
Harnessing the LdCsm RNA Detection Platform for Efficient microRNA Detection



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
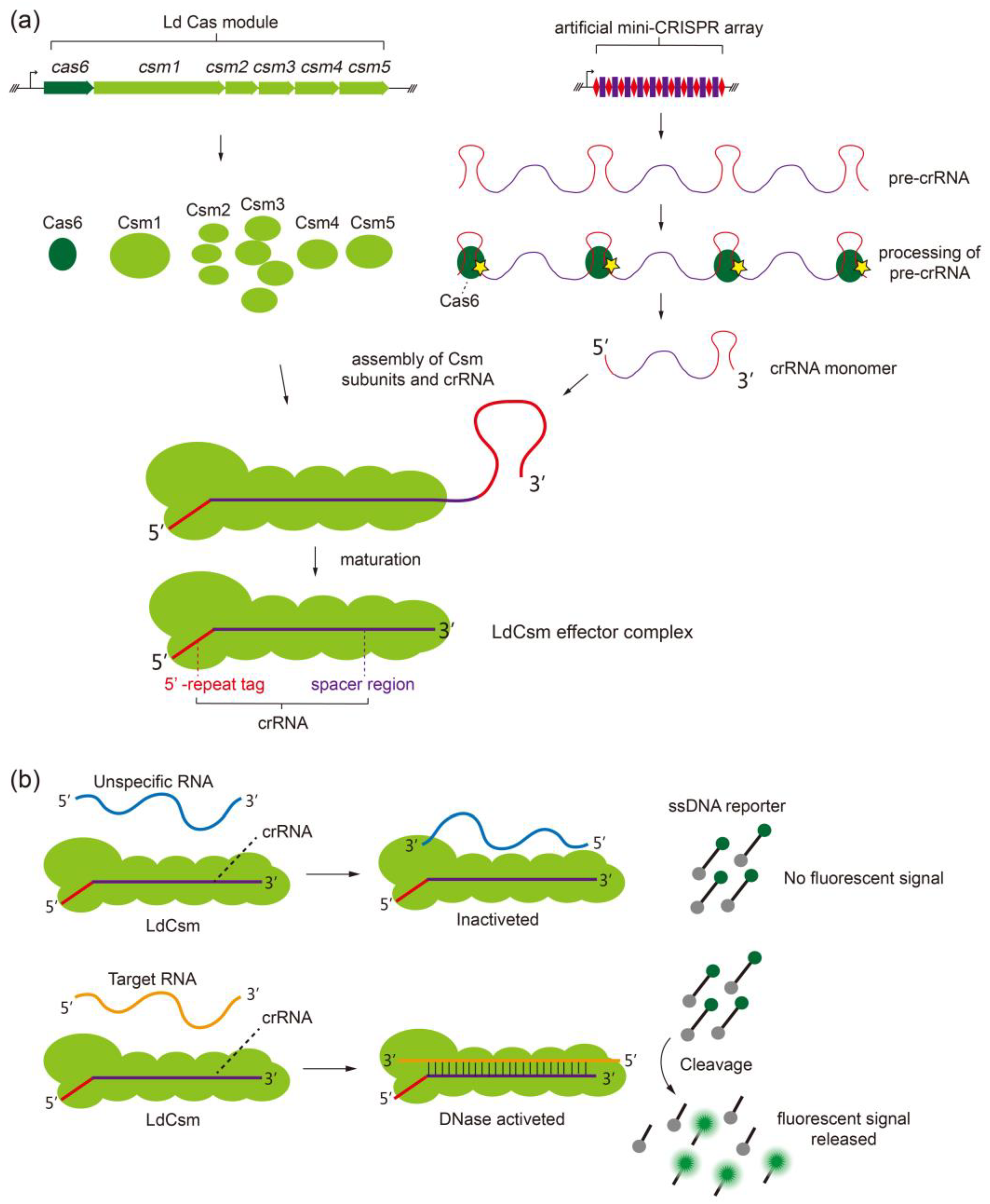
2.1. Developing the LdCsm RNA Detection Platform
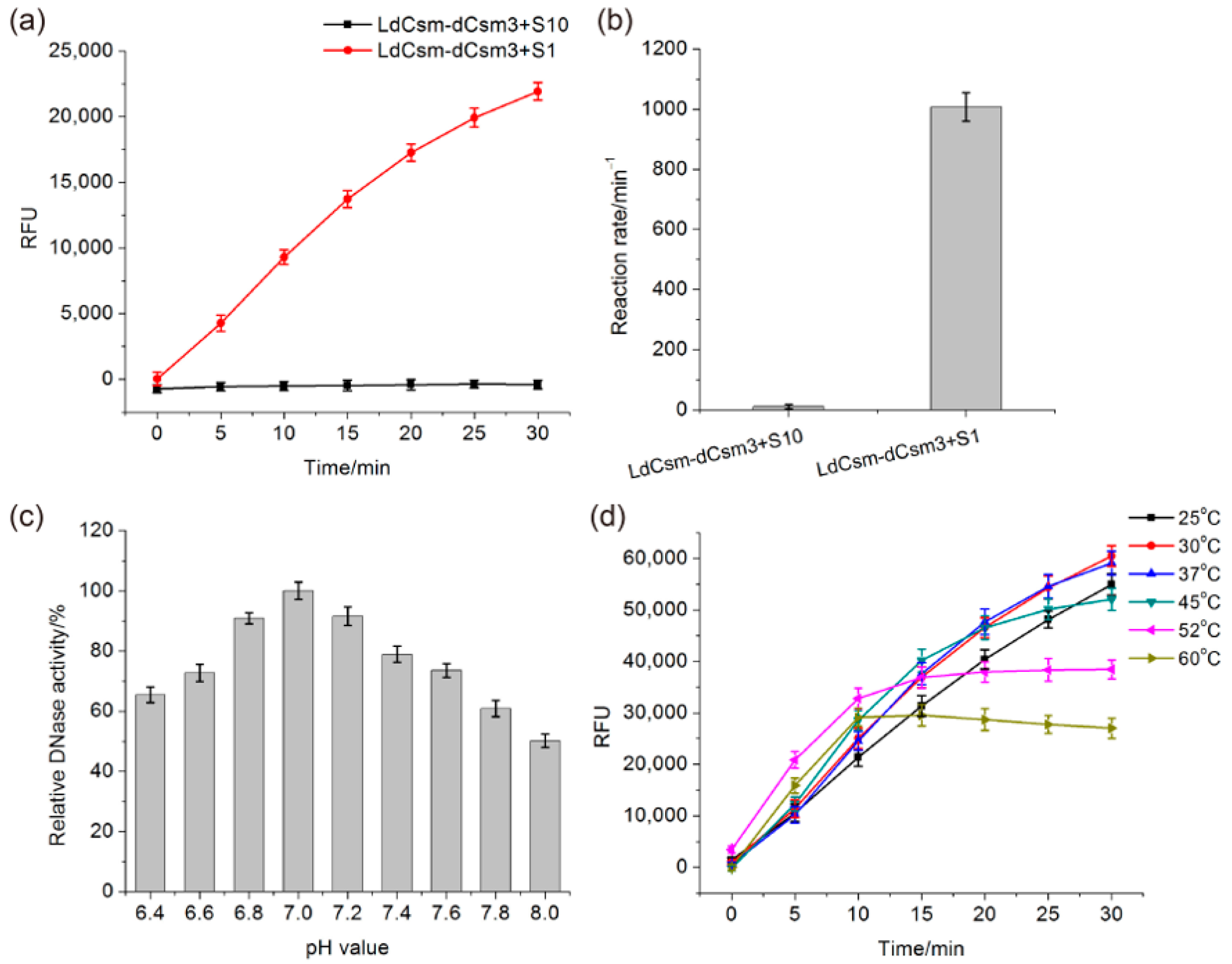
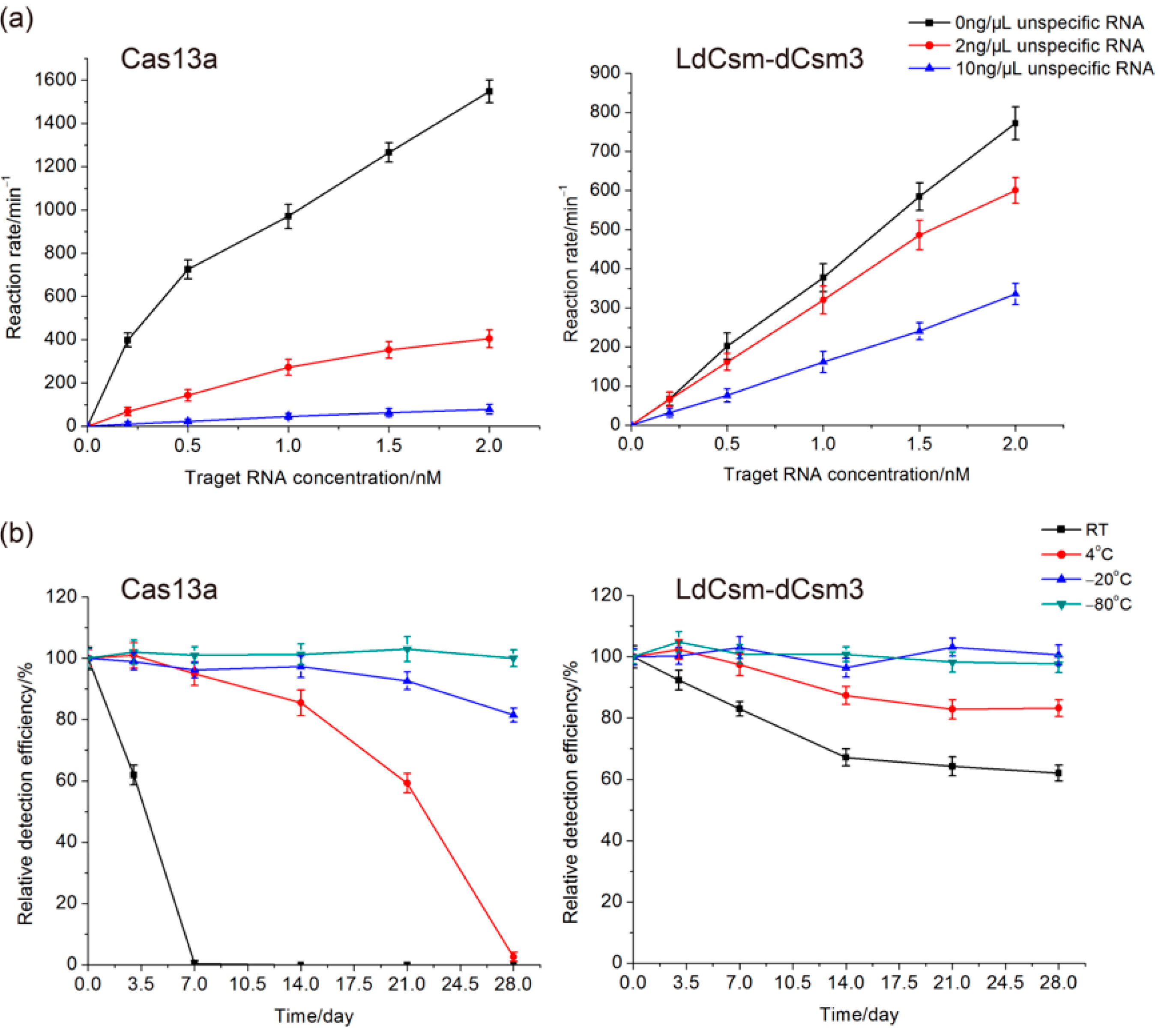
2.2. Comparison of LdCsm and Cas13a Detection Systems
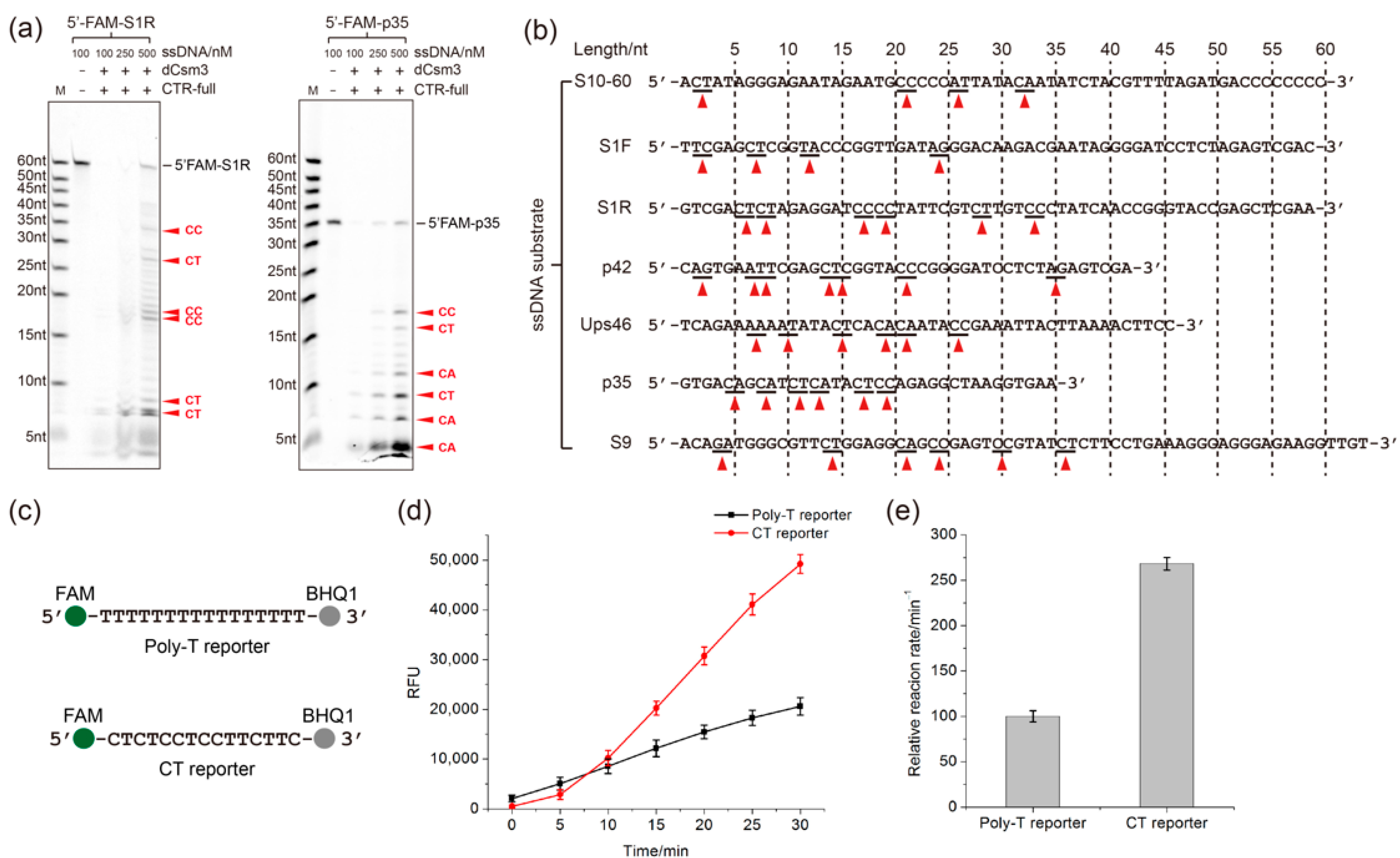
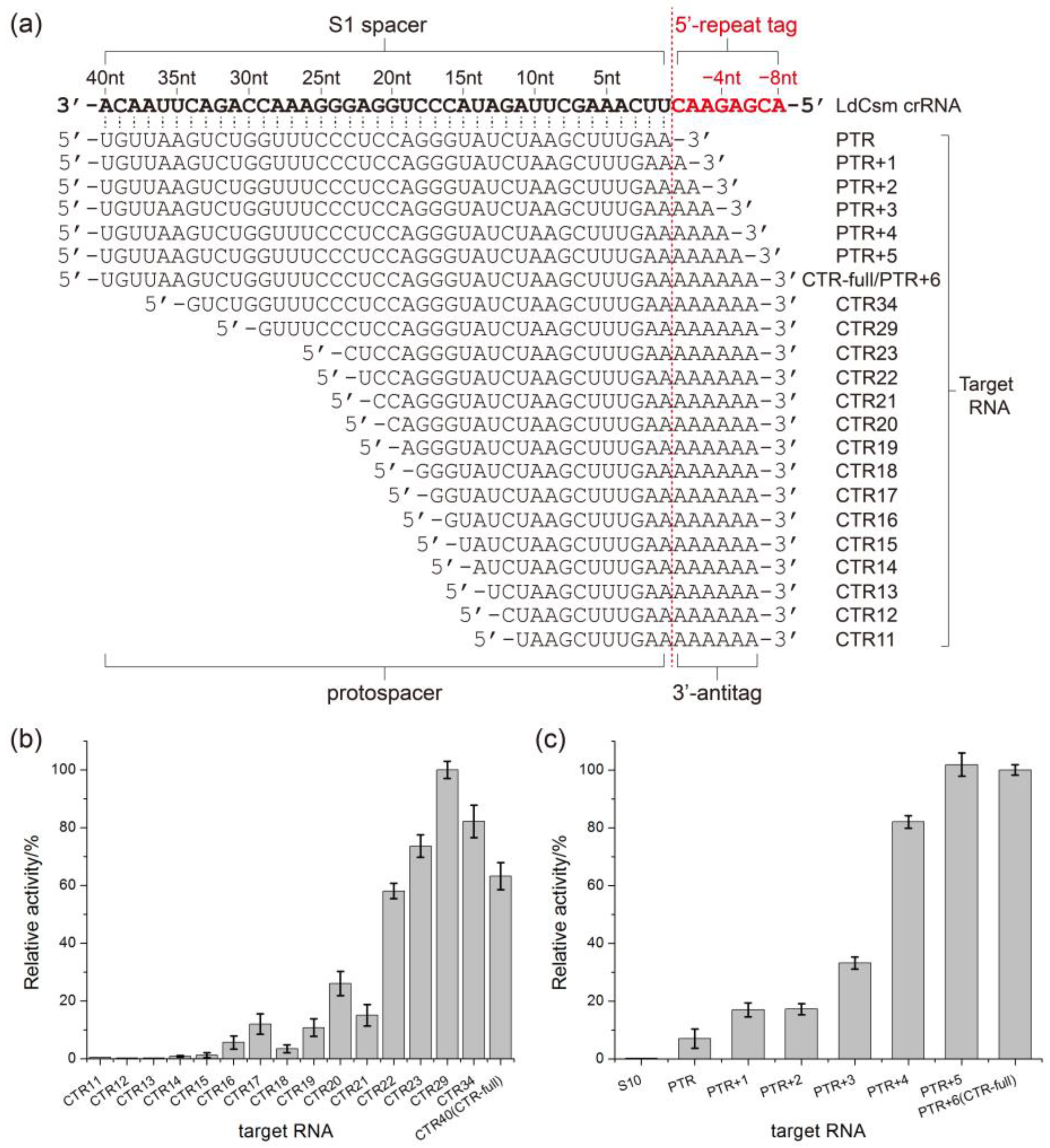
2.3. Determination of the Length Requirement of Activator RNA in the LdCsm-dCsm3 Detection Platform
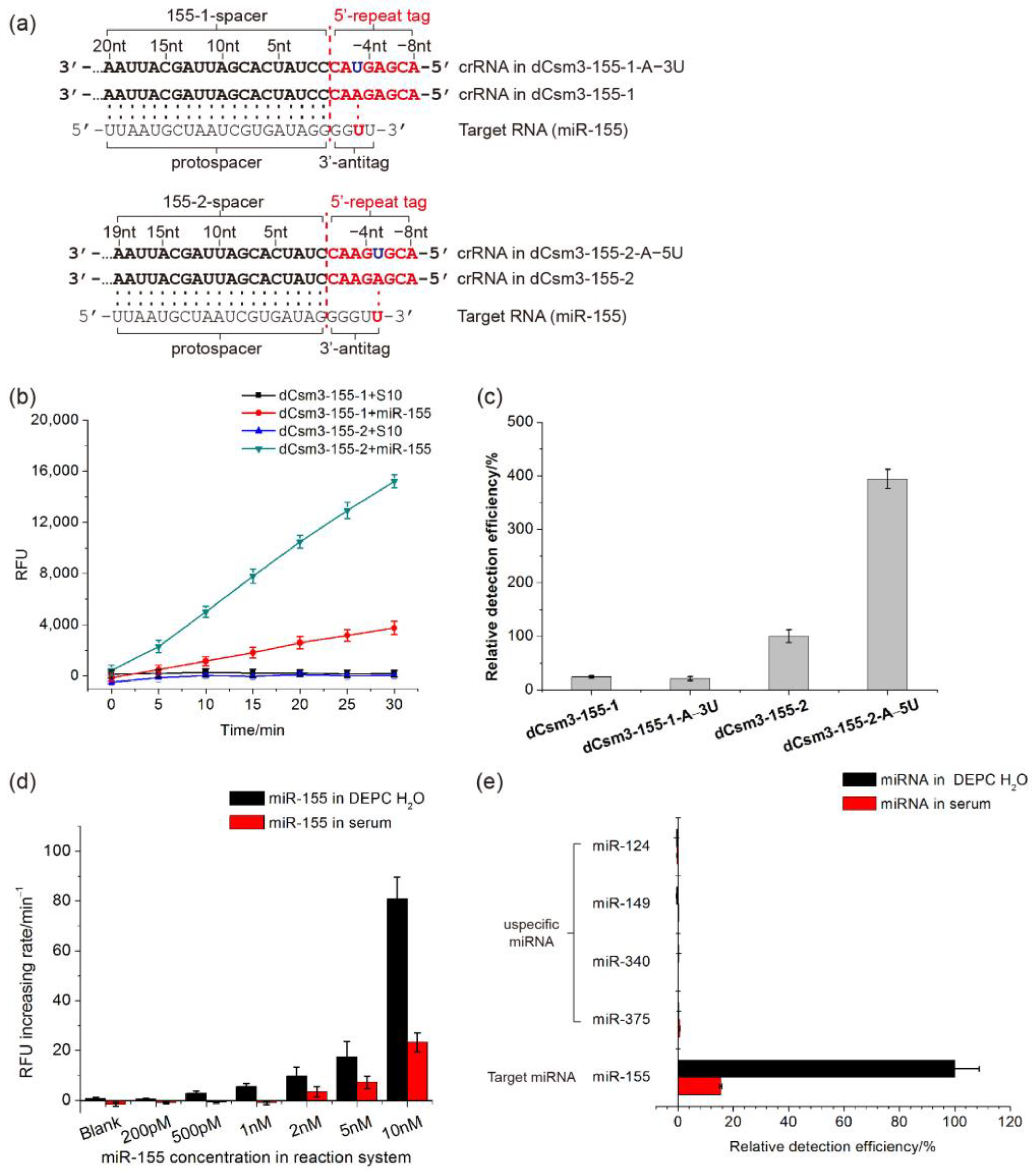
2.4. Developing the LdCsm-dCsm3-Based microRNA Detection System
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Construction of Artificial Mini-CRISPR Plasmids Carrying Different Spacers or Repeats
4.3. Purification of LdCsm Effector Complexes from E. coli
4.4. Collateral Nucleic Acid Cleavage Assay
4.5. Evaluation of Fluorescence DNA Reporter Cleavage Assay/RNA Detection Reaction
4.6. RNA Detection Reaction Using Cas13a
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Prim. 2015, 1, 15035. [Google Scholar] [CrossRef] [PubMed]
- Fauci, A.S.; Touchette, N.A.; Folkers, G.K. Emerging infectious diseases: A 10-year perspective from the National Institute of Allergy and Infectious Diseases. Emerg. Infect. Dis. 2005, 11, 519–525. [Google Scholar] [CrossRef]
- Masmejan, S.; Musso, D.; Vouga, M.; Pomar, L.; Dashraath, P.; Stojanov, M.; Panchaud, A.; Baud, D. Zika Virus. Pathogens 2020, 9, 898. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.T.; Crozier, I.; Fischer, W.A., 2nd; Hewlett, A.; Kraft, C.S.; Vega, M.A.; Soka, M.J.; Wahl, V.; Griffiths, A.; Bollinger, L.; et al. Ebola virus disease. Nat. Rev. Dis. Prim. 2020, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.Q.; Peng, H.J. Characteristics of and Public Health Responses to the Coronavirus Disease 2019 Outbreak in China. J. Clin. Med. 2020, 9, 575. [Google Scholar] [CrossRef]
- Henley, S.J.; Ward, E.M.; Scott, S.; Ma, J.; Anderson, R.N.; Firth, A.U.; Thomas, C.C.; Islami, F.; Weir, H.K.; Lewis, D.R.; et al. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer Res. 2020, 126, 2225–2249. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Tutar, L.; Özgür, A.; Tutar, Y. Involvement of miRNAs and Pseudogenes in Cancer. Methods Mol. Biol. 2018, 1699, 45–66. [Google Scholar]
- Huang, R.; Yu, H.; Zhong, X. Identification of Novel CircRNA-miRNA-mRNA Regulatory Network and Its Prognostic Prediction in Breast Cancer. Evid.-Based Complement. Altern. Med. Ecam 2021, 2021, 2916398. [Google Scholar] [CrossRef]
- Chen, L.; Shan, G. CircRNA in cancer: Fundamental mechanism and clinical potential. Cancer Lett. 2021, 505, 49–57. [Google Scholar] [CrossRef]
- Gurukumar, K.R.; Priyadarshini, D.; Patil, J.A.; Bhagat, A.; Singh, A.; Shah, P.S.; Cecilia, D. Development of real time PCR for detection and quantitation of Dengue Viruses. Virol. J. 2009, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Watzinger, F.; Ebner, K.; Lion, T. Detection and monitoring of virus infections by real-time PCR. Mol. Asp. Med. 2006, 27, 254–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Tan, R.; Wong, L.; Fekete, R.; Halsey, J. Quantitation of microRNAs by real-time RT-qPCR. Methods Mol. Biol. 2011, 687, 113–134. [Google Scholar] [PubMed]
- Abd El Wahed, A.; Sanabani, S.S.; Faye, O.; Pessôa, R.; Patriota, J.V.; Giorgi, R.R.; Patel, P.; Böhlken-Fascher, S.; Landt, O.; Niedrig, M.; et al. Rapid Molecular Detection of Zika Virus in Acute-Phase Urine Samples Using the Recombinase Polymerase Amplification Assay. PLoS Curr. 2017, 9. [Google Scholar] [CrossRef]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase Polymerase Amplification for Diagnostic Applications. Clin. Chem. 2016, 62, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Dao Thi, V.L.; Herbst, K.; Boerner, K.; Meurer, M.; Kremer, L.P.; Kirrmaier, D.; Freistaedter, A.; Papagiannidis, D.; Galmozzi, C.; Stanifer, M.L.; et al. A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples. Sci. Transl. Med. 2020, 12, eabc7075. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, G.; Buss, J.; Barry, A.J.; Patton, G.C.; Tanner, N.A. Enhancing colorimetric loop-mediated isothermal amplification speed and sensitivity with guanidine chloride. BioTechniques 2020, 69, 178–185. [Google Scholar] [CrossRef]
- Lu, W.; Wang, Y.; Song, S.; Chen, C.; Yao, B.; Wang, M. A fishhook probe-based rolling circle amplification (FP-RCA) assay for efficient isolation and detection of microRNA without total RNA extraction. Analyst 2018, 143, 5046–5053. [Google Scholar] [CrossRef]
- Li, J.; Liu, J.; Bi, Y.; Sun, M.; Bai, J.; Zhou, M. Ultrasensitive electrochemiluminescence biosensing platform for miRNA-21 and MUC1 detection based on dual catalytic hairpin assembly. Anal. Chim. Acta 2020, 1105, 87–94. [Google Scholar] [CrossRef]
- Gu, J.; Qiao, Z.; He, X.; Yu, Y.; Lei, Y.; Tang, J.; Shi, H.; He, D.; Wang, K. Enzyme-free amplified detection of miRNA based on target-catalyzed hairpin assembly and DNA-stabilized fluorescent silver nanoclusters. Analyst 2020, 145, 5194–5199. [Google Scholar] [CrossRef]
- Ning, L.; Cheng, H.; Yu, F.; Zhou, Y.; Xie, Y. Construction of simple and sensitive pancreatitis related microRNA detection strategy via self-priming triggered cascade signal amplification. Anal. Bioanal. Chem. 2022, 414, 5837–5844. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Q.; Weng, X.; Du, Y.; Zhou, X. NEase-based amplification for detection of miRNA, multiple miRNAs and circRNA. Anal. Chim. Acta 2021, 1145, 52–58. [Google Scholar] [CrossRef]
- Zhao, G.; Yan, X.; Zhang, Y.; Deng, J.; Liang, X. Sensitive detection of MiRNA and CircRNA through DSN enzyme cooperating NEase assisted dual signal amplification. Anal. Biochem. 2022, 654, 114744. [Google Scholar] [CrossRef] [PubMed]
- Meagher, R.J.; Priye, A.; Light, Y.K.; Huang, C.; Wang, E. Impact of primer dimers and self-amplifying hairpins on reverse transcription loop-mediated isothermal amplification detection of viral RNA. Analyst 2018, 143, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Rolando, J.C.; Jue, E.; Barlow, J.T.; Ismagilov, R.F. Real-time kinetics and high-resolution melt curves in single-molecule digital LAMP to differentiate and study specific and non-specific amplification. Nucleic Acids Res. 2020, 48, e42. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Mason, M.G.; Botella, J.R. Evaluation and improvement of isothermal amplification methods for point-of-need plant disease diagnostics. PloS ONE 2020, 15, e0235216. [Google Scholar] [CrossRef]
- Horvath, P.; Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 2010, 327, 167–170. [Google Scholar] [CrossRef]
- Mohanraju, P.; Makarova, K.S.; Zetsche, B.; Zhang, F.; Koonin, E.V.; van der Oost, J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 2016, 353, aad5147. [Google Scholar] [CrossRef]
- Wright, A.V.; Nunez, J.K.; Doudna, J.A. Biology and Applications of CRISPR Systems: Harnessing Nature’s Toolbox for Genome Engineering. Cell 2016, 164, 29–44. [Google Scholar] [CrossRef]
- Hille, F.; Richter, H.; Wong, S.P.; Bratovič, M.; Ressel, S.; Charpentier, E. The Biology of CRISPR-Cas: Backward and Forward. Cell 2018, 172, 1239–1259. [Google Scholar] [CrossRef]
- Chen, Y.; Zeng, Z.; She, Q.; Han, W. The abortive infection functions of CRISPR-Cas and Argonaute. Trends Microbiol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Samai, P.; Pyenson, N.; Jiang, W.; Goldberg, G.W.; Hatoum-Aslan, A.; Marraffini, L.A. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell 2015, 161, 1164–1174. [Google Scholar] [CrossRef]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef]
- Santiago-Frangos, A.; Hall, L.N.; Nemudraia, A.; Nemudryi, A.; Krishna, P.; Wiegand, T.; Wilkinson, R.A.; Snyder, D.T.; Hedges, J.F.; Cicha, C.; et al. Intrinsic signal amplification by type III CRISPR-Cas systems provides a sequence-specific SARS-CoV-2 diagnostic. Cell Rep. Med. 2021, 2, 100319. [Google Scholar] [CrossRef]
- Steens, J.A.; Zhu, Y.; Taylor, D.W.; Bravo, J.P.K.; Prinsen, S.H.P.; Schoen, C.D.; Keijser, B.J.F.; Ossendrijver, M.; Hofstra, L.M.; Brouns, S.J.J.; et al. SCOPE enables type III CRISPR-Cas diagnostics using flexible targeting and stringent CARF ribonuclease activation. Nat. Commun. 2021, 12, 5033. [Google Scholar] [CrossRef]
- Sridhara, S.; Goswami, H.N.; Whyms, C.; Dennis, J.H.; Li, H. Virus detection via programmable Type III-A CRISPR-Cas systems. Nat. Commun. 2021, 12, 5653. [Google Scholar] [CrossRef]
- Grüschow, S.; Adamson, C.S.; White, M.F. Specificity and sensitivity of an RNA targeting type III CRISPR complex coupled with a NucC endonuclease effector. Nucleic Acids Res. 2021, 49, 13122–13134. [Google Scholar] [CrossRef]
- Peng, S.; Tan, Z.; Chen, S.; Lei, C.; Nie, Z. Integrating CRISPR-Cas12a with a DNA circuit as a generic sensing platform for amplified detection of microRNA. Chem. Sci. 2020, 11, 7362–7368. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Chen, Z.; Lu, J.; Ren, X.; Ma, Y. Ultrasensitive visual detection of miRNA-143 using a CRISPR/Cas12a-based platform coupled with hyperbranched rolling circle amplification. Talanta 2023, 251, 123784. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, J.; He, N.; Zhang, M.; Wu, L.; Chen, X.; Zhu, J.; Ran, F.; Chen, Q.; Zhang, H. CRISPR/Cas12a Coupling with Magnetic Nanoparticles and Cascaded Strand Displacement Reaction for Ultrasensitive Fluorescence Determination of Exosomal miR-21. Molecules 2022, 27, 5338. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.Y.; Zhao, H.L.; Wang, T.; Chen, P.R.; Yin, B.C.; Ye, B.C. A programmable and sensitive CRISPR/Cas12a-based MicroRNA detection platform combined with hybridization chain reaction. Biosens. Bioelectron. 2022, 211, 114382. [Google Scholar] [CrossRef]
- Broto, M.; Kaminski, M.M.; Adrianus, C.; Kim, N.; Greensmith, R.; Dissanayake-Perera, S.; Schubert, A.J.; Stevens, M.M.; Tan, X.; Kim, H.; et al. Nanozyme-catalysed CRISPR assay for preamplification-free detection of non-coding RNAs. Nat. Nanotechnol. 2022, 17, 1120–1126. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, C.; Liu, G.; Zhao, X.; Qian, Q.; Li, S.; Mi, X. Tetrahedral DNA framework based CRISPR electrochemical biosensor for amplification-free miRNA detection. Biosens. Bioelectron. 2022, 217, 114671. [Google Scholar] [CrossRef]
- Lin, J.; Feng, M.; Zhang, H.; She, Q. Characterization of a novel type III CRISPR-Cas effector provides new insights into the allosteric activation and suppression of the Cas10 DNase. Cell Discovry 2020, 6, 29. [Google Scholar] [CrossRef]
- Lin, J.; Shen, Y.; Ni, J.; She, Q. A type III-A CRISPR-Cas system mediates co-transcriptional DNA cleavage at the transcriptional bubbles in close proximity to active effectors. Nucleic Acids Res. 2021, 49, 7628–7643. [Google Scholar] [CrossRef]
- Han, W.; Li, Y.; Deng, L.; Feng, M.; Peng, W.; Hallstrom, S.; Zhang, J.; Peng, N.; Liang, Y.X.; White, M.F.; et al. A type III-B CRISPR-Cas effector complex mediating massive target DNA destruction. Nucleic Acids Res. 2017, 45, 1983–1993. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Myhrvold, C.; Freije, C.A.; Gootenberg, J.S.; Abudayyeh, O.O.; Metsky, H.C.; Durbin, A.F.; Kellner, M.J.; Tan, A.L.; Paul, L.M.; Parham, L.A.; et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science 2018, 360, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Catela Ivkovic, T.; Voss, G.; Cornella, H.; Ceder, Y. microRNAs as cancer therapeutics: A step closer to clinical application. Cancer Lett. 2017, 407, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, R.; Van Roosbroeck, K. miR-155 in cancer drug resistance and as target for miRNA-based therapeutics. Cancer Metastasis Rev. 2018, 37, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, A.M.; Nuzzo, S.; Condorelli, G.; Salvatore, M.; Incoronato, M. Prognostic and Clinicopathological Significance of MiR-155 in Breast Cancer: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 5834. [Google Scholar] [CrossRef] [PubMed]
- Sueta, A.; Yamamoto, Y.; Tomiguchi, M.; Takeshita, T.; Yamamoto-Ibusuki, M.; Iwase, H. Differential expression of exosomal miRNAs between breast cancer patients with and without recurrence. Oncotarget 2017, 8, 69934–69944. [Google Scholar] [CrossRef]
- Zhang, M.; Bai, X.; Zeng, X.; Liu, J.; Liu, F.; Zhang, Z. circRNA-miRNA-mRNA in breast cancer. Clin. Chim. Acta Int. J. Clin. Chem. 2021, 523, 120–130. [Google Scholar] [CrossRef]
- Zhang, X.; An, X. Adaptation by Type III CRISPR-Cas Systems: Breakthrough Findings and Open Questions. Front. Microbiol. 2022, 13, 876174. [Google Scholar] [CrossRef]
- Kazlauskiene, M.; Kostiuk, G.; Siksnys, V.; Tamulaitis, G.; Venclovas, Č. A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science 2017, 357, 605–609. [Google Scholar] [CrossRef]
- Niewoehner, O.; Garcia-Doval, C.; Rostøl, J.T.; Berk, C.; Schwede, F.; Bigler, L.; Hall, J.; Marraffini, L.A.; Jinek, M. Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature 2017, 548, 543–548. [Google Scholar] [CrossRef]
- Han, W.; Stella, S.; Zhang, Y.; Guo, T.; Sulek, K.; Peng-Lundgren, L.; Montoya, G.; She, Q. A Type III-B Cmr effector complex catalyzes the synthesis of cyclic oligoadenylate second messengers by cooperative substrate binding. Nucleic Acids Res. 2018, 46, 10319–10330. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.A.; Zhu, W.; Graham, S.; Rambo, R.; White, M.F.; Gloster, T.M. Structure and mechanism of a Type III CRISPR defence DNA nuclease activated by cyclic oligoadenylate. Nat. Commun. 2020, 11, 500. [Google Scholar] [CrossRef] [PubMed]
- Rostøl, J.T.; Xie, W.; Kuryavyi, V.; Maguin, P.; Kao, K.; Froom, R.; Patel, D.J.; Marraffini, L.A. The Card1 nuclease provides defence during type III CRISPR immunity. Nature 2021, 590, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; McQuarrie, S.; Grüschow, S.; McMahon, S.A.; Graham, S.; Gloster, T.M.; White, M.F. The CRISPR ancillary effector Can2 is a dual-specificity nuclease potentiating type III CRISPR defence. Nucleic Acids Res. 2021, 49, 2777–2789. [Google Scholar] [CrossRef]
- Guo, Z.; Tan, X.; Yuan, H.; Zhang, L.; Wu, J.; Yang, Z.; Qu, K.; Wan, Y. Bis-enzyme cascade CRISPR-Cas12a platform for miRNA detection. Talanta 2023, 252, 123837. [Google Scholar] [CrossRef]
- Wolfinger, R.D.; Beedanagari, S.; Boitier, E.; Chen, T.; Couttet, P.; Ellinger-Ziegelbauer, H.; Guillemain, G.; Mariet, C.; Mouritzen, P.; O’Lone, R.; et al. Two approaches for estimating the lower limit of quantitation (LLOQ) of microRNA levels assayed as exploratory biomarkers by RT-qPCR. BMC Biotechnol. 2018, 18, 6. [Google Scholar] [CrossRef]
- Iguchi, T.; Niino, N.; Tamai, S.; Sakurai, K.; Mori, K. Absolute Quantification of Plasma MicroRNA Levels in Cynomolgus Monkeys, Using Quantitative Real-time Reverse Transcription PCR. J. Vis. Exp. JoVE 2018, 132, e56850. [Google Scholar] [CrossRef]
- Guo, X.; Tian, T.; Deng, X.; Song, Y.; Zhou, X.; Song, E. CRISPR/Cas13a assisted amplification of magnetic relaxation switching sensing for accurate detection of miRNA-21 in human serum. Anal. Chim. Acta 2022, 1209, 339853. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Z.; Xu, J.; She, Q. Harnessing the LdCsm RNA Detection Platform for Efficient microRNA Detection. Int. J. Mol. Sci. 2023, 24, 2857. https://doi.org/10.3390/ijms24032857
Yu Z, Xu J, She Q. Harnessing the LdCsm RNA Detection Platform for Efficient microRNA Detection. International Journal of Molecular Sciences. 2023; 24(3):2857. https://doi.org/10.3390/ijms24032857
Chicago/Turabian StyleYu, Zhenxiao, Jianan Xu, and Qunxin She. 2023. "Harnessing the LdCsm RNA Detection Platform for Efficient microRNA Detection" International Journal of Molecular Sciences 24, no. 3: 2857. https://doi.org/10.3390/ijms24032857
APA StyleYu, Z., Xu, J., & She, Q. (2023). Harnessing the LdCsm RNA Detection Platform for Efficient microRNA Detection. International Journal of Molecular Sciences, 24(3), 2857. https://doi.org/10.3390/ijms24032857