
Intermolecular-Type Conical Intersections in Benzene Dimer


Abstract
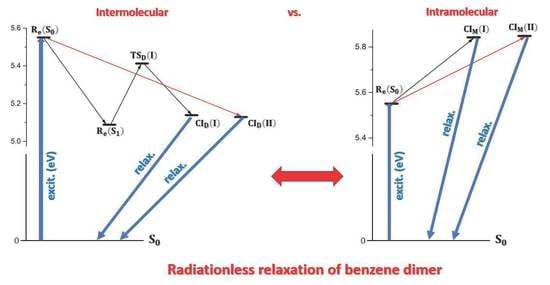
:
1. Introduction
2. Results and Discussion
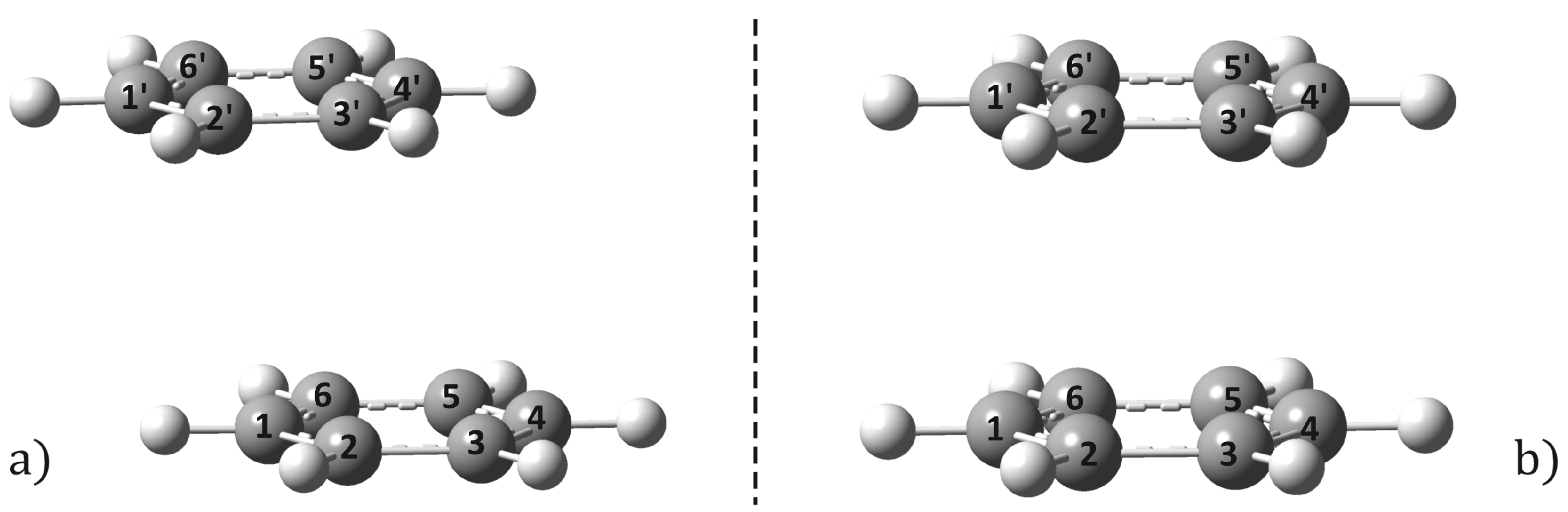
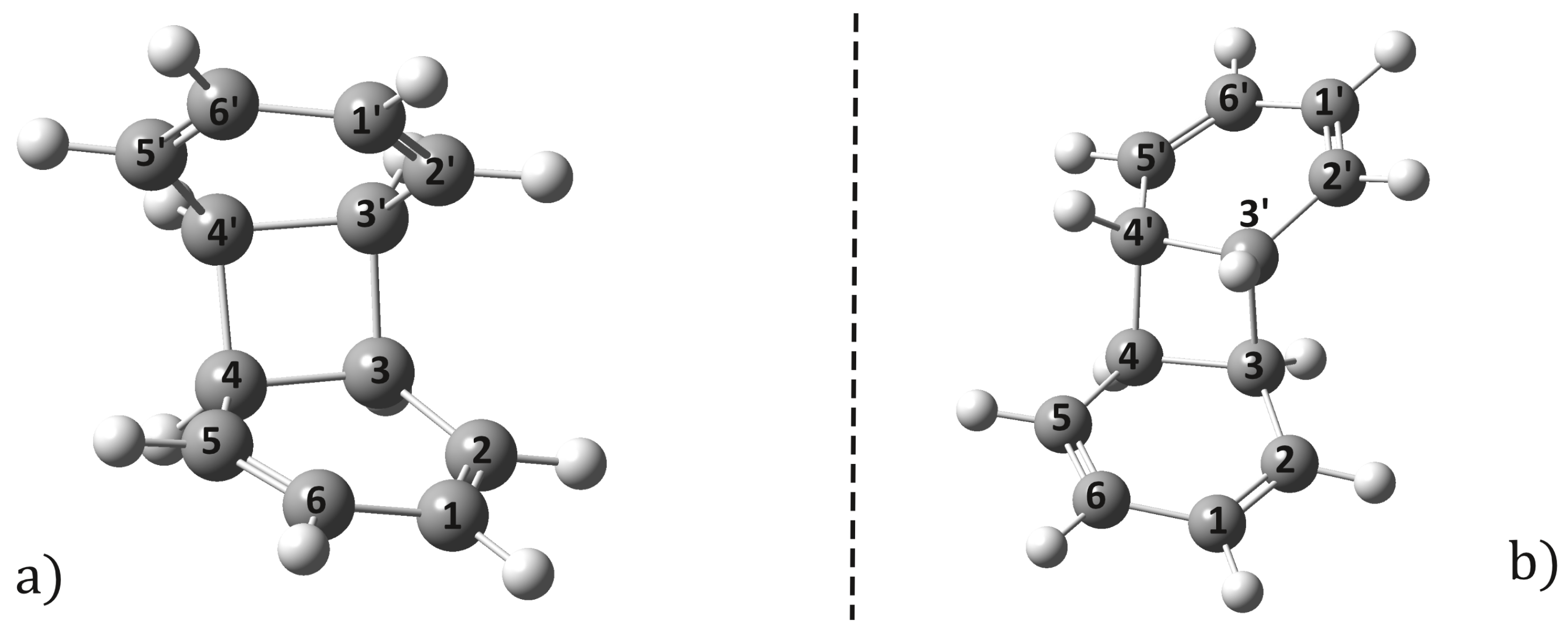
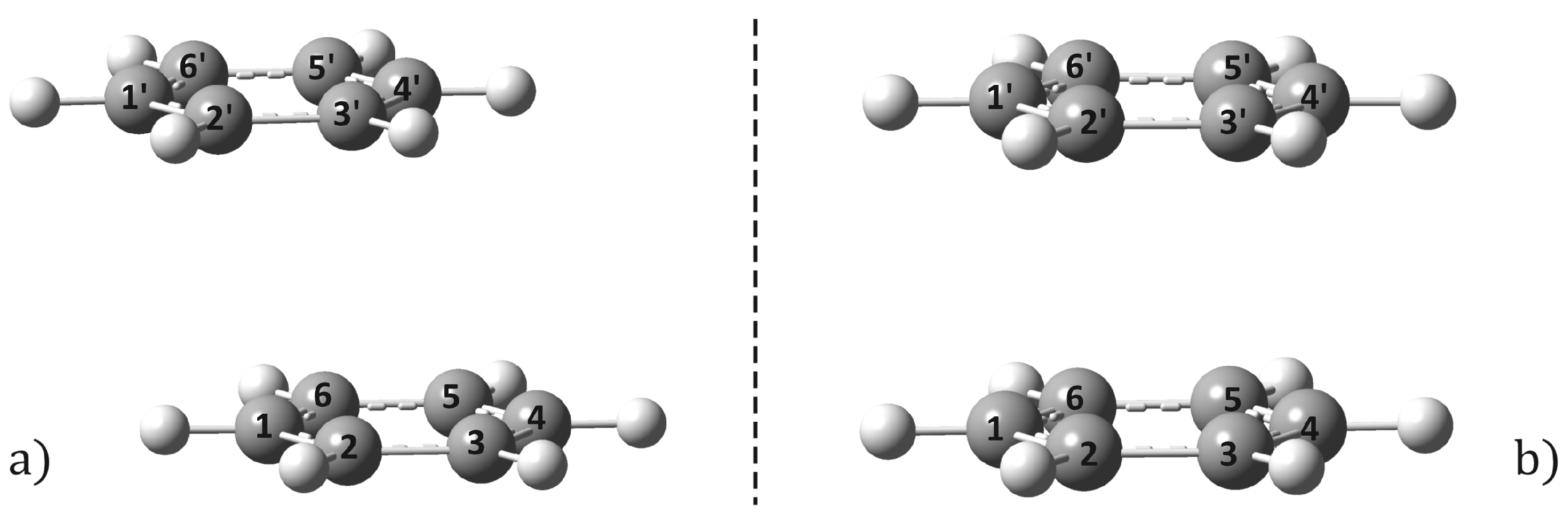
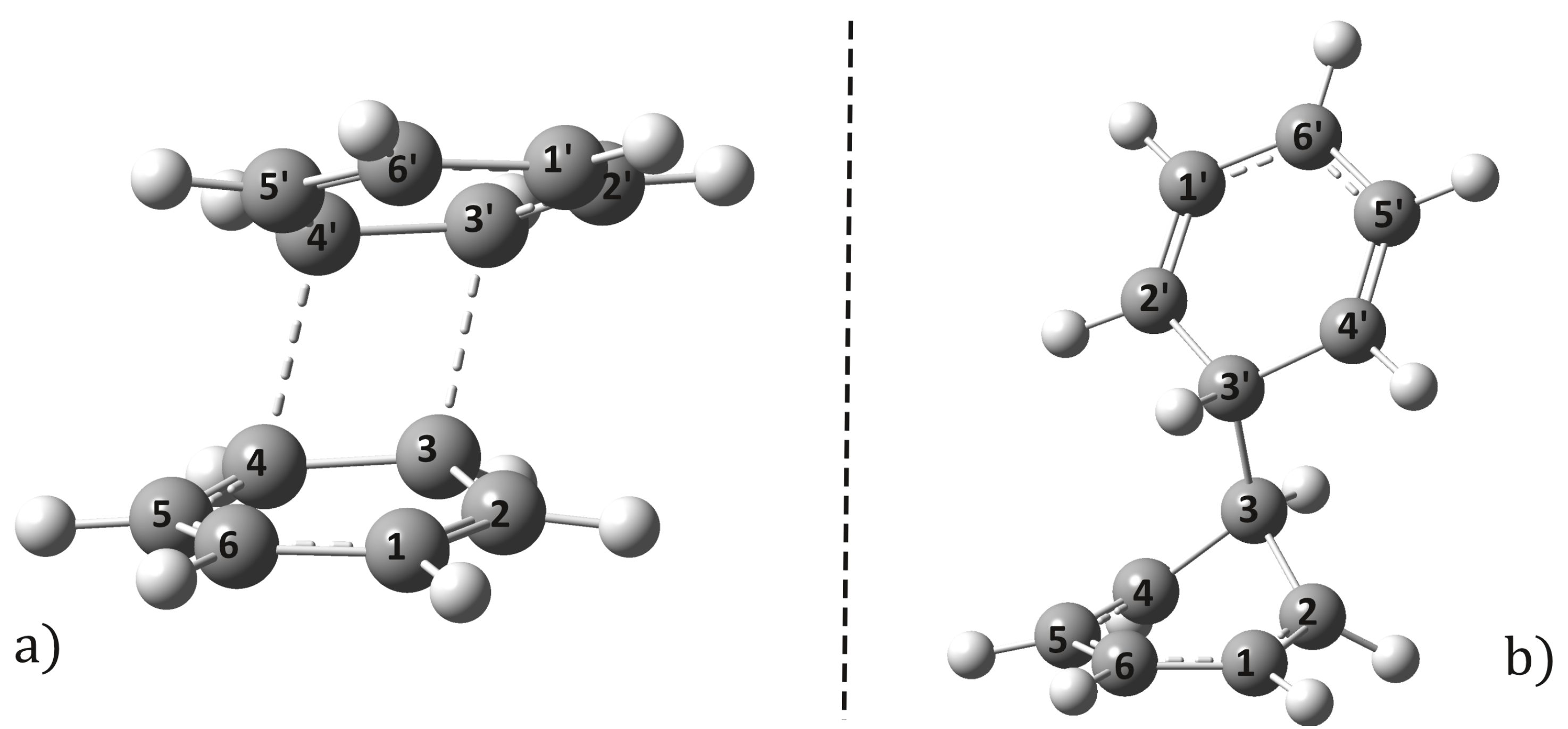
2.1. Equilibrium Geometries
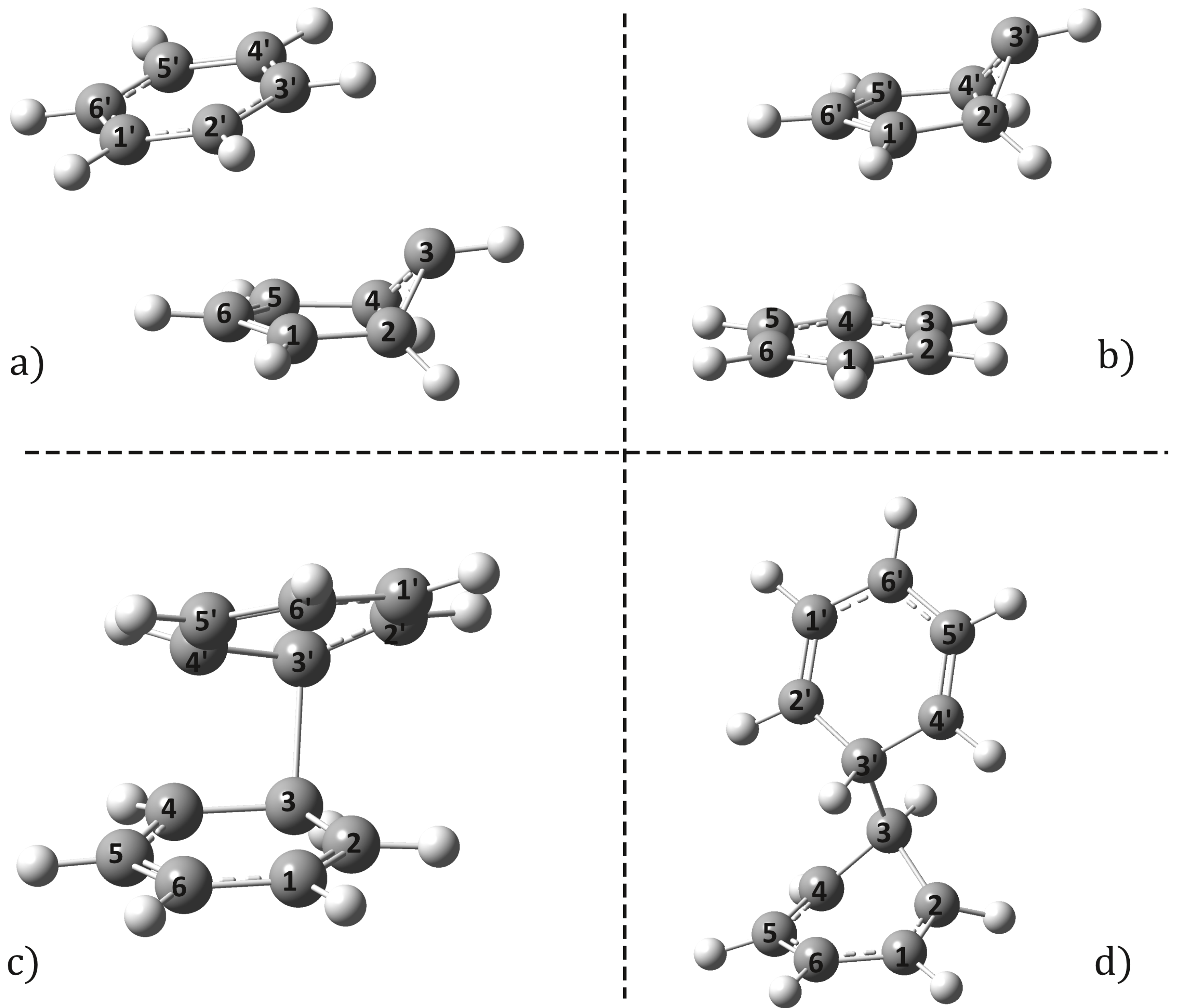
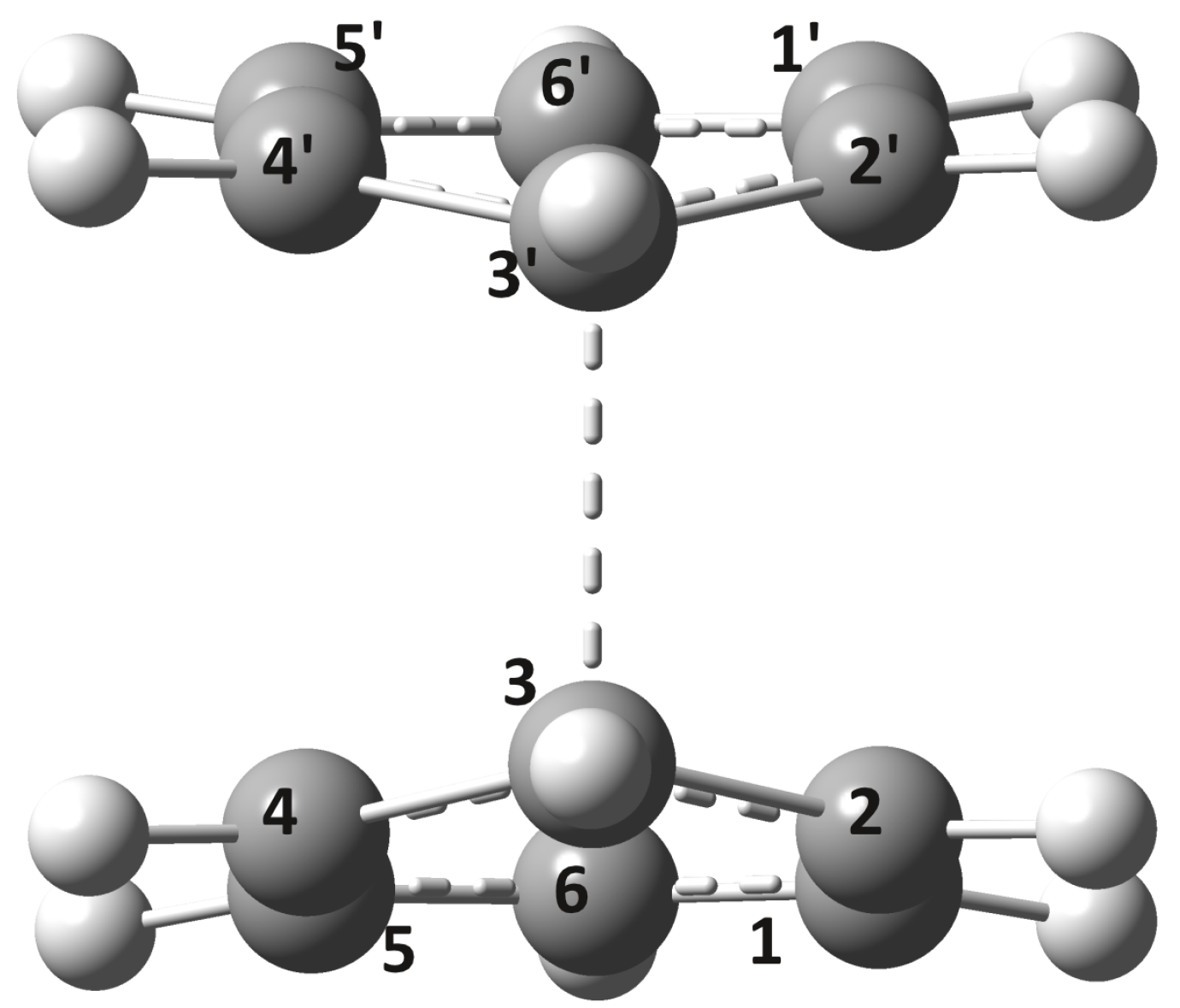
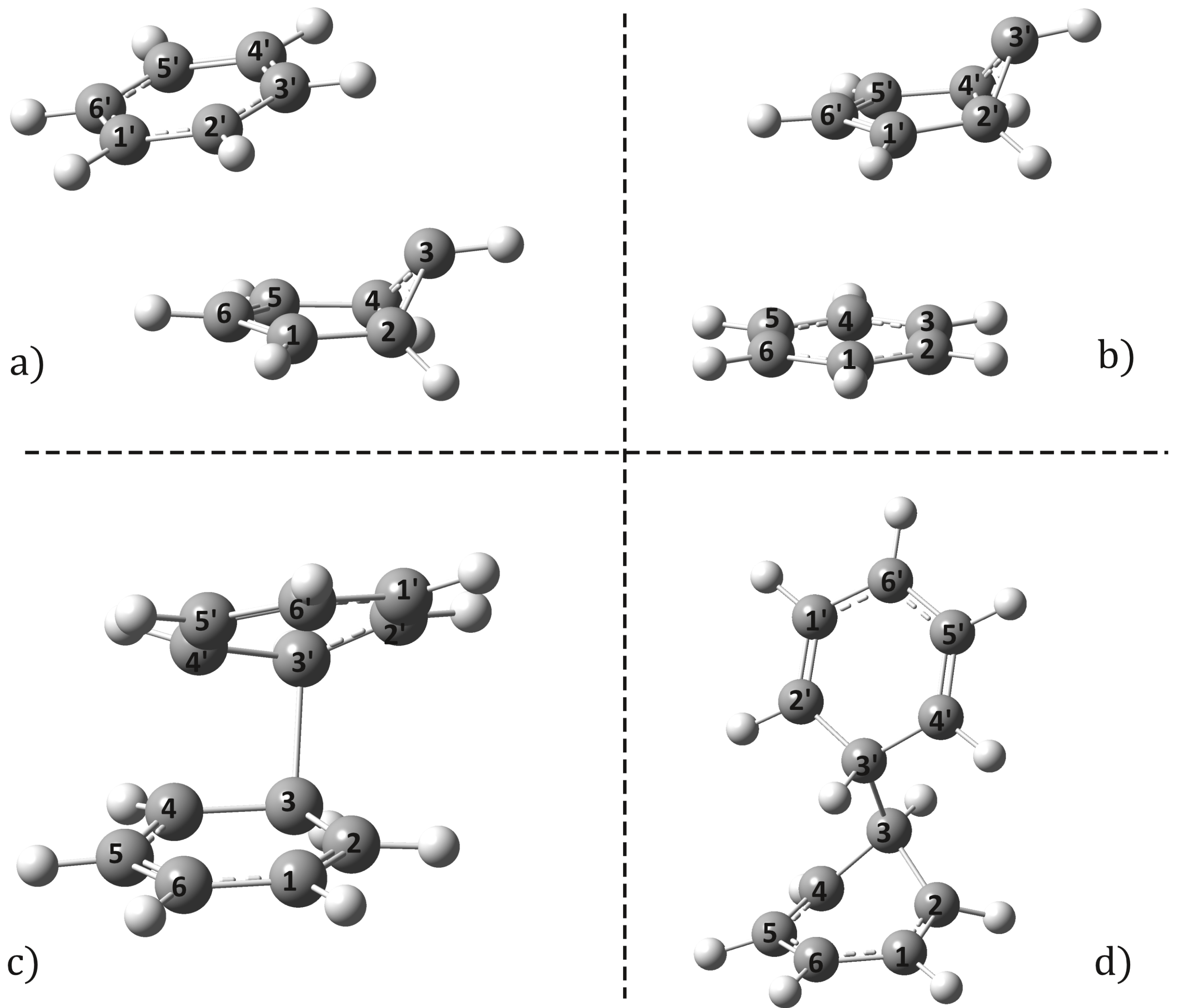
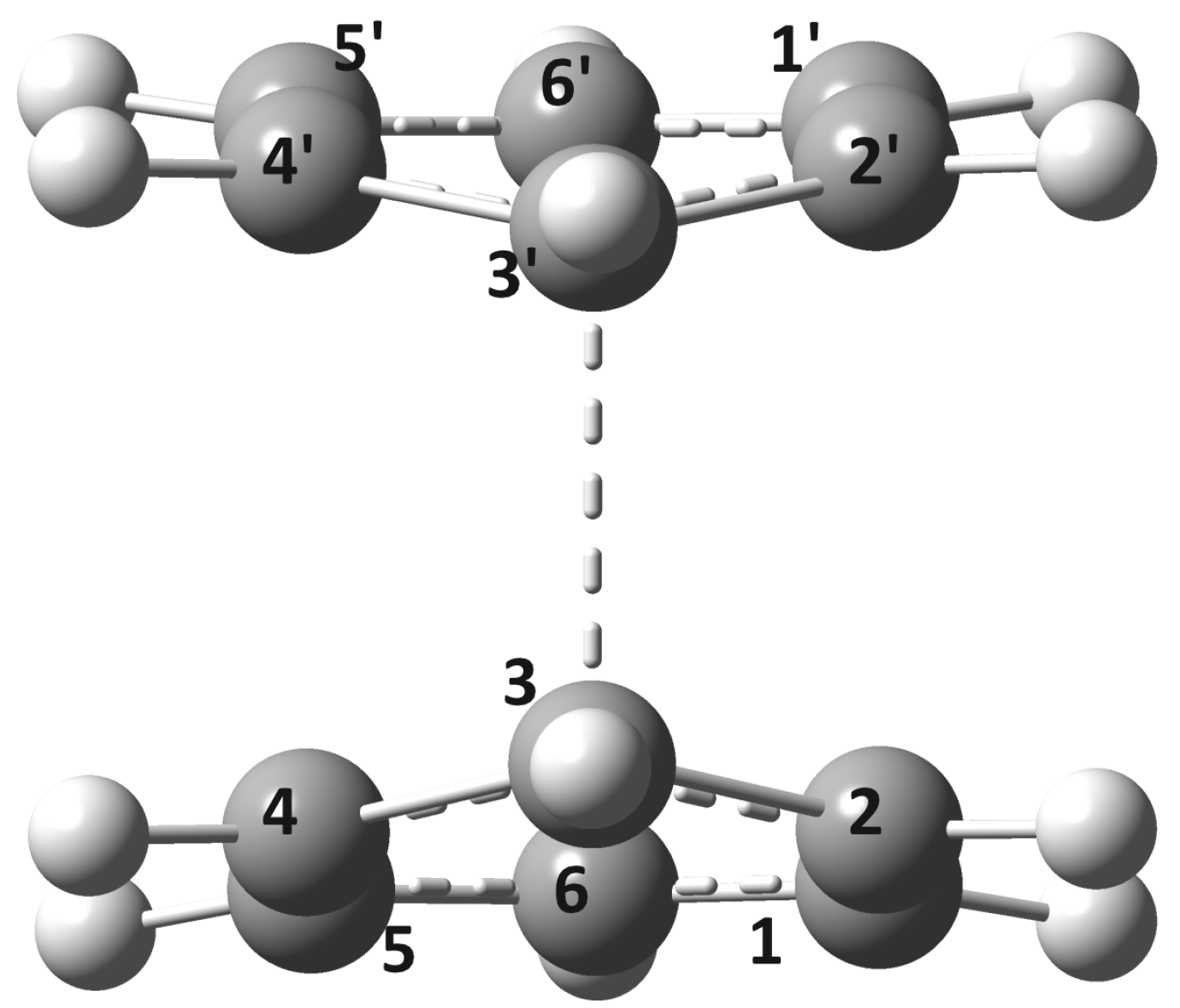
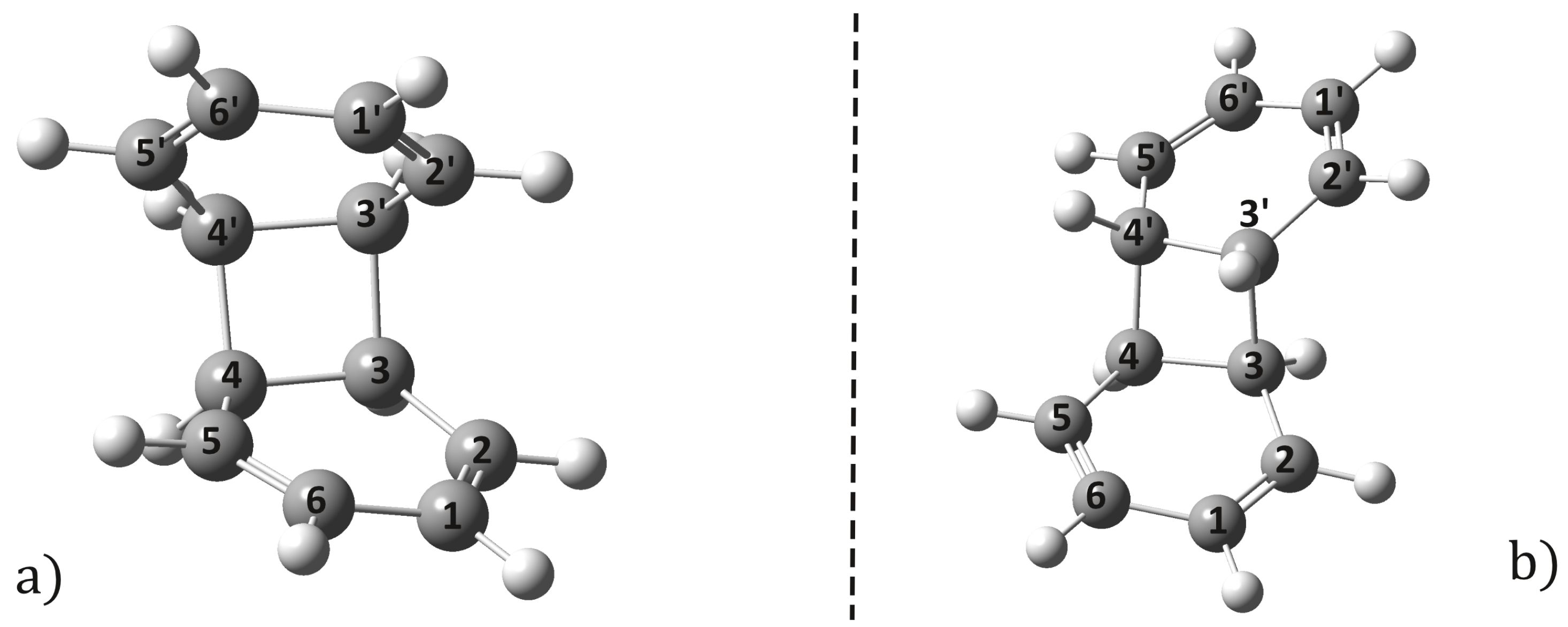
2.2. Conical Intersection Geometries
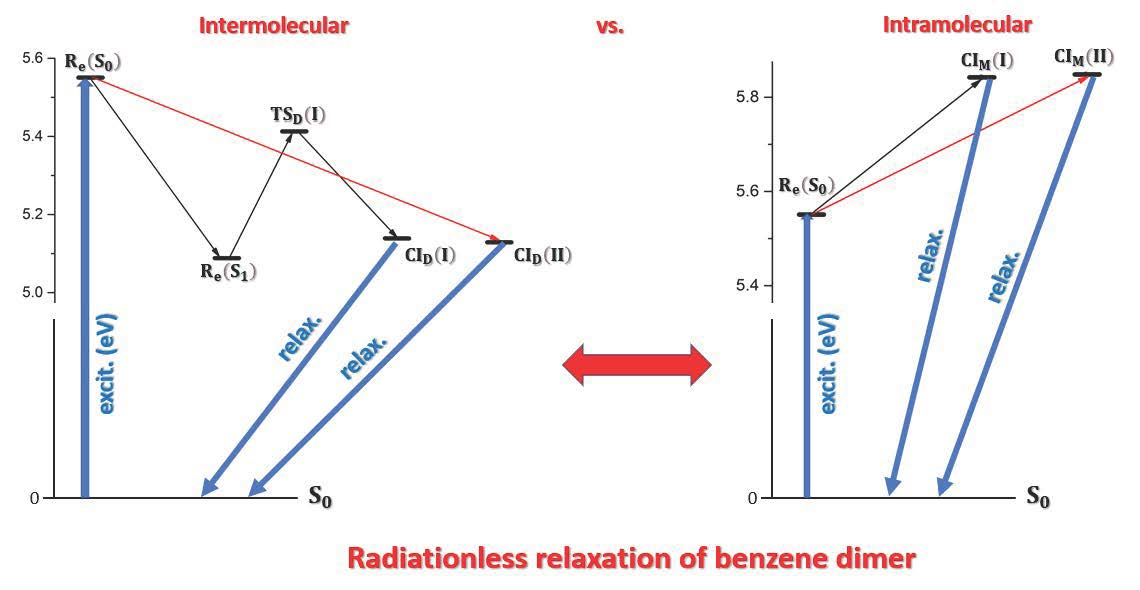
2.3. Possible Relaxation Scenarios
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klessinger, M.; Michl, J. Excited States and Photochemistry of Organic Molecules; VCH Publishers, Inc.: New York, NY, USA, 1995. [Google Scholar]
- Teller, E. The Crossing of Potential Surfaces. J. Phys. Chem. 1937, 41, 109–116. [Google Scholar] [CrossRef]
- Robb, M.A.; Bernardi, F.; Olivucci, M. Conical intersections as a mechanistic feature of organic photochemistry. Pure Appl. Chem. 1995, 67, 783–789. [Google Scholar] [CrossRef]
- Bernardi, F.; Olivucci, M.; Robb, M.A. Potential energy surface crossings in organic photochemistry. Chem. Soc. Rev. 1996, 25, 321–328. [Google Scholar] [CrossRef]
- Worth, G.A.; Cederbaum, L.S. BEYOND BORN-OPPENHEIMER: Molecular Dynamics Through a Conical Intersection. Annu. Rev. Phys. Chem. 2004, 55, 127–158. [Google Scholar] [CrossRef]
- Matsika, S.; Krause, P. Nonadiabatic Events and Conical Intersections. Annu. Rev. Phys. Chem. 2011, 62, 621–643. [Google Scholar] [CrossRef]
- Schuurman, M.S.; Stolow, A. Dynamics at Conical Intersections. Annu. Rev. Phys. Chem. 2018, 69, 17. [Google Scholar] [CrossRef] [PubMed]
- Radloff, W.; Stert, V.; Freudenberg, T.; Hertel, I.V.; Jouvet, C.; Dedonder-Lardeux, C.; Solgadi, D. Internal conversion in highly excited benzene and benzene dimer: Femtosecond time-resolved photoelectron spectroscopy. Chem. Phys. Lett. 1997, 281, 20–26. [Google Scholar] [CrossRef]
- Parker, D.S.N.; Minns, R.S.; Penfold, T.J.; Worth, G.A.; Fielding, H.H. Ultrafast dynamics of the S1 excited state of benzene. Chem. Phys. Lett. 2009, 469, 43–47. [Google Scholar] [CrossRef]
- Adachi, S.; Suzuki, T. Methyl substitution effects on the non-adiabatic dynamics of benzene: Lifting three-state quasi-degeneracy at conical intersections. Phys. Chem. Chem. Phys. 2020, 22, 2814–2818. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, A.; Thompson, A.L.; Martínez, T.J. Excited state direct dynamics of benzene with reparameterized multi-reference semiempirical configuration interaction methods. Chem. Phys. 2004, 304, 133–145. [Google Scholar] [CrossRef]
- Thompson, A.L.; Martínez, T.J. Time-resolved photoelectron spectroscopy from first principles: Excited state dynamics of benzene. Faraday Discuss. 2011, 150, 293–311. [Google Scholar] [CrossRef]
- Palmer, I.J.; Ragazos, N.I.; Bernardi, F.; Olivucci, M.; Robb, M.A. An MC-SCF study of the S1 and S2 photochemical reactions of benzene. J. Am. Chem. Soc. 1993, 115, 673–682. [Google Scholar] [CrossRef]
- Smith, B.R.; Bearpark, M.J.; Robb, M.A.; Bernardi, F.; Olivucci, M. ’Classical wavepacket’ dynamics through a conical intersection. Application to the S1/S 0 photochemistry of benzene. Chem. Phys. Lett. 1995, 242, 27–32. [Google Scholar] [CrossRef]
- Lasorne, B.; Sicilia, F.; Bearpark, M.J.; Robb, M.A.; Worth, G.A.; Blancafort, L. Automatic generation of active coordinates for quantum dynamics calculations: Application to the dynamics of benzene photochemistry. J. Chem. Phys. 2008, 128, 124307. [Google Scholar] [CrossRef] [PubMed]
- Lasorne, B.; Bearpark, M.J.; Robb, M.A.; Worth, G.A. Controlling S1/S0 Decay and the Balance between Photochemistry and Photostability in Benzene: A Direct Quantum Dynamics Study. J. Phys. Chem. A 2008, 112, 13017–13027. [Google Scholar] [CrossRef]
- Penfold, T.J.; Worth, G.A. A model Hamiltonian to simulate the complex photochemistry of benzene II. J. Chem. Phys. 2009, 131, 064303. [Google Scholar] [CrossRef]
- Li, Q.; Mendive-Tapia, D.; Paterson, M.J.; Migani, A.; Bearpark, M.J.; Robb, M.A.; Blancafort, L. A global picture of the S1/S0 conical intersection seam of benzene. Chem. Phys. 2010, 377, 60–65. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
- Nishimoto, Y. Locating conical intersections using the quasidegenerate partially and strongly contracted NEVPT2 methods. Chem. Phys. Lett. 2020, 744, 137219. [Google Scholar] [CrossRef]
- Birks, J.B. Excimers. Rep. Prog. Phys. 1975, 38, 903–974. [Google Scholar] [CrossRef]
- Cardozo, T.M.; Galliez, A.P.; Jr, I.B.; Plasser, F.; Aquino, A.J.A.; Barbatti, M.; Lischka, H. Dynamics of benzene excimer formation from the parallel-displaced dimer. Phys. Chem. Chem. Phys. 2019, 21, 13916–13924. [Google Scholar] [CrossRef]
- Rocha-Rinza, T.; Vico, L.D.; Veryazov, V.; Roos, B.O. A theoretical study of singlet low-energy excited states of the benzene dimer. Chem. Phys. Lett. 2006, 426, 268–272. [Google Scholar] [CrossRef]
- Diaz-Andres, A.; Casanova, D. Benzene Excimer and Excited Multimers: Electronic Character, Interaction Nature, and Aromaticity. J. Phys. Chem. Lett. 2021, 12, 7400–7408. [Google Scholar] [CrossRef]
- Diri, K.; Krylov, A.I. Electronic States of the Benzene Dimer: A Simple Case of Complexity. J. Phys. Chem. A 2012, 116, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam Iyer, E.S.; Sadybekov, A.; Lioubashevski, O.; Krylov, A.I.; Ruhman, S. Rewriting the Story of Excimer Formation in Liquid Benzene. J. Phys. Chem. A 2017, 121, 1962–1975. [Google Scholar] [CrossRef]
- Shinohara, H.; Nishi, N. Excited state lifetimes and appearance potentials of benzene dimer and trimer. J. Chem. Phys. 1989, 91, 6743–6751. [Google Scholar] [CrossRef]
- Gan, H.; Horner, M.G.; Hrnjez, B.J.; McCormack, T.A.; King, J.L.; Gasyna, Z.; Chen, G.; Gleiter, R.; Yang, N. Chemistry of syn-o,o’-Dibenzene. J. Am. Chem. Soc. 2000, 122, 12098–12111. [Google Scholar] [CrossRef]
- Shu, K.; Zhao, J.; Yuan, S.; Dou, Y.; Lo, G.V. Mechanisms of Laser-Induced Reactions of Stacked Benzene Molecules: A Semiclassical Dynamics Simulation and CASSCF Calculation. Int. J. Photoenergy 2014, 2014, 874102. [Google Scholar] [CrossRef]
- Boggio-Pasqua, M.; Groenhof, G.; Schäfer, L.V.; Grubmüller, H.; Robb, M.A. Ultrafast Deactivation Channel for Thymine Dimerization. J. Am. Chem. Soc. 2007, 129, 10996–10997. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Douki, T.; Improta, R.; Gustavsson, T.; Onidas, D.; Vayá, I.; Perron, M.; Markovitsi, D. Electronic Excited States Responsible for Dimer Formation upon UV Absorption Directly by Thymine Strands: Joint Experimental and Theoretical Study. J. Am. Chem. Soc. 2012, 134, 14834–14845. [Google Scholar] [CrossRef]
- Wang, H.; Chen, X. How Does Thymine DNA Survive Ultrafast Dimerization Damage? Molecules 2017, 22, 60. [Google Scholar] [CrossRef]
- Bende, A.; Farcas, A.A.; Toşa, V. Theoretical study of light-induced crosslinking reaction between pyrimidine DNA bases and aromatic amino acids. Front. Bioeng. Biotechnol. 2022, 9, 806415. [Google Scholar] [CrossRef] [PubMed]
- Bende, A.; Farcaş, A.A.; Falamaş, A.; Petran, A. New insight to the catechol photochemistry: The role of different monomer and dimer configurations in radiationless decay of S1 electronic excited state. Phys. Chem. Chem. Phys. 2022, 24, 29165–29175. [Google Scholar] [CrossRef]
- Keal, T.W.; Koslowski, A.; Thiel, W. Comparison of algorithms for conical intersection optimisation using semiempirical methods. Theor. Chem. Acc. 2007, 118, 837–844. [Google Scholar] [CrossRef]
- Minezawa, N.; Gordon, M.S. Optimizing Conical Intersections by Spin-Flip Density Functional Theory: Application to Ethylene. J. Phys. Chem A 2009, 113, 12749–12753. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Kato, S. Dynamic electron correlation effect on conical intersections in photochemical ring-opening reaction of cyclohexadiene: MS-CASPT2 study. Chem. Phys. Lett. 2009, 476, 97–100. [Google Scholar] [CrossRef]
- Aldaz, C.; Kammeraad, J.A.; Zimmerman, P.M. Discovery of conical intersection mediated photochemistry with growing string methods. Phys. Chem. Chem. Phys. 2018, 20, 27394–27405. [Google Scholar] [CrossRef]
- Harabuchi, Y.; Hatanaka, M.; Maeda, S. Exploring approximate geometries of minimum energy conical intersections by TDDFT calculations. Chem. Phys. Lett. 2019, 737S, 100007. [Google Scholar] [CrossRef]
- Winslow, M.; Cross, W.B.; Robinson, D. Comparison of Spin-Flip TDDFT-Based Conical Intersection Approaches with XMS-CASPT2. J. Chem. Theory Comput. 2020, 16, 3253–3263. [Google Scholar] [CrossRef]
- Levine, B.G.; Ko, C.; Quenneville, J.; Martínez, T.J. Conical intersections and double excitations in time-dependent density functional theory. Mol. Phys. 2006, 104, 1039–1051. [Google Scholar] [CrossRef]
- Huix-Rotllant, M.; Nikiforov, A.; Thiel, W.; Filatov, M. Description of Conical Intersections with Density Functional Methods. In Density-Functional Methods for Excited States; Ferré, N., Filatov, M., Huix-Rotllant, M., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2016; pp. 445–476. [Google Scholar] [CrossRef]
- Huix-Rotllant, M.; Ferré, N.; Barbatti, M. Time-Dependent Density Functional Theory. In Quantum Chemistry and Dynamics of Excited States; González, L., Lindh, R., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2020; Chapter 2; pp. 13–46. [Google Scholar] [CrossRef]
- Schnell, M.; Erlekam, U.; Bunker, P.R.; von Helden, G.; Grabow, J.U.; Meijer, G.; van der Avoird, A. Structure of the Benzene Dimer-Governed by Dynamics. Angew. Chem. Int. Ed. 2013, 52, 5180–5183. [Google Scholar] [CrossRef]
- Sinnokrot, M.O.; Sherrill, C.D. Highly Accurate Coupled Cluster Potential Energy Curves for the Benzene Dimer: Sandwich, T-Shaped, and Parallel-Displaced Configurations. J. Phys. Chem. A 2004, 108, 10200–10207. [Google Scholar] [CrossRef]
- Hill, J.G.; Platts, J.A.; Werner, H.J. Calculation of intermolecular interactions in the benzene dimer using coupled-cluster and local electron correlation methods. Phys. Chem. Chem. Phys. 2006, 8, 4072–4078. [Google Scholar] [CrossRef]
- Falamaş, A.; Petran, A.; Hada, A.M.; Bende, A. Dopamine Photochemical Behaviour under UV Irradiation. Int. J. Mol. Sci. 2022, 23, 5483. [Google Scholar] [CrossRef] [PubMed]
- De Sio, A.; Sommer, E.; Nguyen, X.T.; Groß, L.; Popović, D.; Nebgen, B.T.; Fernandez-Alberti, S.; Pittalis, S.; Rozzi, C.A.; Molinari, E.; et al. Intermolecular conical intersections in molecular aggregates. Nat. Nanotechnol. 2021, 16, 63–68. [Google Scholar] [CrossRef]
- Dawson, W.R.; Windsor, M.W. Fluorescence yields of aromatic compounds. J. Phys. Chem. 1968, 72, 3251–3260. [Google Scholar] [CrossRef]
- Selinger, B.K.; Ware, W.R. Fluorescence Lifetime of Benzene and Benzene-d6 Vapor Excited to Single Vibronic Levels. J. Chem. Phys. 1970, 53, 3160–3168. [Google Scholar] [CrossRef]
- Luria, M.; Ofran, M.; Stein, G. Natural and experimental fluorescence lifetimes of benzene in various solvents. J. Phys. Chem. 1974, 78, 1904–1909. [Google Scholar] [CrossRef]
- Quina, F.H.; Carroll, F.A. Radiative and nonradiative transitions in solution. First excited singlet state of benzene and its methyl derivatives. J. Am. Chem. Soc. 1976, 98, 6–9. [Google Scholar] [CrossRef]
- de Souza, B.; Neese, F.; Izsák, R. On the theoretical prediction of fluorescence rates from first principles using the path integral approach. J. Chem. Phys. 2018, 148, 034104. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Lin, Y.S.; Li, G.D.; Mao, S.P.; Chai, J.D. Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections. J. Chem. Theory Comput. 2013, 9, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Xu, X.; Truhlar, D.G. Minimally augmented Karlsruhe basis sets. Theor. Chem. Accounts 2011, 128, 295–305. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley INterdisciplinary-Rev.-Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A ’chain-of-spheres’ algorithm for the Hartree-Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Hellweg, A.; Hättig, C.; Höfener, S.; Klopper, W. Optimized accurate auxiliary basis sets for RI-MP2 and RI-CC2 calculations for the atoms Rb to Rn. Theor. Chem. Accounts 2007, 117, 587–597. [Google Scholar] [CrossRef]
- Rinkevicius, Z.; Vahtras, O.; Ågren, H. Spin-flip time dependent density functional theory applied to excited states with single, double, or mixed electron excitation character. J. Chem. Phys. 2010, 133, 114104. [Google Scholar] [CrossRef]
- Mei, Y.; Yang, W. Excited-State Potential Energy Surfaces, Conical Intersections, and Analytical Gradients from Ground-State Density Functional Theory. J. Phys. Chem. Lett. 2019, 10, 2538–2545. [Google Scholar] [CrossRef]
- Casanova, D.; Krylov, A.I. Spin-flip methods in quantum chemistry. Phys. Chem. Chem. Phys. 2020, 22, 4326–4342. [Google Scholar] [CrossRef] [PubMed]
- Matsika, S. Electronic Structure Methods for the Description of Nonadiabatic Effects and Conical Intersections. Chem. Rev. 2021, 121, 9407–9449. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Werner, H.; Knowles, P.J. A second order multiconfiguration SCF procedure with optimum convergence. J. Chem. Phys. 1985, 82, 5053–5063. [Google Scholar] [CrossRef]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Korona, T.; Lindh, R.; Mitrushenkov, A.; Rauhut, G.; et al. MOLPRO, 2012.1, A Package of Ab Initio Programs. Available online: https://www.molpro.net (accessed on 9 September 2022).
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Ásgeirsson, V.; Birgisson, B.O.; Bjornsson, R.; Becker, U.; Neese, F.; Riplinger, C.; Jónsson, H. Nudged Elastic Band Method for Molecular Reactions Using Energy-Weighted Springs Combined with Eigenvector Following. J. Chem. Theor. Comput. 2021, 17, 4929–4945. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I. Charge, bond order and valence in the ab initio scf theory. Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar] [CrossRef]
- Mayer, I. Bond order and valence: Relations to Mulliken’s population analysis. Int. J. Quantum Chem. 1984, 26, 151–154. [Google Scholar] [CrossRef]
- Allouche, A.R. Gabedit–A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennington, R.; Keith, T.; Millam, J. Gaussview 5.0.9; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Geom. | Benzene Monomer’s C–C Bonds | Dimer | Energy | |||||
|---|---|---|---|---|---|---|---|---|
| C–C | C–C | C–C | C–C | C–C | C–C | C···C | ref: | |
| C–C | C–C | C–C | C–C | C–C | C–C | R(S) | ||
| DFT: R(S), R(S), R(S), TS(I); TDDFT: R(S); SF-TDDFT: CI(I or II), CI(I or II) | ||||||||
| (using the ωB97X-D3/ma-def2-TZVPP computation scheme) | ||||||||
| R(S) | 1.387 | 1.387 | 1.387 | 1.387 | 1.387 | 1.387 | 3.579 | |
| 1.387 | 1.387 | 1.387 | 1.387 | 1.387 | 1.387 | |||
| R(S) | 1.330 | 1.497 | 1.553 | 1.495 | 1.468 | 1.387 | 1.563 | |
| 1.330 | 1.495 | 1.554 | 1.498 | 1.330 | 1.468 | 1.564 | ||
| R(S) | 1.331 | 1.493 | 1.554 | 1.497 | 1.331 | 1.468 | 1.562 | |
| 1.331 | 1.493 | 1.554 | 1.496 | 1.331 | 1.468 | 1.554 | ||
| R(S) | 1.401 | 1.401 | 1.401 | 1.401 | 1.401 | 1.401 | 3.073 | 0.000 |
| 1.401 | 1.401 | 1.401 | 1.401 | 1.401 | 1.401 | |||
| CI(I) | 1.454 | 1.440 | 1.440 | 1.454 | 1.381 | 1.381 | 3.764 | 0.757 |
| 1.388 | 1.387 | 1.387 | 1.386 | 1.388 | 1.387 | (17.46) | ||
| CI(II) | 1.387 | 1.387 | 1.387 | 1.386 | 1.387 | 1.387 | 3.602 | 0.763 |
| 1.458 | 1.437 | 1.440 | 1.451 | 1.384 | 1.378 | (17.60) | ||
| CI(I) | 1.363 | 1.449 | 1.492 | 1.400 | 1.371 | 1.418 | 1.844 | 0.051 |
| 1.359 | 1.431 | 1.486 | 1.418 | 1.369 | 1.427 | (1.18) | ||
| CI(II) | 1.406 | 1.482 | 1.479 | 1.407 | 1.397 | 1.399 | 1.585 | 0.043 |
| 1.359 | 1.482 | 1.482 | 1.359 | 1.408 | 1.408 | (1.00) | ||
| CI(60°) | 1.366 | 1.464 | 1.541 | 1.407 | 1.374 | 1.408 | 1.753 | 0.621 |
| 1.364 | 1.428 | 1.457 | 1.413 | 1.373 | 1.433 | (14.32) | ||
| CI(100°) | 1.415 | 1.489 | 1.435 | 1.370 | 1.418 | 1.370 | 1.930 | 0.302 |
| 1.369 | 1.420 | 1.481 | 1.429 | 1.364 | 1.422 | (6.98) | ||
| TS(I) | 1.366 | 1.439 | 1.438 | 1.365 | 1.408 | 1.408 | 2.301 | 0.327 |
| 1.365 | 1.438 | 1.439 | 1.365 | 1.408 | 1.408 | (7.54) | ||
| CASSCF(12,11)/6–31G(d,p) | ||||||||
| CI(I) | 1.358 | 1.466 | 1.479 | 1.426 | 1.363 | 1.446 | 2.010 | |
| 1.364 | 1.426 | 1.481 | 1.452 | 1.346 | 1.445 | 2.049 | ||
| CI(II) | 1.446 | 1.497 | 1.492 | 1.407 | 1.397 | 1.399 | 1.546 | |
| 1.365 | 1.509 | 1.509 | 1.366 | 1.424 | 1.425 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bende, A.; Farcaş, A.-A. Intermolecular-Type Conical Intersections in Benzene Dimer. Int. J. Mol. Sci. 2023, 24, 2906. https://doi.org/10.3390/ijms24032906
Bende A, Farcaş A-A. Intermolecular-Type Conical Intersections in Benzene Dimer. International Journal of Molecular Sciences. 2023; 24(3):2906. https://doi.org/10.3390/ijms24032906
Chicago/Turabian StyleBende, Attila, and Alex-Adrian Farcaş. 2023. "Intermolecular-Type Conical Intersections in Benzene Dimer" International Journal of Molecular Sciences 24, no. 3: 2906. https://doi.org/10.3390/ijms24032906
APA StyleBende, A., & Farcaş, A.-A. (2023). Intermolecular-Type Conical Intersections in Benzene Dimer. International Journal of Molecular Sciences, 24(3), 2906. https://doi.org/10.3390/ijms24032906