
Helicobacter pylori and Gastric Cancer: Pathogenetic Mechanisms

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Role of Chronic Inflammation
2.1. Innate Immune Response and Oxidative Stress
2.2. Adaptive Immune Response
3. H. pylori Virulence Factors
3.1. Cytotoxin-Associated Gene A (CagA) and the Cytotoxin-Associated Gene Pathogenicity Island (cagPAI)
3.2. Vacuolating Cytotoxin A (VacA)
4. Genomic Instability
4.1. H. pylori Infection and the MMR Pathway
4.2. H. pylori Infection and the BER Pathway
4.3. H. pylori Infection and other Repair Pathways
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Karimi, P.; Islami, F.; Anandasabapathy, S.; Freedman, N.D.; Kamangar, F. Gastric cancer: Descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol. Biomark. Prev. 2014, 23, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef]
- Polk, D.B.; Peek, R.M., Jr. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.B.; Gold, B.D. The disease spectrum of Helicobacter pylori: The immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu. Rev. Microbiol. 2000, 54, 615–640. [Google Scholar] [CrossRef]
- Machado, A.M.; Figueiredo, C.; Seruca, R.; Rasmussen, L.J. Helicobacter pylori infection generates genetic instability in gastric cells. Biochim. Biophys. Acta 2010, 1806, 58–65. [Google Scholar] [CrossRef]
- Shimizu, T.; Chiba, T.; Marusawa, H. Helicobacter pylori-Mediated Genetic Instability and Gastric Carcinogenesis. Curr. Top. Microbiol. Immunol. 2017, 400, 305–323. [Google Scholar] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Lamb, A.; Chen, L.F. Role of the Helicobacter pylori-induced inflammatory response in the development of gastric cancer. J. Cell. Biochem. 2013, 114, 491–497. [Google Scholar] [CrossRef]
- White, J.R.; Winter, J.A.; Robinson, K. Differential inflammatory response to Helicobacter pylori infection: Etiology and clinical outcomes. J. Inflamm. Res. 2015, 8, 137–147. [Google Scholar]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fialkow, L.; Wang, Y.; Downey, G.P. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free. Radic. Biol. Med. 2007, 42, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Kalisperati, P.; Spanou, E.; Pateras, I.S.; Korkolopoulou, P.; Varvarigou, A.; Karavokyros, I.; Gorgoulis, V.G.; Vlachoyiannopoulos, P.G.; Sougioultzis, S. Inflammation, DNA Damage, Helicobacter pylori and Gastric Tumorigenesis. Front. Genet. 2017, 8, 20. [Google Scholar] [CrossRef]
- Butcher, L.D.; den Hartog, G.; Ernst, P.B.; Crowe, S.E. Oxidative Stress Resulting from Helicobacter pylori Infection Contributes to Gastric Carcinogenesis. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 316–322. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Cheng, Y.; Asim, M.; Bussiere, F.I.; Xu, H.; Gobert, A.P.; Hacker, A.; Casero, R.A., Jr.; Wilson, K.T. Induction of polyamine oxidase 1 by Helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization. J. Biol. Chem. 2004, 279, 40161–40173. [Google Scholar] [CrossRef]
- D′Elios, M.M.; Manghetti, M.; De Carli, M.; Costa, F.; Baldari, C.T.; Burroni, D.; Telford, J.L.; Romagnani, S.; Del Prete, G. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J. Immunol. 1997, 158, 962–967. [Google Scholar] [CrossRef]
- Monteleone, G.; Del Vecchio Blanco, G.; Palmieri, G.; Vavassori, P.; Monteleone, I.; Colantoni, A.; Battista, S.; Spagnoli, L.G.; Romano, M.; Borrelli, M.; et al. Induction and regulation of Smad7 in the gastric mucosa of patients with Helicobacter pylori infection. Gastroenterology 2004, 126, 674–682. [Google Scholar] [CrossRef]
- Caruso, R.; Fina, D.; Peluso, I.; Fantini, M.C.; Tosti, C.; Del Vecchio Blanco, G.; Paoluzi, O.A.; Caprioli, F.; Andrei, F.; Stolfi, C.; et al. IL-21 is highly produced in Helicobacter pylori-infected gastric mucosa and promotes gelatinases synthesis. J. Immunol. 2007, 178, 5957–5965. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zeng, Z.; Xu, L.; Yu, J.; Cao, Q.; Chen, M.; Sung, J.J.; Hu, P. Increased expression of IL17A in human gastric cancer and its potential roles in gastric carcinogenesis. Tumour Biol. 2014, 35, 5347–5356. [Google Scholar] [CrossRef]
- Borrego, S.; Vazquez, A.; Dasi, F.; Cerda, C.; Iradi, A.; Tormos, C.; Sanchez, J.M.; Bagan, L.; Boix, J.; Zaragoza, C.; et al. Oxidative Stress and DNA Damage in Human Gastric Carcinoma: 8-Oxo-7′8-dihydro-2′-deoxyguanosine (8-oxo-dG) as a Possible Tumor Marker. Int. J. Mol. Sci. 2013, 14, 3467–3486. [Google Scholar] [CrossRef]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–620. [Google Scholar] [CrossRef] [Green Version]
- Futagami, S.; Hiratsuka, T.; Shindo, T.; Horie, A.; Hamamoto, T.; Suzuki, K.; Kusunoki, M.; Miyake, K.; Gudis, K.; Crowe, S.E.; et al. Expression of apurinic/apyrimidinic endonuclease-1 (APE-1) in H. pylori-associated gastritis, gastric adenoma, and gastric cancer. Helicobacter 2008, 13, 209–218. [Google Scholar] [CrossRef]
- Caruso, R.; Pallone, F.; Monteleone, G. Emerging role of IL-23/IL-17 axis in H pylori-associated pathology. World J. Gastroenterol. 2007, 13, 5547–5551. [Google Scholar] [CrossRef]
- Li, S.; Lu, A.P.; Zhang, L.; Li, Y.D. Anti-Helicobacter pylori immunoglobulin G (IgG) and IgA antibody responses and the value of clinical presentations in diagnosis of H. pylori infection in patients with precancerous lesions. World J. Gastroenterol. 2003, 9, 755–758. [Google Scholar] [CrossRef]
- Gorelik, L.; Constant, S.; Flavell, R.A. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J. Exp. Med. 2002, 195, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, G.; Kumberova, A.; Croft, N.M.; McKenzie, C.; Steer, H.W.; MacDonald, T.T. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J. Clin. Investig. 2001, 108, 601–609. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, H.; Wu, X.; Bian, Z.; Gao, Q. Interleukin 17A promotes gastric cancer invasiveness via NF-kappaB mediated matrix metalloproteinases 2 and 9 expression. PLoS ONE 2014, 9, e96678. [Google Scholar]
- Caruso, R.; Fina, D.; Paoluzi, O.A.; Del Vecchio Blanco, G.; Stolfi, C.; Rizzo, A.; Caprioli, F.; Sarra, M.; Andrei, F.; Fantini, M.C.; et al. IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur. J. Immunol. 2008, 38, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Morey, P.; Pfannkuch, L.; Pang, E.; Boccellato, F.; Sigal, M.; Imai-Matsushima, A.; Dyer, V.; Koch, M.; Mollenkopf, H.J.; Schlaermann, P.; et al. Helicobacter pylori Depletes Cholesterol in Gastric Glands to Prevent Interferon Gamma Signaling and Escape the Inflammatory Response. Gastroenterology 2018, 154, 1391–1404.e9. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Sheu, B.S.; Wu, J.J. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed. J. 2016, 39, 14–23. [Google Scholar] [CrossRef]
- Mobley, H.L.; Garner, R.M.; Bauerfeind, P. Helicobacter pylori nickel-transport gene nixA: Synthesis of catalytically active urease in Escherichia coli independent of growth conditions. Mol. Microbiol. 1995, 16, 97–109. [Google Scholar] [CrossRef]
- Weeks, D.L.; Eskandari, S.; Scott, D.R.; Sachs, G. A H+-gated urea channel: The link between Helicobacter pylori urease and gastric colonization. Science 2000, 287, 482–485. [Google Scholar] [CrossRef] [Green Version]
- Kao, C.Y.; Sheu, B.S.; Sheu, S.M.; Yang, H.B.; Chang, W.L.; Cheng, H.C.; Wu, J.J. Higher motility enhances bacterial density and inflammatory response in dyspeptic patients infected with Helicobacter pylori. Helicobacter 2012, 17, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Ilver, D.; Arnqvist, A.; Ogren, J.; Frick, I.M.; Kersulyte, D.; Incecik, E.T.; Berg, D.E.; Covacci, A.; Engstrand, L.; Boren, T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998, 279, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Polenghi, A.; Bossi, F.; Fischetti, F.; Durigutto, P.; Cabrelle, A.; Tamassia, N.; Cassatella, M.A.; Montecucco, C.; Tedesco, F.; de Bernard, M. The neutrophil-activating protein of Helicobacter pylori crosses endothelia to promote neutrophil adhesion in vivo. J. Immunol. 2007, 178, 1312–1320. [Google Scholar] [CrossRef]
- Zhao, Y.; Yokota, K.; Ayada, K.; Yamamoto, Y.; Okada, T.; Shen, L.; Oguma, K. Helicobacter pylori heat-shock protein 60 induces interleukin-8 via a Toll-like receptor (TLR)2 and mitogen-activated protein (MAP) kinase pathway in human monocytes. J. Med. Microbiol. 2007, 56 Pt 2, 154–164. [Google Scholar] [CrossRef]
- Tanaka, A.; Kamada, T.; Yokota, K.; Shiotani, A.; Hata, J.; Oguma, K.; Haruma, K. Helicobacter pylori heat shock protein 60 antibodies are associated with gastric cancer. Pathol. Res. Pract. 2009, 205, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Roesler, B.M.; Rabelo-Goncalves, E.M.; Zeitune, J.M. Virulence Factors of Helicobacter pylori: A Review. Clin. Med. Insights Gastroenterol. 2014, 7, 9–17. [Google Scholar] [CrossRef]
- Hatakeyama, M. Helicobacter pylori CagA and gastric cancer: A paradigm for hit-and-run carcinogenesis. Cell Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Kodama, T.; Gutierrez, O.; Kim, J.G.; Kashima, K.; Graham, D.Y. Relationship between Helicobacter pylori iceA, cagA, and vacA status and clinical outcome: Studies in four different countries. J. Clin. Microbiol. 1999, 37, 2274–2279. [Google Scholar] [CrossRef]
- Odenbreit, S.; Puls, J.; Sedlmaier, B.; Gerland, E.; Fischer, W.; Haas, R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000, 287, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Fujita, A.; Yamazaki, S.; Asaka, M.; Azuma, T.; Hatakeyama, M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc. Natl. Acad. Sci. USA 2002, 99, 14428–14433. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Mimuro, H.; Kiga, K.; Fukumatsu, M.; Ishijima, N.; Morikawa, H.; Nagai, S.; Koyasu, S.; Gilman, R.H.; Kersulyte, D.; et al. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe 2009, 5, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Kang, G.H.; Lee, H.J.; Hwang, K.S.; Lee, S.; Kim, J.H.; Kim, J.S. Aberrant CpG island hypermethylation of chronic gastritis, in relation to aging, gender, intestinal metaplasia, and chronic inflammation. Am. J. Pathol. 2003, 163, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, J. DNA methyltransferases and their roles in tumorigenesis. Biomark. Res. 2017, 5, 1. [Google Scholar] [CrossRef]
- Zhang, B.G.; Hu, L.; Zang, M.D.; Wang, H.X.; Zhao, W.; Li, J.F.; Su, L.P.; Shao, Z.; Zhao, X.; Zhu, Z.G.; et al. Helicobacter pylori CagA induces tumor suppressor gene hypermethylation by upregulating DNMT1 via AKT-NFkappaB pathway in gastric cancer development. Oncotarget 2016, 7, 9788–9800. [Google Scholar] [CrossRef] [PubMed]
- Fu, V.; Plouffe, S.W.; Guan, K.L. The Hippo pathway in organ development, homeostasis, and regeneration. Curr. Opin. Cell Biol. 2017, 49, 99–107. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef]
- Li, N.; Feng, Y.; Hu, Y.; He, C.; Xie, C.; Ouyang, Y.; Artim, S.C.; Huang, D.; Zhu, Y.; Luo, Z.; et al. Helicobacter pylori CagA promotes epithelial mesenchymal transition in gastric carcinogenesis via triggering oncogenic YAP pathway. J. Exp. Clin. Cancer Res. 2018, 37, 280. [Google Scholar] [CrossRef]
- Szabo, I.; Brutsche, S.; Tombola, F.; Moschioni, M.; Satin, B.; Telford, J.L.; Rappuoli, R.; Montecucco, C.; Papini, E.; Zoratti, M. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 1999, 18, 5517–5527. [Google Scholar] [CrossRef]
- Terebiznik, M.R.; Vazquez, C.L.; Torbicki, K.; Banks, D.; Wang, T.; Hong, W.; Blanke, S.R.; Colombo, M.I.; Jones, N.L. Helicobacter pylori VacA toxin promotes bacterial intracellular survival in gastric epithelial cells. Infect. Immun. 2006, 74, 6599–6614. [Google Scholar] [CrossRef]
- Yamasaki, E.; Wada, A.; Kumatori, A.; Nakagawa, I.; Funao, J.; Nakayama, M.; Hisatsune, J.; Kimura, M.; Moss, J.; Hirayama, T. Helicobacter pylori vacuolating cytotoxin induces activation of the proapoptotic proteins Bax and Bak, leading to cytochrome c release and cell death, independent of vacuolation. J. Biol. Chem. 2006, 281, 11250–11259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akazawa, Y.; Isomoto, H.; Matsushima, K.; Kanda, T.; Minami, H.; Yamaghchi, N.; Taura, N.; Shiozawa, K.; Ohnita, K.; Takeshima, F.; et al. Endoplasmic reticulum stress contributes to Helicobacter pylori VacA-induced apoptosis. PLoS ONE 2013, 8, e82322. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Xue, J.; Zhang, Z.J.; Jia, Y.P.; Tong, Y.N.; Han, D.; Li, Q.; Xiang, Y.; Mao, X.H.; Tang, B. Helicobacter pylori VacA induces autophagic cell death in gastric epithelial cells via the endoplasmic reticulum stress pathway. Cell Death Dis. 2017, 8, 3207. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Kidane, D. Molecular Mechanisms of H. pylori-Induced DNA Double-Strand Breaks. Int. J. Mol. Sci. 2018, 19, 2891. [Google Scholar] [CrossRef]
- Fishel, R.; Lescoe, M.K.; Rao, M.R.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Koopman, M.; Kortman, G.A.; Mekenkamp, L.; Ligtenberg, M.J.; Hoogerbrugge, N.; Antonini, N.F.; Punt, C.J.; van Krieken, J.H. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br. J. Cancer 2009, 100, 266–273. [Google Scholar] [CrossRef]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef]
- Kadyrova, L.Y.; Gujar, V.; Burdett, V.; Modrich, P.L.; Kadyrov, F.A. Human MutLgamma, the MLH1-MLH3 heterodimer, is an endonuclease that promotes DNA expansion. Proc. Natl. Acad. Sci. USA 2020, 117, 3535–3542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrantonio, F.; Miceli, R.; Raimondi, A.; Kim, Y.W.; Kang, W.K.; Langley, R.E.; Choi, Y.Y.; Kim, K.M.; Nankivell, M.G.; Morano, F.; et al. Individual Patient Data Meta-Analysis of the Value of Microsatellite Instability as a Biomarker in Gastric Cancer. J. Clin. Oncol. 2019, 37, 3392–3400. [Google Scholar] [CrossRef] [PubMed]
- Leung, W.K.; Kim, J.J.; Kim, J.G.; Graham, D.Y.; Sepulveda, A.R. Microsatellite instability in gastric intestinal metaplasia in patients with and without gastric cancer. Am. J. Pathol. 2000, 156, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Tao, H.; Carloni, E.; Leung, W.K.; Graham, D.Y.; Sepulveda, A.R. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology 2002, 123, 542–553. [Google Scholar] [CrossRef]
- Park, D.I.; Park, S.H.; Kim, S.H.; Kim, J.W.; Cho, Y.K.; Kim, H.J.; Sohn, C.I.; Jeon, W.K.; Kim, B.I.; Cho, E.Y.; et al. Effect of Helicobacter pylori infection on the expression of DNA mismatch repair protein. Helicobacter 2005, 10, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Mirzaee, V.; Molaei, M.; Shalmani, H.M.; Zali, M.R. Helicobacter pylori infection and expression of DNA mismatch repair proteins. World J. Gastroenterol. 2008, 14, 6717–6721. [Google Scholar] [CrossRef] [PubMed]
- Machado, A.M.; Figueiredo, C.; Touati, E.; Maximo, V.; Sousa, S.; Michel, V.; Carneiro, F.; Nielsen, F.C.; Seruca, R.; Rasmussen, L.J. Helicobacter pylori infection induces genetic instability of nuclear and mitochondrial DNA in gastric cells. Clin. Cancer Res. 2009, 15, 2995–3002. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.Z.; O′Hara, A.M.; Denning, T.L.; Dirden-Kramer, B.; Mifflin, R.C.; Reyes, V.E.; Ryan, K.A.; Elliott, S.N.; Izumi, T.; Boldogh, I.; et al. Helicobacter pylori and H2O2 increase AP endonuclease-1/redox factor-1 expression in human gastric epithelial cells. Gastroenterology 2004, 127, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Koeppel, M.; Garcia-Alcalde, F.; Glowinski, F.; Schlaermann, P.; Meyer, T.F. Helicobacter pylori Infection Causes Characteristic DNA Damage Patterns in Human Cells. Cell Rep. 2015, 11, 1703–1713. [Google Scholar] [CrossRef]
- Hartung, M.L.; Gruber, D.C.; Koch, K.N.; Gruter, L.; Rehrauer, H.; Tegtmeyer, N.; Backert, S.; Muller, A.H. pylori-Induced DNA Strand Breaks Are Introduced by Nucleotide Excision Repair Endonucleases and Promote NF-kappaB Target Gene Expression. Cell Rep. 2015, 13, 70–79. [Google Scholar] [CrossRef]
- Han, T.; Jing, X.; Bao, J.; Zhao, L.; Zhang, A.; Miao, R.; Guo, H.; Zhou, B.; Zhang, S.; Sun, J.; et al. H. pylori infection alters repair of DNA double-strand breaks via SNHG17. J. Clin. Investig. 2020, 130, 3901–3918. [Google Scholar] [CrossRef]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic Base Excision Repair: New Approaches Shine Light on Mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Mechanism of interaction between human 8-oxoguanine-DNA glycosylase and AP endonuclease. DNA Repair 2007, 6, 317–328. [Google Scholar] [CrossRef]
- Mokkapati, S.K.; Wiederhold, L.; Hazra, T.K.; Mitra, S. Stimulation of DNA glycosylase activity of OGG1 by NEIL1: Functional collaboration between two human DNA glycosylases. Biochemistry 2004, 43, 11596–11604. [Google Scholar] [CrossRef] [PubMed]
- Glassner, B.J.; Rasmussen, L.J.; Najarian, M.T.; Posnick, L.M.; Samson, L.D. Generation of a strong mutator phenotype in yeast by imbalanced base excision repair. Proc. Natl. Acad. Sci. USA 1998, 95, 9997–10002. [Google Scholar] [CrossRef]
- Toller, I.M.; Neelsen, K.J.; Steger, M.; Hartung, M.L.; Hottiger, M.O.; Stucki, M.; Kalali, B.; Gerhard, M.; Sartori, A.A.; Lopes, M.; et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14944–14949. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Uchida, T.; Tsukamoto, Y.; Watada, M.; Yamaguchi, N.; Yamamoto, K.; Shiota, S.; Moriyama, M.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori infection introduces DNA double-strand breaks in host cells. Infect. Immun. 2014, 82, 4182–4189. [Google Scholar] [CrossRef]
- Cui, J.; Cui, H.; Yang, M.; Du, S.; Li, J.; Li, Y.; Liu, L.; Zhang, X.; Li, S. Tongue coating microbiome as a potential biomarker for gastritis including precancerous cascade. Protein Cell 2019, 10, 496–509. [Google Scholar] [CrossRef]
- Yan, L.; Chen, Y.; Chen, F.; Tao, T.; Hu, Z.; Wang, J.; You, J.; Wong, B.C.Y.; Chen, J.; Ye, W. Effect of Helicobacter pylori Eradication on Gastric Cancer Prevention: Updated Report from a Randomized Controlled Trial With 26.5 Years of Follow-up. Gastroenterology 2022, 163, 154–162.e3. [Google Scholar] [CrossRef] [PubMed]
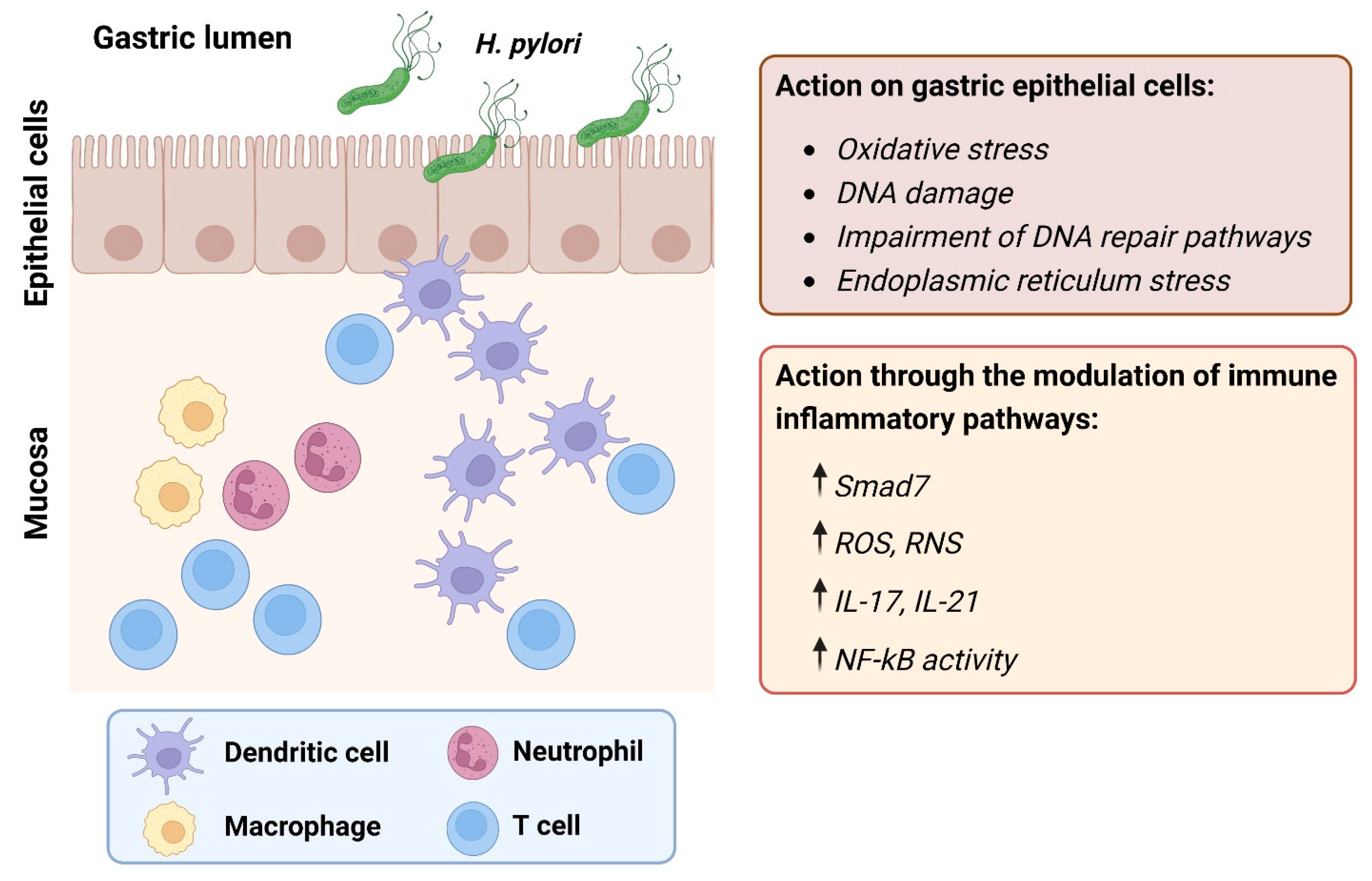


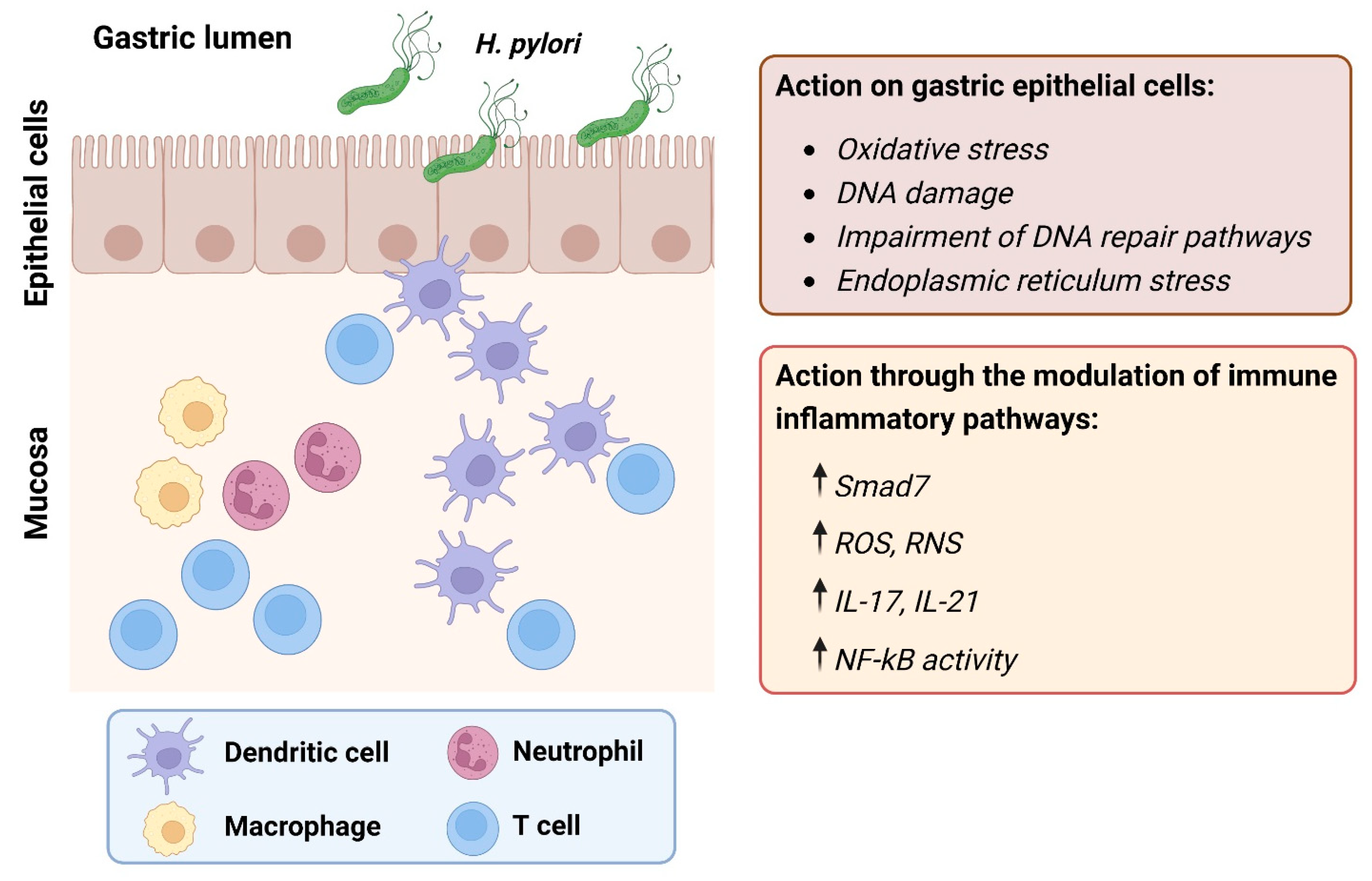{kind=link}
{kind=link}
| Category | Target | Effects | Disease Course | Ref |
|---|---|---|---|---|
| Oxidative stress | Neutrophils/macrophages | Increased production of ROS and RNS. | Onset | [14] |
| SMOX | Increased production of H2O2 as a by-product of conversion of spermine to spermidine. | Onset | [15] | |
| Adaptive immune response | Th1/Th17 cells | Increased synthesis of IFN-γ, TNF-α, and IL-12. | Onset, development | [16] |
| SMAD7 | Prevents endogenous TGF-β1 from dampening the ongoing tissue-damaging Th1 response. | Development | [17] | |
| IL-21 | Increased production of MMP-2 and MMP-9 in a NF-ĸB-dependent fashion. | Development | [18] | |
| IL-17A | Increased production of IL-1β, IL-6, TNF-α, and MMPs. | Development | [19] |
| Repair Pathway | Target | H. pylori–Associated Events | Disease Course | Ref |
|---|---|---|---|---|
| MMR | MLH1, PMS1, PMS2, MSH2, and MSH6 | Decreased level following H. pylori infection | Onset, development | [65] |
| MLH1 and MSH2 | Increased expression after H. pylori eradication | Onset, development | [65,66] | |
| MLH1 | Decreased fraction of MLH1-positive epithelial cell nuclei in H. pylori-infected patients | Onset, development | [67] | |
| MMR genes | Decreased expression and activity after H. pylori infection both in vitro and in vivo | Onset, development | [68] | |
| BER | APE1 | Decreased expression after H. pylori infection resulting in an imbalance between the generation and repair of AP sites, which is highly mutagenic | Onset, development | [68] |
| APE1 | Increased levels in both cultured cells and in primary gastric epithelial cells during H. pylori infection | / | [22] | |
| APE1 | Upregulation during H. pylori infection, downregulation after bacterial eradication | / | [69] | |
| HR and NHEJ | ATR, ATRIP, RAD51, RPA1, MRE11, and NBS1 | Decreased expression following H. pylori infection | Onset, development | [70] |
| NHEJ-related genes | Increased expression after H. pylori infection | Onset, development | [71] | |
| SNHG17 | Shifting of the DSB repair balance from HR toward NHEJ | Onset, development | [72] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvatori, S.; Marafini, I.; Laudisi, F.; Monteleone, G.; Stolfi, C. Helicobacter pylori and Gastric Cancer: Pathogenetic Mechanisms. Int. J. Mol. Sci. 2023, 24, 2895. https://doi.org/10.3390/ijms24032895
Salvatori S, Marafini I, Laudisi F, Monteleone G, Stolfi C. Helicobacter pylori and Gastric Cancer: Pathogenetic Mechanisms. International Journal of Molecular Sciences. 2023; 24(3):2895. https://doi.org/10.3390/ijms24032895
Chicago/Turabian StyleSalvatori, Silvia, Irene Marafini, Federica Laudisi, Giovanni Monteleone, and Carmine Stolfi. 2023. "Helicobacter pylori and Gastric Cancer: Pathogenetic Mechanisms" International Journal of Molecular Sciences 24, no. 3: 2895. https://doi.org/10.3390/ijms24032895
APA StyleSalvatori, S., Marafini, I., Laudisi, F., Monteleone, G., & Stolfi, C. (2023). Helicobacter pylori and Gastric Cancer: Pathogenetic Mechanisms. International Journal of Molecular Sciences, 24(3), 2895. https://doi.org/10.3390/ijms24032895