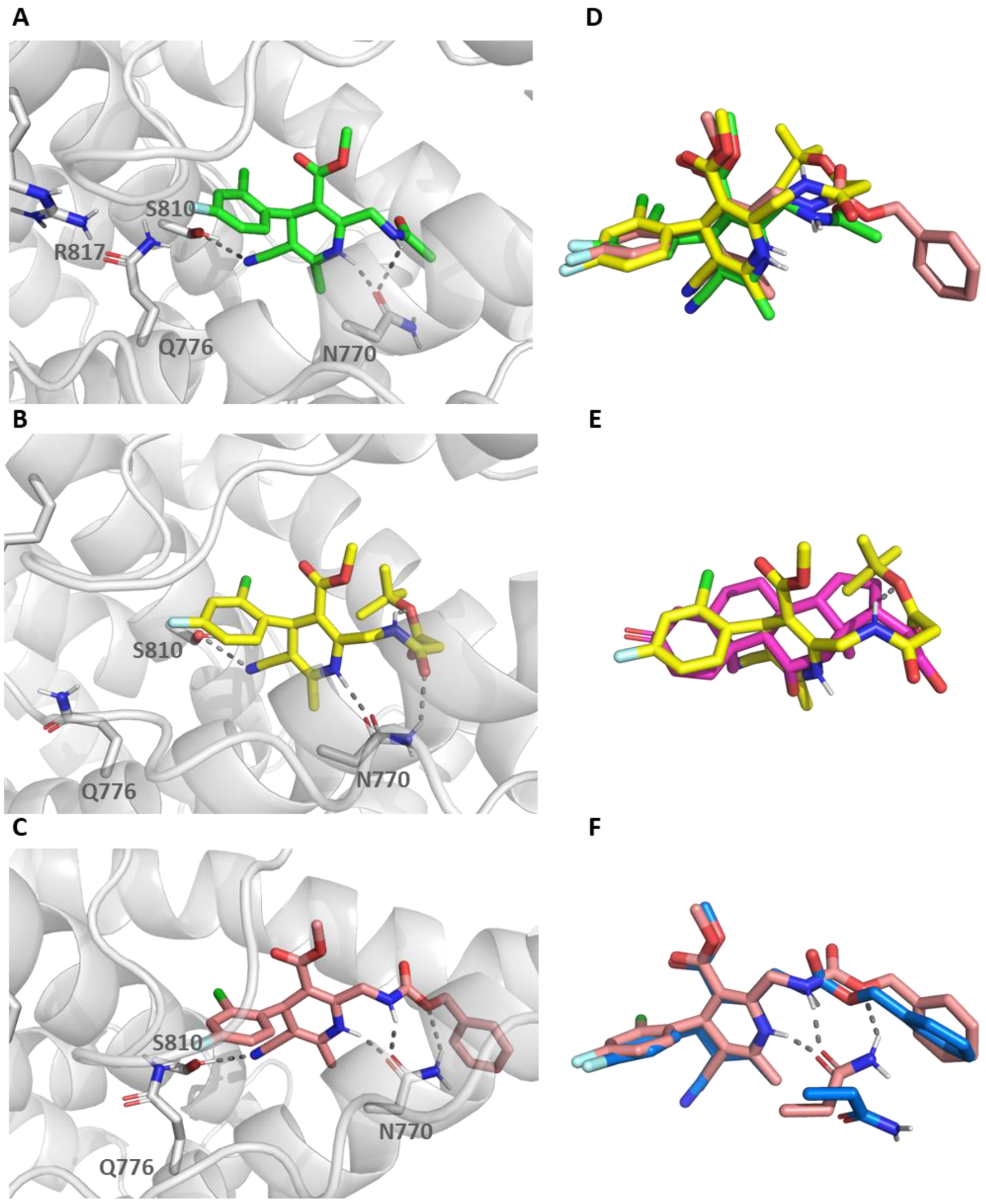
2.4. Molecular Dynamic Simulations
3. Conclusions
4. Materials and Methods
4.1. Molecular Modelling Studies
4.1.1. Induced Fit Docking Studies
4.1.2. Molecular Dynamic Studies
4.2. Chemistry
4.2.1. Synthesis of Dihydropiridines
4.2.2. Acylation of Primary Amine
4.3. Biological Studies
4.3.1. Plasmid Constructs and Cell Culture
4.3.2. Ligand-Receptor Binding Assays
4.3.3. Nuclear Receptor Transcriptional Activity Assays
4.3.4. Ligand-Induced Nuclear Translocation Assays
4.3.5. Selectivity against a Panel of Nuclear Receptors
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arriza, J.L.; Weinberger, C.; Cerelli, G.; Glaser, T.M.; Handelin, B.L.; Housman, D.E.; Evans, R.M. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 1987, 237, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, E.; Gomez-Sanchez, C.E. The multifaceted mineralocorticoid receptor. Compr. Physiol. 2014, 4, 965–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossmann, C.; Almeida-Prieto, B.; Nolze, A.; Alvarez de la Rosa, D. Structural and molecular determinants of mineralocorticoid receptor signalling. Br. J. Pharm. 2022, 179, 3103–3118. [Google Scholar] [CrossRef] [PubMed]
- Farman, N.; Rafestin-Oblin, M.E. Multiple aspects of mineralocorticoid selectivity. Am. J. Physiol. Ren. Physiol. 2001, 280, F181–F192. [Google Scholar] [CrossRef] [Green Version]
- Chapman, K.; Holmes, M.; Seckl, J. 11beta-hydroxysteroid dehydrogenases: Intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Sanchez, E.P. Third-generation Mineralocorticoid Receptor Antagonists: Why Do We Need a Fourth? J. Cardiovasc. Pharm. 2016, 67, 26–38. [Google Scholar] [CrossRef] [Green Version]
- Barrera-Chimal, J.; Bonnard, B.; Jaisser, F. Roles of Mineralocorticoid Receptors in Cardiovascular and Cardiorenal Diseases. Annu. Rev. Physiol. 2022, 84, 585–610. [Google Scholar] [CrossRef]
- Bauersachs, J.; Lopez-Andres, N. Mineralocorticoid receptor in cardiovascular diseases-Clinical trials and mechanistic insights. Br. J. Pharm. 2022, 179, 3119–3134. [Google Scholar] [CrossRef]
- van der Heijden, C.; Bode, M.; Riksen, N.P.; Wenzel, U.O. The role of the mineralocorticoid receptor in immune cells in cardiovascular disease. Br. J. Pharm. 2022, 179, 3135–3151. [Google Scholar] [CrossRef]
- Feraco, A.; Marzolla, V.; Scuteri, A.; Armani, A.; Caprio, M. Mineralocorticoid Receptors in Metabolic Syndrome: From Physiology to Disease. Trends Endocrinol. Metab. 2020, 31, 205–217. [Google Scholar] [CrossRef]
- Schreier, B.; Zipprich, A.; Uhlenhaut, H.; Gekle, M. Mineralocorticoid receptors in non-alcoholic fatty liver disease. Br. J. Pharm. 2022, 179, 3165–3177. [Google Scholar] [CrossRef]
- Perez, P. The mineralocorticoid receptor in skin disease. Br. J. Pharm. 2022, 179, 3178–3189. [Google Scholar] [CrossRef]
- Behar-Cohen, F.; Zhao, M. Mineralocorticoid pathway in retinal health and diseases. Br. J. Pharm. 2022, 179, 3190–3204. [Google Scholar] [CrossRef]
- Paul, S.N.; Wingenfeld, K.; Otte, C.; Meijer, O.C. Brain mineralocorticoid receptor in health and disease: From molecular signalling to cognitive and emotional function. Br. J. Pharm. 2022, 179, 3205–3219. [Google Scholar] [CrossRef]
- Jaisser, F.; Farman, N. Emerging Roles of the Mineralocorticoid Receptor in Pathology: Toward New Paradigms in Clinical Pharmacology. Pharm. Rev. 2016, 68, 49–75. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, R.; Kolkhof, P.; Bakris, G.; Bauersachs, J.; Haller, H.; Wada, T.; Zannad, F. Steroidal and non-steroidal mineralocorticoid receptor antagonists in cardiorenal medicine. Eur. Heart J. 2021, 42, 152–161. [Google Scholar] [CrossRef]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef]
- Zannad, F.; McMurray, J.J.; Krum, H.; van Veldhuisen, D.J.; Swedberg, K.; Shi, H.; Vincent, J.; Pocock, S.J.; Pitt, B.; Emphasis-Hf Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N. Engl. J. Med. 2011, 364, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Lother, A.; Jaisser, F.; Wenzel, U.O. Emerging fields for therapeutic targeting of the aldosterone-mineralocorticoid receptor signaling pathway. Br. J. Pharm. 2022, 179, 3099–3102. [Google Scholar] [CrossRef]
- Pitt, B.; Bakris, G.; Ruilope, L.M.; DiCarlo, L.; Mukherjee, R.; Investigators, E. Serum potassium and clinical outcomes in the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS). Circulation 2008, 118, 1643–1650. [Google Scholar] [CrossRef] [Green Version]
- Kolkhof, P.; Borden, S.A. Molecular pharmacology of the mineralocorticoid receptor: Prospects for novel therapeutics. Mol. Cell. Endocrinol. 2012, 350, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Armanini, D.; Goland, G.J.; Adam, W.R.; Funder, J.W. Cyproheptadine and mineralocorticoid effector mechanisms. J. Clin. Endocrinol. Metab. 1983, 56, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Clarisse, D.; Deng, L.; de Bosscher, K.; Lother, A. Approaches towards tissue-selective pharmacology of the mineralocorticoid receptor. Br. J. Pharm. 2022, 179, 3235–3249. [Google Scholar] [CrossRef] [PubMed]
- Kintscher, U.; Bakris, G.L.; Kolkhof, P. Novel non-steroidal mineralocorticoid receptor antagonists in cardiorenal disease. Br. J. Pharm. 2022, 179, 3220–3234. [Google Scholar] [CrossRef]
- Martin-Martinez, M.; Perez-Gordillo, F.L.; Alvarez de la Rosa, D.; Rodriguez, Y.; Gerona-Navarro, G.; Gonzalez-Muniz, R.; Zhou, M.M. Modulating Mineralocorticoid Receptor with Non-steroidal Antagonists. New Opportunities for the Development of Potent and Selective Ligands without Off-Target Side Effects. J. Med. Chem. 2017, 60, 2629–2650. [Google Scholar] [CrossRef]
- Dietz, J.D.; Du, S.; Bolten, C.W.; Payne, M.A.; Xia, C.; Blinn, J.R.; Funder, J.W.; Hu, X. A number of marketed dihydropyridine calcium channel blockers have mineralocorticoid receptor antagonist activity. Hypertension 2008, 51, 742–748. [Google Scholar] [CrossRef] [Green Version]
- Brandish, P.; Fraley, M.E.; Hershey, J.C.; Steen, J.T. Mineralocorticoid Receptor Modulators. International Patent. WO2009/078934A1, 25 June 2009. [Google Scholar]
- Arhancet, G.B.; Woodard, S.S.; Iyanar, K.; Case, B.L.; Woerndle, R.; Dietz, J.D.; Garland, D.J.; Collins, J.T.; Payne, M.A.; Blinn, J.R.; et al. Discovery of novel cyanodihydropyridines as potent mineralocorticoid receptor antagonists. J. Med. Chem. 2010, 53, 5970–5978. [Google Scholar] [CrossRef]
- Barfacker, L.; Kuhl, A.; Hillisch, A.; Grosser, R.; Figueroa-Perez, S.; Heckroth, H.; Nitsche, A.; Erguden, J.K.; Gielen-Haertwig, H.; Schlemmer, K.H.; et al. Discovery of BAY 94-8862: A nonsteroidal antagonist of the mineralocorticoid receptor for the treatment of cardiorenal diseases. ChemMedChem 2012, 7, 1385–1403. [Google Scholar] [CrossRef]
- Arhancet, G.B.; Woodard, S.S.; Dietz, J.D.; Garland, D.J.; Wagner, G.M.; Iyanar, K.; Collins, J.T.; Blinn, J.R.; Numann, R.E.; Hu, X.; et al. Stereochemical requirements for the mineralocorticoid receptor antagonist activity of dihydropyridines. J. Med. Chem. 2010, 53, 4300–4304. [Google Scholar] [CrossRef]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Fagart, J.; Hillisch, A.; Huyet, J.; Barfacker, L.; Fay, M.; Pleiss, U.; Pook, E.; Schafer, S.; Rafestin-Oblin, M.E.; Kolkhof, P. A new mode of mineralocorticoid receptor antagonism by a potent and selective nonsteroidal molecule. J. Biol. Chem. 2010, 285, 29932–29940. [Google Scholar] [CrossRef] [Green Version]
- Alajarin, R.; Jordan, P.; Vaquero, J.J.; Alvarez-Builla, J. Synthesis of unsymmetrically substituted 1,4-dihydropyridines and analogous calcium antagonists by microwave heating. Synthesis 1995, 4, 389–391. [Google Scholar] [CrossRef]
- Öhberg, L.; Westman, J. An efficient and fast procedure for the Hantzsch dihydropyridine synthesis under microwave conditions. Synlett 2001, 8, 1296–1298. [Google Scholar] [CrossRef]
- Cory, R.T.; Charles, K.Z. Chain extension of amino acid skeletons: Preparation of ketomethylene isosteres. Tetrahedron 2003, 59, 1521–1527. [Google Scholar]
- Hashiguchi, S.; Kawada, A.; Natsugari, H. Baker’s Yeast Reduction of N-Protected Methyl 4-Amino-3-oxobutanoates and 3-oxopentanoates. Synthesis 1992, 1992, 403–408. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Wang, Z.; Hall, C.D. Synthesis of Achiral and Chiral N-Protected γ-Amino-β-Ketones and β-Ketoesters. ARKIVOC 2007, 10, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Richard, D.J.; Portonovo, P.S.; Joullie, M.M. Total syntheses and biological investigations of tamandarins A and B and tamandarin A analogs. J Am Chem Soc 2001, 123, 4469–4474. [Google Scholar] [CrossRef]
- Perez-Faginas, P.; Aranda, M.T.; Garcia-Lopez, M.T.; Infantes, L.; Fernandez-Carvajal, A.; Gonzalez-Ros, J.M.; Ferrer-Montiel, A.; Gonzalez-Muniz, R. Highly functionalized 1,2-diamino compounds through reductive amination of amino acid-derived beta-keto esters. PLoS ONE 2013, 8, e53231. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Fan, W.; Li, W.; Ma, X.; Zhu, L.; Xie, X.; Zhang, Z. Synthesis of (S)-7-amino-5-azaspiro[2.4]heptane via highly enantioselective hydrogenation of protected ethyl 1-(2-aminoaceto)cyclopropanecarboxylates. J. Org. Chem. 2011, 76, 2807–2813. [Google Scholar] [CrossRef]
- Arai, K.; Homma, T.; Morikawa, Y.; Ubukata, N.; Tsuruoka, H.; Aoki, K.; Ishikawa, H.; Mizuno, M.; Sada, T. Pharmacological profile of CS-3150, a novel, highly potent and selective non-steroidal mineralocorticoid receptor antagonist. Eur. J. Pharm. 2015, 761, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Rogerson, F.M.; Yao, Y.; Smith, B.J.; Fuller, P.J. Differences in the determinants of eplerenone, spironolactone and aldosterone binding to the mineralocorticoid receptor. Clin. Exp. Pharm. Physiol. 2004, 31, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.G.; Gernert, D.L.; Grese, T.A.; Belvo, M.D.; Borromeo, P.S.; Kelley, S.A.; Kennedy, J.H.; Kolis, S.P.; Lander, P.A.; Richey, R.; et al. (S)-N-3-[1-cyclopropyl-1-(2,4-difluoro-phenyl)-ethyl]-1H-indol-7-yl-methanesulfonamide: A potent, nonsteroidal, functional antagonist of the mineralocorticoid receptor. J. Med. Chem. 2007, 50, 6443–6445. [Google Scholar] [CrossRef] [PubMed]
- Amazit, L.; Le Billan, F.; Kolkhof, P.; Lamribet, K.; Viengchareun, S.; Fay, M.R.; Khan, J.A.; Hillisch, A.; Lombes, M.; Rafestin-Oblin, M.E.; et al. Finerenone Impedes Aldosterone-dependent Nuclear Import of the Mineralocorticoid Receptor and Prevents Genomic Recruitment of Steroid Receptor Coactivator-1. J. Biol. Chem. 2015, 290, 21876–21889. [Google Scholar] [CrossRef] [Green Version]
- Bledsoe, R.K.; Madauss, K.P.; Holt, J.A.; Apolito, C.J.; Lambert, M.H.; Pearce, K.H.; Stanley, T.B.; Stewart, E.L.; Trump, R.P.; Willson, T.M.; et al. A ligand-mediated hydrogen bond network required for the activation of the mineralocorticoid receptor. J. Biol. Chem. 2005, 280, 31283–31293. [Google Scholar] [CrossRef] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Fagart, J.; Wurtz, J.M.; Souque, A.; Hellal-Levy, C.; Moras, D.; Rafestin-Oblin, M.E. Antagonism in the human mineralocorticoid receptor. EMBO J. 1998, 17, 3317–3325. [Google Scholar] [CrossRef] [Green Version]
- Ouvrard-Pascaud, A.; Puttini, S.; Sainte-Marie, Y.; Athman, R.; Fontaine, V.; Cluzeaud, F.; Farman, N.; Rafestin-Oblin, M.E.; Blot-Chabaud, M.; Jaisser, F. Conditional gene expression in renal collecting duct epithelial cells: Use of the inducible Cre-lox system. Am. J. Physiol. Ren. Physiol. 2004, 286, F180–F187. [Google Scholar] [CrossRef]
- Hellal-Levy, C.; Couette, B.; Fagart, J.; Souque, A.; Gomez-Sanchez, C.; Rafestin-Oblin, M. Specific hydroxylations determine selective corticosteroid recognition by human glucocorticoid and mineralocorticoid receptors. FEBS Lett. 1999, 464, 9–13. [Google Scholar] [CrossRef]
- Bai, S.; He, B.; Wilson, E.M. Melanoma antigen gene protein MAGE-11 regulates androgen receptor function by modulating the interdomain interaction. Mol. Cell. Biol. 2005, 25, 1238–1257. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, Z.; Lonard, D.M.; Smith, C.L.; Lev-Lehman, E.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol. Cell. Biol. 1999, 19, 1182–1189. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Diaz, I.; Giraldez, T.; Arnau, M.R.; Smits, V.A.; Jaisser, F.; Farman, N.; Alvarez de la Rosa, D. The mineralocorticoid receptor is a constitutive nuclear factor in cardiomyocytes due to hyperactive nuclear localization signals. Endocrinology 2010, 151, 3888–3899. [Google Scholar] [CrossRef]
- Aguilar-Sanchez, C.; Hernandez-Diaz, I.; Lorenzo-Diaz, F.; Navarro, J.F.; Hughes, T.E.; Giraldez, T.; Alvarez de la Rosa, D. Identification of permissive insertion sites for generating functional fluorescent mineralocorticoid receptors. Endocrinology 2012, 153, 3517–3525. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Diaz, I.; Giraldez, T.; Morales, S.; Hernandez, G.; Salido, E.; Canessa, C.M.; Alvarez de la Rosa, D. Heterogeneous nuclear ribonucleoprotein A2/B1 is a tissue-specific aldosterone target gene with prominent induction in the rat distal colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G122–G131. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Canino, R.; Fernandes, M.X.; Alvarez de la Rosa, D. Phosphorylation of Mineralocorticoid Receptor Ligand Binding Domain Impairs Receptor Activation and Has a Dominant Negative Effect over Non-phosphorylated Receptors. J. Biol. Chem. 2016, 291, 19068–19078. [Google Scholar] [CrossRef] [Green Version]
- Mani, O.; Nashev, L.G.; Livelo, C.; Baker, M.E.; Odermatt, A. Role of Pro-637 and Gln-642 in human glucocorticoid receptors and Ser-843 and Leu-848 in mineralocorticoid receptors in their differential responses to cortisol and aldosterone. J. Steroid Biochem. Mol. Biol. 2016, 159, 31–40. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Comp. | R1 | R2 | R3 | R4 | R5 | R6 | Ki (nM) | 95% CI (nM) |
| EPLE | 175 | 96–320 | ||||||
| 11 | H | CH3 | F | Cl | CN | Cbz | 510 | 359–727 |
| 12a | CH3 | CH3 | F | Cl | CN | Cbz | 566 | 307–1106 |
| 12b | CH3 | CH3 | F | Cl | CN | Cbz | 1359 | 564–3950 |
| 13ab | (CH3)2CH | CH3 | F | Cl | CN | Cbz | 1414 | 1024–1969 |
| 14 | H | CH3CH2 | F | Cl | CN | Cbz | 355 | 134–915 |
| 15 | H | (CH3)3C | F | Cl | CN | Cbz | 1068 | 620–1902 |
| 16 | H | CH3 | CN | OCH3 | CN | Cbz | 262 | 125–565 |
| 17 | H | CH3 | F | Cl | CONH2 | Cbz | 1643 | 778–4022 |
| 19 | H | CH3 | F | Cl | CN | COCH3 | 152 | 95–236 |
| 20 | H | CH3 | F | Cl | CN | COCH2CO2tBu | 858 | 489–1547 |
| 23a | H | CH3 | F | Cl | CN | Glu | 740 | 107–1817 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Gordillo, F.L.; Serrano-Morillas, N.; Acosta-García, L.M.; Aranda, M.T.; Passeri, D.; Pellicciari, R.; Pérez de Vega, M.J.; González-Muñiz, R.; Alvarez de la Rosa, D.; Martín-Martínez, M. Novel 1,4-Dihydropyridine Derivatives as Mineralocorticoid Receptor Antagonists. Int. J. Mol. Sci. 2023, 24, 2439. https://doi.org/10.3390/ijms24032439
Pérez-Gordillo FL, Serrano-Morillas N, Acosta-García LM, Aranda MT, Passeri D, Pellicciari R, Pérez de Vega MJ, González-Muñiz R, Alvarez de la Rosa D, Martín-Martínez M. Novel 1,4-Dihydropyridine Derivatives as Mineralocorticoid Receptor Antagonists. International Journal of Molecular Sciences. 2023; 24(3):2439. https://doi.org/10.3390/ijms24032439
Chicago/Turabian StylePérez-Gordillo, Felipe Luis, Natalia Serrano-Morillas, Luz Marina Acosta-García, María Teresa Aranda, Daniela Passeri, Roberto Pellicciari, María Jesús Pérez de Vega, Rosario González-Muñiz, Diego Alvarez de la Rosa, and Mercedes Martín-Martínez. 2023. "Novel 1,4-Dihydropyridine Derivatives as Mineralocorticoid Receptor Antagonists" International Journal of Molecular Sciences 24, no. 3: 2439. https://doi.org/10.3390/ijms24032439
APA StylePérez-Gordillo, F. L., Serrano-Morillas, N., Acosta-García, L. M., Aranda, M. T., Passeri, D., Pellicciari, R., Pérez de Vega, M. J., González-Muñiz, R., Alvarez de la Rosa, D., & Martín-Martínez, M. (2023). Novel 1,4-Dihydropyridine Derivatives as Mineralocorticoid Receptor Antagonists. International Journal of Molecular Sciences, 24(3), 2439. https://doi.org/10.3390/ijms24032439