Abstract
Even with modern therapy, patients with heart failure only have a 50% five-year survival rate. To improve the development of new therapeutic strategies, preclinical models of disease are needed to properly emulate the human condition. Determining the most appropriate model represents the first key step for reliable and translatable experimental research. Rodent models of heart failure provide a strategic compromise between human in vivo similarity and the ability to perform a larger number of experiments and explore many therapeutic candidates. We herein review the currently available rodent models of heart failure, summarizing their physiopathological basis, the timeline of the development of ventricular failure, and their specific clinical features. In order to facilitate the future planning of investigations in the field of heart failure, a detailed overview of the advantages and possible drawbacks of each model is provided.
1. Introduction
Thanks to the advances in medical therapies for heart failure (HF), as well as the persistent improvements in cardiovascular surgical techniques and devices technologies, many previously fatal cardiac diseases are now chronically managed with satisfactory medium- and long-term prognosis. However, once the cardiac performance is severely affected, a progressively declining syndrome is established, whose definitive fate is often end-stage organ failure [1]. Demographic projections estimate that by the next decade one out of every thirty-three people in the US will be affected by HF [2]. In the highly complex population of pediatric patients with structural heart defects, the number of patients palliated with the Fontan procedure is expected to double in the next 20 years [3]. Currently, more than 90% of children with congenital heart defects survive to adulthood [4], indicating a substantial increase in demand for HF-related services as this population ages into adulthood with expected evolving cardiac dysfunction.
Current treatment options for end-stage HF entail organ replacement with human grafts, artificial devices, or xenografts. However, none these strategies are definitively curative (incorporating graft failure) and are limited by the availability of donors [5], chronic immune rejection [6,7], high rates of graft vascular disease, and abnormal activation of coagulation cascades and platelets at the blood–device interface [8,9]. Conversely, regenerative medicine techniques may represent a new frontier in the management of end-stage HF. Cardiac progenitor stem cells and derived proteins [10,11], molecularly targeted drugs [12], and reinvented surgical procedures [13,14] aim at stimulating the intrinsic repair ability of the human heart [15,16]. Genome editing technologies are emerging as potential therapeutic strategies able to address specific causative monogenetic disorders associated with the development of cardiomyopathy and HF. Through base editing, the expression of key proteins that are dysregulated in rare genetic forms of dilated cardiomyopathies (DCM), such as dystrophin in Duchenne muscular dystrophy [17] and Rbm20 [18], can be restored, representing a promising therapeutic concept.
To test novel putative therapeutic approaches to HF in a controlled manner, viable animal models of HF are paramount. Due to an advantageous compromise between adequate body size and low housing and maintenance costs, rodent models of HF have been extensively used by basic and translational scientists for decades [19,20,21]. Particularly, rats offer sufficient dimensions to perform cardiac surgical procedures and invasive hemodynamic measurements safely, shortening learning curves for operators. Moreover, they expedite advanced imaging measurements and provide a 10-fold greater myocardial mass for subsequent histopathological or molecular analyses, when compared to mice.
Many methods have been tested and validated to induce progressive HF and DCM in rats, mimicking the different etiologies of HF in humans. With the present review, we illustrate the available rat models of HF (Figure 1), highlighting their strengths and drawbacks. A comprehensive description of their mechanisms and timelines for developing HF is provided, as well as their documented or potential purposes for translational research and experimental surgery.
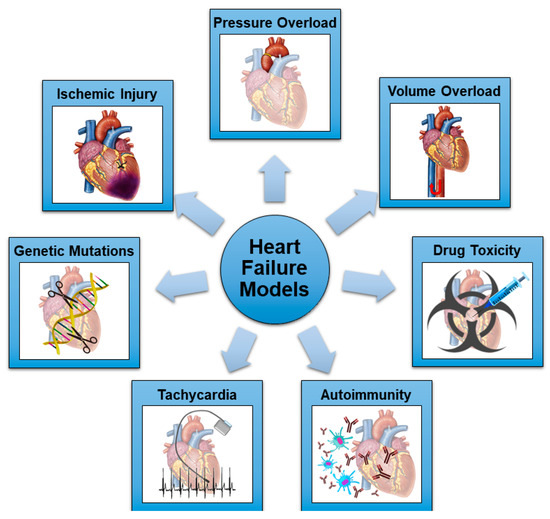
Figure 1.
Schematic representation of the most common mechanisms utilized in rodent models of HF.
2. Rats and Mice as Animal Models of Heart Failure
Rats and mice share the benefits of small mammalian models, in terms of ease of handling and housing, short breeding cycle, and the number of recruitable animals to improve the statistical power of experiments. The small physical size reduces the costs of novel therapeutic agents and molecules, whose administration is usually calculated based on body weight. However, some differences apply between these two species, starting with the size of the animals. Despite a similar 2–3 years life span, adult (6–8 week-old) rats weigh 250 to 550 g, while adult mice are generally 10 times smaller (25–30 g) [22]. This aspect becomes particularly valuable in the setting of experimental surgery. Animal intubation and ventilation, access to anatomical structures of interest, and performing specific procedures are easier in larger rodents, shortening learning times for operators and increasing animal survival. Moreover, invasive micromanometer catheterization can be accomplished more safely in rats, and the spectrum of applicable noninvasive echocardiographic and magnetic resonance imaging techniques mimics the one available in the clinical setting [23,24]. Finally, a greater number of different postmortem analyses can be accomplished on the same rat, thanks to the larger size of tissues and higher intravascular blood volume.
Rodent cardiac physiology, contraction patterns, and energetics resemble the human equivalents. Rodent and human myocardial tissue share similar functions for many proteins, although rodent cardiomyocytes exhibit a predominance of alpha-myosin heavy chains (compared to beta-myosin heavy chains in humans), which are characterized by a rapid ATPase activity to facilitate the extremely high heart rate and the short cardiac cycle. Mice present a resting heart rate of 500 to 600 bpm, with 350 bpm for rats. Although still far from human values, the de-escalation from mice to rats in terms of cellular bioenergetics and mitochondrial efficiency contributes to translational value [25]. These differences should be taken into account particularly in the setting of preventative and reparative molecular therapies for HF since they could influence the likelihood of moving findings into human clinical practice [21]. An intermediate validation in large animal models is mandatory for this purpose (i.e., porcine).
The mechanisms of HF are often multifactorial, involving several risk factors or acquired conditions that induce or accelerate myocardial damage, with or without a predisposing genetic background. Small animal models are particularly useful in the study of specific risk factors that promote HF, avoiding the confounding effects of external variables present in the clinical setting. Single pathophysiological pathways can be investigated: myocardial ischemia, pressure and volume overload, toxins, diabetes mellitus, atherosclerotic disease, and several metabolic syndromes [20,26]. In this setting, rat models are broadly adopted to test therapeutic hypotheses and target specific sensitive pathways [19]. Moreover, rat models were the first to be used for the study of the combination of different HF mechanisms in the same animal [27].
The high degree of genetic similarity between rodents and humans allowed for the creation of relevant transgenic and knockout strains with an HF phenotype [21]. Easier manipulation of the mouse genome explains the extremely large number of transgenic mouse strains currently available for target analysis. Mouse models obtained from mutations in cardiac myosin light chain, lamin, troponin, and other extra or intrasarcomeric proteins have been addressed in previous reviews [28,29,30,31]. Conversely, genetic rat models of HF are infrequent [21]. The translation of molecular mediators resulting from genetically modulated rodents into human studies requires a cautious approach. Genetic mutations that cause idiopathic DCM in humans are mostly undiscovered [32]. Although many proteins share similar functions, their expression levels and the final organ and biological effects can differ substantially between rodents and larger mammals and humans [19,21]. As an example, dystrophin-deficient mice exhibit a normal life span and mild cardiomyopathy, which is in contrast with the human phenotype of Duchenne muscular dystrophy [33]. Testing genetic hypotheses in large models is therefore recommended before human trials.
3. Rat Models: General Considerations
The ideal animal model should be able to reproduce the typical echocardiographic, histological, and clinical features of the desired type of HF. Reflecting the higher prevalence of left-sided HF in the general population, left ventricular (LV) dysfunction models leading to HF and eventually DCM are more widely utilized. Human DCM is defined as a spectrum of myocardial diseases which share ventricular dilatation and depressed contractility [34]. The key phenotype is characterized by a progressive LV dilatation, together with a ventricular shape transition from its original ellipsoid shape to a more spherical one, wall thinning, and a global reduction in contractility, which is revealed by a decrease in stroke volume, cardiac index, and increased strain parameters [32,35]. These features differentiate DCM from other cardiomyopathies, such as hypertrophic cardiomyopathy (where increased LV wall thickness and normal or even supranormal contractility is noted [36]), restrictive cardiomyopathy (in which ventricular chamber dimensions are reduced, impairing LV filling and creating a primary diastolic dysfunction [37]), and arrhythmogenic right ventricular cardiomyopathy (characterized by typical electrocardiographic anomalies and an often pathognomonic fibrous-fatty myocardial replacement [38]). The available animal models of hypertrophic, restrictive, and arrhythmogenic cardiomyopathies have been extensively reviewed in previous publications [39,40,41,42,43,44].
In DCM, associated diastolic dysfunction can occur, and the combination of increased LV filling pressures and ventricular dilatation often generates functional mitral regurgitation. Magnetic resonance imaging can detect areas of late gadolinium enhancement, which represent the process of diffuse fibrosis that is common to many DCM etiologies (the slower heart rate of the rat also makes cardiac MRI more feasible, although specialized instrumentation is still needed). Endomyocardial biopsy typically reveals morphological alterations in DCM: myocardial disarray, fibrosis, cell death, cardiomyocyte hypertrophy, scar formation, and inflammatory infiltration [45]. Importantly, this phenotype could characterize only the LV or present with biventricular involvement, depending on the etiology and severity of the disease. This aspect requires particular attention when choosing the most adequate animal model for the desired experimentation.
To date, a benchmark rat model of DCM is not available. Existing animal models are not able to respond to all of the above-mentioned requisites of the ideal model. The main reason lies in the fundamental difference in the development of human DCM versus the induction of experimental DCM. As previously stated, DCM in humans has a wide spectrum of causes, but in most of the cases a leading factor cannot be detected, often with evolution over significant time periods. Experimental DCM models rely mostly on a single pathogenic pathway with relatively rapid onset, which can consequently reproduce only some specific features of the DCM phenotype.
Recently, scientific interest has moved towards other variants and causes of HF, generating the need for new, representative animal models. Heart failure with preserved ejection has been recognized as an independent clinical entity with specific etiologies and pathogenic pathways involved [46]. Right ventricular (RV) failure is a considerable issue in patients who have survived surgical correction of congenital heart defects during infancy, as well as those children and adolescents palliated with a Fontan circulation utilizing a systemic RV [47]. In this view, novel rodent models of HF not progressing to DCM or specifically involving the RV have been developed. To summarize, there are seven main detrimental stimuli which can induce HF in preclinical models: ischemic injury; pressure overload; volume overload; drug toxicity; autoimmunity; rapid pacing; and genetic mutations (Figure 1 and Table 1). All these models will be discussed in the following sections.

Table 1.
Rat Models of HF.
4. Ischemic Injury Models
Ischemic injury in rats has been induced in different ways: subcutaneous injections of isoproterenol [49], direct damage using electrocautery [50], arterial ligation, or cryogenic damage with a metal probe cooled in liquid nitrogen [51]. In 1979, Pfeffer et al. introduced the left anterior descending (LAD) artery ligation model [55], which now has become the preferred ischemic model. A direct correlation exists between the infarct size and the severity of LV dilatation and contractile function impairment, which ranges from completely preserved if the scar involves <30% of the LV circumference to congestive HF if >46% [55]. However, the predictability of the infarcted myocardial area after LAD ligation is less consistent and varies depending on the level where the suture is placed and anatomical variation [56]. Coronary anatomy in rats presents significant variability between animals: the single septal branch may originate from the proximal left coronary artery in 60% of cases and from the proximal right coronary in the remaining 40%; the circumflex artery branches distally from a long left main coronary artery in 66% of animals, while it arises more proximally from a short left main coronary artery or the main septal branch in 34% [56]. As a result, a distal LAD ligation always creates an infarction only in the LV anterior wall, while a proximal ligation (just below the left atrial appendage) can create a wider anterolateral infarction (64% of cases) or an only anterior infarction (36%). Standardized surgical protocols are now widely available to guide the investigator [53,54], helping to reduce model animal-to-animal variability.
An alternative ischemic model consists of ligation of the circumflex artery, which is demonstrated to produce a significant infarct zone (40% of LV diameter) [52]. However, this procedure is less validated and, being technically more demanding, should be adopted as a secondary option to the LAD ligation model.
A general concern regarding the ischemic rat model of HF is that the LAD ligation procedure is usually performed in young (4–8-week-old) rats, while ischemic diseases affect an older and multidiseased population. Since the regenerative potential of the human and mammalian heart is age dependent [15,134], and the causes of idiopathic DCM rely only marginally on an ischemic substrate, the transition from the LAD ligation model to clinical practice necessitates careful verifications in this specific context. On the other hand, the leading cause of congestive HF in humans remains coronary arterial disease, which the LAD ligation model clearly exemplifies. Thanks to the advancement of emergency care, most of the patients that present with an acute myocardial infarction with ST-elevation can now benefit from a prompt revascularization strategy [135]. Despite successful revascularization, up to 20% of patients surviving an ST-elevation myocardial infarction are hospitalized with HF in the first year after the event [136]. After revascularization, the underlying pathophysiological basis of myocardial damage switches from irreversible ischemia and cell necrosis to transient ischemia and ischemia-reperfusion injury. To account for these mechanisms, the LAD ligation procedure has evolved from a permanent ligation to a temporary (typically 30 min) ligation, followed by controlled reperfusion [57]. This approach allowed for the investigation of molecular pathways involved in the protective role of the ischemic preconditioning process [58]. Moreover, Petters et al. proposed an innovative model of transient LAD ligation followed by temporary (60 min) cardiopulmonary bypass support [59]. This model ingeniously mimics advanced extracorporeal membrane oxygenation or ventricular assist device protection during high-risk percutaneous or surgical coronary interventions and cardiogenic shock.
With the LAD or circumflex ligation models, it is clear that the subsequent impairment of myocardial performance will be limited to the LV. Although most of the reports using isoproterenol-induced HF describe changes in the LV, parallel hypertrophy in the RV has been observed, supporting a biventricular effect of this drug [49]. Electrocautery or cryogenic-based direct damage to the myocardium can theoretically be applied on every surface of the heart, potentially inducing isolated RV, isolated LV, or biventricular failure. While these methods generate predictable and controllable damage, these models are least similar to human physiology and are not preferred.
Despite the mentioned limitations of the ischemic rodent model, it is indisputable that this model represents a primary reference platform for the study of ischemic HF. As an example of translational ability, ischemic injury models have permitted the study of the reverse remodeling properties of angiotensin-converting enzyme inhibitors and angiotensin II receptor antagonists on LV volumes and performance [19], which now constitute the mainstays of medical therapy for HF [46]. Table 1 summarizes the advantages and drawbacks of each ischemic model of DCM.
5. Pressure Overload Models
Pressure overload can be generated in rats using surgical or non-surgical methods (Table 1). Surgical techniques usually entail an abrupt augmentation of the ventricular afterload by placing a tight band around the aorta or the pulmonary artery (PA).
Aortic banding can be achieved at different levels. In the original procedure described by Rockman et al. in a mouse model [60], a suture is placed around the transverse aortic arch and tightened against a 27 G needle. The same technique has been implemented in rats (using an 18–22 G needle) and modified by applying a more proximal banding of the ascending aorta [62,63], which mimics the pathophysiological features of LV failure induced by severe aortic stenosis. Finally, the abdominal aorta can be banded to investigate the mechanisms of ventricular remodeling by creating a slower-developing hypertensive HF profile [68].
Tightening of the aorta or PA can be accomplished using a suture tied against a needle, with the needle rapidly removed to restore antegrade blood flow. Given the need for complete vessel mobilization and the related risk of fatal bleedings, alternative techniques have been proposed, such as half-closed surgical clips [69], which generate even higher pressure gradients across the PA with minimal mobilization of the vessel, or O-rings of predetermined diameters, which guarantee reproducible grades of aortic banding in mice [61].
Using these surgical approaches, delineated RV failure can be achieved 7 weeks from PA banding, as demonstrated by a reduced ejection fraction and cardiac output and severely dilated right chambers [70], with temporal sex-related differences in developing the HF phenotype (observed less contractile and diastolic dysfunction and fibrosis in females) [71]. The grade of PA banding contributes directly to the timing of the transition to a decompensated RV response, which can start manifesting after 1–3 weeks from the procedure in case of severe PA constriction [70,72]. On the other hand, aortic banding in rats produces an initial compensated hypertrophic response, with preserved ejection fraction and LV diameters 4 weeks after banding [64], which can last until 18–20 weeks before LV systo-diastolic failure occurs [62,65,66]. Interestingly, this adaptation to pressure overload is generally absent in mice, which develop very early LV failure and DCM [64]. Understanding the timing of progression from hypertrophy to cardiac decompensation in rat models is mandatory to guide the interventional and analytical planning of experiments.
Although technically challenging and with not insignificant mortality rates [66], rats can undergo aortic de-banding to investigate the antiremodeling effects of LV unloading which are accomplished by aortic valve replacement or ventricular assist devices in the clinical setting. After 6 to 9 weeks of aortic banding, LV unloading (i.e., de-banding) promotes the regression of hypertrophy and the recovery of diastolic function [73], supported by the restoration of mitochondrial energetics [74] and a reduction of pro-fibrotic factors [67]. Interestingly, aortic banding triggers significant systo-diastolic impairment and activation of prohypertrophic and profibrotic pathways also in the RV, which persist partially altered even after de-banding [63].
Surgical hypertensive models have been utilized to test reverse remodeling pharmacological strategies to treat HF, such as angiotensin-converting enzyme inhibitors and angiotensin II receptor antagonists [12,68,137] and, more recently, to guide targeted anti-fibrotic therapeutic molecules [67]. Interesting insights into the most appropriate timing to plan aortic valve replacement in the setting of asymptomatic aortic stenosis may be derived from these models in the future.
Non-surgical hypertensive models of LV failure include angiotensin II infusion by osmotic minipumps implanted subcutaneously [75], Dahl salt-sensitive rats fed with a high-salt diet [76], and spontaneously hypersensitive rats [77]. These models produce a reliable phenotype of slow-developing cardiac hypertrophy and diastolic dysfunction, which perfectly parallels the pathophysiological progression of HF with preserved ejection fraction in humans. However, the progression to decompensated HF can take up to 25 weeks, where a sudden drop in Dahl sensitive rats survival is noticed [76], or even 12 months for the spontaneously hypersensitive rats [78], increasing maintenance and housing costs when adopting these models.
Several methods have been described to induce RV failure secondary to severe pulmonary hypertension (in primis monocrotaline infusion and sugen/hypoxia models), which have been extensively reviewed by Andersen et al. [79]. Interestingly, angio-proliferative pulmonary hypertension generates more profound fibrotic changes, capillary rarefaction, and contractile impairment in the RV than isolated pressure overload (i.e., from PA banding) [80]. The proposed failure in antioxidant defenses may account for the early development of RV failure 6 weeks after monocrotaline infusion, which differs substantially from the compensated hypertrophic response in the PA banding model, which can be sustained up to 22 weeks [80]. Monocrotaline is proven to induce diffuse sclerosis in other organs, leading to hepatic sinusoidal obstruction [81], renal fibrosis [82], and cachexia [83], which must be taken into account when considering the most appropriate model for HF investigation. The intense systemic oxidative stress and cytokine dysregulation do not represent the usual pathogenic pathways involved in human HF (except for specific myocarditis-based conditions).
Injection of the vascular endothelial growth factor receptor (VEGFR) antagonist SU5416 (known as sugen), followed by a 2–3 week exposure to hypoxia, determines a paradoxical angio-obliterative pulmonary hypertension in rats, due to lung endothelial cell apoptosis [84]. Interbreed differences exist in the RV response to the sugen/hypoxia pulmonary hypertension model. Fisher rats display an early maladaptive RV remodeling leading to exitus by 5 weeks, while Sprague Dawley rats can show preserved RV performance and survival even beyond 9 weeks [86]. Proteomics analysis revealed that a significant downregulation of adenylate kinase 1 (ADK1), which translates into inefficient cardiac energetics, might be the molecular substrate for the premature deterioration of RV function in Fisher rats [86]. In Sprague Dawley rats, RV hypertrophy and increased filling pressures are present after 5 weeks, with a subsequent RV enlargement by 8 weeks, but still preserved RV stroke volume and ejection fraction [85]. Rodent hypoxia-based pulmonary hypertension models have been exhaustively addressed by several reviews [79,84,138].
6. Volume Overload Models
Both extracardiac and intracardiac surgical methods have been described to induce volume overload in rats leading to HF (Table 1). The extracardiac approach is the most frequently adopted and entails the creation of an aorto-caval fistula by puncturing the aorta with an 18G needle, which is advanced until perforating the adjacent inferior vena cava. The result is an arterial-venous shunt which creates a high cardiac output HF with volume overload and increased filling pressures into the right chambers. Compensated biventricular hypertrophy is noticed 8 weeks after the procedure, which progresses towards ventricular dilatation in the following weeks [88]. At 12 weeks, pro-arrhythmogenic electrophysiological changes are noticed in the right atrium [89], as well as an increase in LV myocardial stiffness, which is a reliable predictor of animal mortality [90]. After 24 weeks, biventricular failure is established, with reduced contractile function and severe dilatation of both ventricles, although more profoundly in the RV, as reflected by a predominant upregulation of stress and metabolic markers in the RV [87]. Concomitantly, an escalation in animal mortality is evident, whose median survival after the aorto-caval fistula is 43 weeks (in Wistar rats) [87]. The main drawbacks of this technique are the resulting high cardiac output profile, which is a rare cause of HF in the clinical setting, and the artificial mixing of oxygenated and venous blood, which is seen only in HF secondary to congenital heart defects with left-to-right shunt.
Closure of the aorto-caval fistula can be accomplished through a re-laparotomy using a hemoclip [91]. Adopting this temporary volume overloaded model, the reversibility of echocardiographic, histological, and metabolomic changes has been investigated, revealing that, once LV contractile function is compromised (16 weeks after the fistula creation), the correction of the volume overload does not revert LV systolic impairment or repair the defective glycolysis and fatty acid oxidation metabolic pathways [91]. Important considerations from this model might be translated into the clinical decision making for the treatment of aortic and mitral valve regurgitation.
Intracardiac surgical methods include damaging the mitral, pulmonary, or aortic leaflets to generate acute valvular insufficiency and volume overload. The mitral valve is accessed through the LV apex, where a purse-string suture is placed and a 23G needle is advanced under echocardiographic guidance, puncturing the anterior mitral leaflet. After 2 weeks, a regurgitant fraction of 40% is reached, which determinates a progressive enlargement of the LV chamber (end-diastolic volume +28% at 2 weeks, +65% at 10 weeks, +87% at 20 weeks, and +100% at 40 weeks) [92]. A drop in ejection fraction is noticed at 14 weeks with myocyte and extracellular matrix remodeling and a transcriptomic shift illustrating upregulation of oxidative stress pathways [92]. Cardiomyocyte elongation, cytoskeletal disorganization, and loss of mitochondrial networking represent the ultrastructural bases of this remodeling [93]. The low-pressure volume overloaded mitral regurgitation model can be combined with the LAD ligation-based ischemic model to better reproduce the chronic ischemic DCM profile [94]. The additional volume overload accelerates LV dilatation, confirming that mitral regurgitation is a potent driver of adverse cardiac remodeling after a myocardial infarction [95].
Aortic regurgitation is obtained by puncturing the valve with a fixed-core guidewire inserted from the surgically exposed right carotid artery [96]. The temporal sequence in LV remodeling parallels that seen in the mitral regurgitation model, with initial eccentric hypertrophy and dilatation of left-sided chambers (at 8 weeks), followed by a reduction in fractional shortening (at 12 weeks) [97]. Different from the PA banding model, female rats experience a faster progression to LV spherical dilation and wall thickening with reduced circumferential strain, when compared to males [98].
Only one rat model of pulmonary regurgitation is currently available in the scientific literature. By lacerating the pulmonary leaflets with a 22.5 G needle inserted into the main PA through a purse-string suture, pulmonary regurgitation was achieved by Akazawa et al. [99]. In this model, RV dilatation manifested after only 2 weeks, while progressive RV systolic dysfunction occurred after 4 weeks. Due to negative ventricular–ventricular interactions, RV dilatation contributes to diastolic LV compression and impaired relaxation. Interestingly, cardiomyocyte hypertrophy and myocardial fibrosis affected the RV exclusively [99], underlying differential biological pathways supporting ventricular–ventricular interactions in response to RV volume and pressure overload.
Finally, multivalvular regurgitation involving all four cardiac valves has been produced by long-term pergolide and serotonin administration in rats [100], simulating carcinoid heart disease in advanced neuroendocrine tumors.
7. Drug Toxicity Models
Drug administration can induce HF in experimental rat models by direct toxicity of myocardial tissue (doxorubicin, ethanol, and homocysteine), producing myocardial ischemia (isoproterenol), type 1 diabetes mellitus (streptozotocin [139]), or pulmonary hypertension (monocrotaline [79]).
Repeated intraperitoneal doxorubicin injections represent one of the most validated rodent models of HF progressing to DCM [140]. By generating mitochondrial dysfunction and intense oxidative stress [101], doxorubicin promotes swelling and vacuolization of cardiomyocytes, disorganization of myofibrils, and intense interstitial fibrosis [102]. Different administration protocols have been described, although a long-term scheme with intraperitoneal weekly injections for a total of 9 weeks (cumulative dose of 18mg/kg) has been found to be more effective to create LV dilatation and systolic impairment [102]. Dose-dependent cardiotoxicity is present, together with a regionalized effect on myocardial contraction, whose earliest impairment is noticed in the basal LV segments by speckle tracking imaging [103].
While experimental papers focus mainly on doxorubicin’s effects on the LV, clinical experience in cancer survivors demonstrated that anthracycline treatment has a detrimental effect also on right-sided chambers [141]. Recent findings suggest increased free radical production in the RV and the conduction system of doxorubicin-treated rats [104], underlining its biventricular toxicity. Given its molecular mechanisms, extracardiac side-effects of doxorubicin are numerous, including hepatic toxicity [105], nephrotic syndrome [106], and bone marrow suppression [107].
Hyperhomocysteinemia in rats is achieved by specific amino acid-defined control diets. After 6 to 10 weeks of treatment, histopathological analysis reveals biventricular hypertrophy of cardiomyocytes, myocardial fiber disarrangement, perivascular and interstitial fibrosis, and inflammatory infiltrate [108,109]. In vivo findings range from LV hypertrophy with preserved systolic function [108] to LV dilatation and loss of contractile function [109].
Acute intravenous administration of ethanol provokes myocardial dysfunction and hypotension, sustained by an abrupt increase in oxidative stress [142]. Chronic moderate ethanol ingestion (8 months of treatment) results in myocyte loss, LV wall thinning, dilatation, and depressed contractile performance [111], while the RV seems to be spared from ethanol toxicity [110].
Several rodent models of diabetes mellitus are available, recently reviewed by Pandey et al. [143]. These models have contributed to the discovery and validation of many antidiabetic molecules and the investigation of diabetes-related multi-organ complications. The intraperitoneal injection of streptozotocin causes a reproducible chemical ablation of pancreatic beta cells, inducing irreversible diabetes in rats. Early signs of diabetic cardiomyopathy can be documented 2–3 weeks after the injection, such as prolonged contraction and relaxation times in isolated cardiomyocytes and enlarged left atrium and impaired systolic parameters with echocardiography [112,113]. Interestingly, these functional alterations appear before the occurrence of morphological changes in the structure of the myocardium and microvasculature [112]. Subsequently, the activation of pro-apoptotic pathways determines the depletion of contractile myocardium which is replaced by fibrotic tissue [114], resulting in LV systo-diastolic dysfunction [116]. Moreover, chronic hyperglycemia triggers secondary pulmonary hypertension which contributes to RV hypertrophy and late systolic dysfunction (12 weeks from disease induction) [115].
8. Autoimmune-Mediated Models
Activation of the inflammatory cascade in response to cardiac injury is a common mechanism of ventricular remodeling in many models of HF. After an insult, damaged cardiomyocytes release molecular signals that trigger an acute inflammatory response, digesting necrotic cell debris and promoting subsequent healing pathways [144]. Monocytes and tissue-resident macrophages then acquire a resolution (anti-inflammatory) profile and catalyze the differentiation of myofibroblasts from quiescent fibroblasts [145,146,147]. Myofibroblasts produce the extracellular matrix components that form the basis of scar formation, although if dysregulated, myofibroblasts may also promote a negative fibrotic remodeling process [148]. The inflammatory system initiates the detrimental stimulus which sustains myocardial damage in the setting of acute myocarditis or chronic inflammatory cardiomyopathy [149].
Acute autoimmune myocarditis has been reproduced in rats by injecting porcine myocardial myosin and generating a cross-reactivity with native cardiomyocytes. Using a two-stage protocol of subcutaneous footpad injection of purified porcine cardiac myosin supplemented with complete Freund adjuvant, cardiomyocyte injury, inflammatory infiltrate, and replacement fibrosis are achieved even 18 days after the completion of the injection protocol [117]. Extensive myocardial damage is sustained by several modalities of cell death (i.e., necroptosis, apoptosis, and autophagy) [118]. After 3 weeks, early signs of LV contractile impairment can be detected using cardiac magnetic resonance tissue tracking [119]. Four weeks after the completion of the protocol, LV dilatation and a drop in ejection fraction are evident with standard echocardiography [117]. Interestingly, the RV seems to be almost spared from foci of late gadolinium enhancement with magnetic resonance imaging, suggesting preferential effect on the LV from the acute myocarditis (inflammatory) process [120].
9. Rapid Ventricular Pacing Models
Due to practical reasons, rapid pacing-induced HF models usually involve larger mammals, in whom a pacemaker with a pacing lead in the RV apex is implanted subcutaneously and used to maintain supranormal heart rates for several weeks [150]. Rapid pacing has also been adopted in rats to investigate the intracellular effects of chronic tachycardia. Zhou et al. surgically implanted an electrode on the epicardial surface of the RV apex of rats and used it for rapid pacing (550 bpm) for 4 weeks with an external pacemaker [122]. Tachycardia and rapid pacing promoted rat myocyte apoptotic pathways, increased the intracellular levels of reactive oxygen species [121], and dysregulated calcium signaling, especially at higher pacing rates [123]. These cellular mechanisms translated into the development of an apoptotic-based HF profile, with reduced LV contractile function and increased LV end-diastolic pressure after 4 weeks of rapid pacing [122]. Given the need for extracorporeal devices, rapid pacing rat models might be more suitable for the in vitro study of single myocardial fiber response to chronic electric stimulation than whole animal in vivo experiments.
10. Genetic Models
Although murine models of HF induced by specific genetic mutations are widely available, few genetically based rat models have been reported. Greaser et al. identified a rat strain with an autosomal dominant mutation of the gene encoding the RNA binding motif protein 20 (Rbm20), which alters the isoform expression of the sarcomeric protein titin (TTN) [124]. Titin is a cardiac and skeletal muscle protein involved in sarcomere assembly and protection from overstretching. Both heterozygous and homozygous Rbm20-deficient rats exhibit a phenotype of DCM with dilated LV, increased subendocardial fibrosis, but initially preserved contractile function [125]. Starting from 3 months of age, progressive LV wall thinning and a decrease in fractional shortening and cardiac output are observed [126]. As seen in humans with hereditary DCM caused by Rbm20 mutations, fibrosis is accompanied by electrical abnormalities that predispose to arrhythmias and sudden cardiac death, which start at 10 months of age in rats [125]. Interestingly, the correction of the cardiac transcriptional profile via base editing can restore cardiac function and revert the HF process in Rbm20 mutant mice [18].
Given its central role in controlling the mechanical properties of the sarcomere, truncating variants of TTN lead to a wide number of inherited myopathies and cardiac disorders [151], ranging from hypertrophic to dilated ventricular profiles. Several mouse models carrying variants in the TTN gene have been developed to understand the structural role of TTN and the clinical correlates in patients. All of the available animal models have been excellently summarized by Marcello and colleagues in a very recent review exclusively focused on TTN pathophysiology [152].
The clinical phenotype of Duchenne muscular dystrophy includes the development of progressive DCM, the leading cause of death in this population [153]. Using CRISPR/Cas9 genome editing of the dystrophin gene, Sugihara and colleagues recently described the first rat model of Duchenne dystrophy with heart involvement [127]. At 10 months of age, mutant rats developed biventricular systolic impairment and myocardial fibrosis. As in the clinical setting, animals also displayed systemic muscular atrophy, with a non-negligible mortality rate. This model provided a suitable platform for the investigation of the potential therapeutic role of a ketogenic diet with medium-chain triglycerides (although the diet was documented to ameliorate skeletal muscle function [154], it triggered a paradoxical exacerbation of the cardiac disorder [128]).
Ling et al. developed a promising heterozygote knockout rat for the myocardium-specific Isca1, a causal gene for multiple mitochondrial dysfunction syndromes with cardiac dysplasia [129]. From 3 months of age, rats manifest an echocardiographic and histopathological phenotype of DCM, characterized by LV wall thinning and dilatation, contractile impairment, myocardial lysis and fibrosis. The subcellular mechanisms of mitochondrial dysfunction syndromes were clearly represented in this model, with swollen mitochondria containing damaged membrane structure and partial absence of crests, together with reduced expression levels of key enzymes for ATP synthesis and iron homeostasis [129,130].
Recently, a novel rat model that relies on chemo–genetic interactions was established by Steinhorn et al., injecting rats with adeno-associated virus type 9 carrying a cardiac-specific recombinant D-amino acid oxidase, which produces hydrogen peroxidase during the conversion of D-amino acids into alpha-keto acids [131]. When rats are fed with D-alanine (the substrate of the enzyme D-amino acid oxidase), the acute generation of intracellular hydrogen peroxidase induces a state of oxidative stress in cardiomyocytes. After 4 weeks of the specific diet, rats present HF with reduced LV contractile function and an enlarged LV. Interestingly, LV thickness is maintained and histopathological analysis confirms the absence of fibrosis (at 4 weeks, fibrosis develops later at 8 weeks) [131,133]. Cardiac transcriptome and metabolome analysis revealed marked alterations in mitochondrial function, cardiac energetics, redox homeostasis, amino acid metabolism, cytoskeletal and extracellular matrix organization, and antioxidant systems pathways [132]. This HF model was noticeably reverted by the administration of an angiotensin II receptor blocker, which normalized the echocardiographic, morphologic, and metabolomic markers of disease [132]. Oxidative stress is a common finding in different etiologies of HF, and this model may facilitate the testing of new molecular targets potentially involved in the process of ventricular remodeling.
Finally, several other genetic-based murine models of hereditary cardiomyopathy have been developed. Homozygous desmin knockout and knockin mice display the typical cardiac involvement (LV thinning, fibrosis, and systolic dysfunction) of autosomal-recessive desminopathies [155]. Progressive DCM sustained by cardiomyocyte nuclear envelope disarrangement (envelopathy) can be produced by a knockin mutation of the LEDM2 gene in mice [156]. Manipulating the expression of desmosomal genes (especially desmocollin-2 and desmoglein-2) causes the development of severe biventricular cardiomyopathy in mice, triggered by an acute inflammatory process leading to cardiomyocyte necrosis and fibrosis [157]. Previous reviews have specifically addressed these topics [31,43,158].
11. Conclusions
Adopting the most representative animal model provides the basis for reliable experimental research that translates to clinical practice. Rats provide a strategic compromise between sufficient body size for several morphological evaluations/ease of interventions and low maintenance costs/short breeding cycles. Understanding the advantages and possible drawbacks of each specific rat model can expedite experiments, improve reproducibility, and strengthen reliability. In the present review, we summarize the main characteristics of the currently available rat models of HF and describe their timelines for the development of ventricular failure and phenotypic features to facilitate the standardization and optimization of future investigations in the field of HF.
Author Contributions
Conceptualization, investigation, data curation, writing—original draft preparation, M.P.; writing—review and editing, supervision, J.G.C. and J.T.M. All authors have read and agreed to the published version of the manuscript.
Funding
SickKids Foundation through the Curtis Joseph and Harold Groves Chair in Anesthesia and Pain Medicine (JTM) and the Department of Anesthesiology and Pain Medicine, University of Toronto through a Merit Award (JTM). Labatt Family Heart Centre, The Hospital for Sick Children, Toronto (MP and JGC).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Truby, L.K.; Rogers, J.G. Advanced Heart Failure: Epidemiology, Diagnosis, and Therapeutic Approaches. JACC. Heart Fail. 2020, 8, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Albert, N.M.; Allen, L.A.; Bluemke, D.A.; Butler, J.; Fonarow, G.C.; Ikonomidis, J.S.; Khavjou, O.; Konstam, M.A.; Maddox, T.M.; et al. Forecasting the Impact of Heart Failure in the United States: A Policy Statement from the American Heart Association. Circ. Heart Fail. 2013, 6, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Schilling, C.; Dalziel, K.; Nunn, R.; Du Plessis, K.; Shi, W.Y.; Celermajer, D.; Winlaw, D.; Weintraub, R.G.; Grigg, L.E.; Radford, D.J.; et al. The Fontan Epidemic: Population Projections from the Australia and New Zealand Fontan Registry. Int. J. Cardiol. 2016, 219, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Moons, P.; Bovijn, L.; Budts, W.; Belmans, A.; Gewillig, M. Temporal Trends in Survival to Adulthood among Patients Born with Congenital Heart Disease from 1970 to 1992 in Belgium. Circulation 2010, 122, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Rossano, J.W.; Cherikh, W.S.; Chambers, D.C.; Goldfarb, S.; Hayes, D.J.; Khush, K.K.; Kucheryavaya, A.Y.; Toll, A.E.; Levvey, B.J.; Meiser, B.; et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Twenty-First Pediatric Heart Transplantation Report-2018; Focus Theme: Multiorgan Transplantation. J. Hear. Lung Transplant. Off. Publ. Int. Soc. Hear. Transplant. 2018, 37, 1184–1195. [Google Scholar] [CrossRef]
- Zangwill, S. Five Decades of Pediatric Heart Transplantation: Challenges Overcome, Challenges Remaining. Curr. Opin. Cardiol. 2017, 32, 69–77. [Google Scholar] [CrossRef]
- Kirk, R.; Dipchand, A.I.; Davies, R.R.; Miera, O.; Chapman, G.; Conway, J.; Denfield, S.; Gossett, J.G.; Johnson, J.; McCulloch, M.; et al. ISHLT Consensus Statement on Donor Organ Acceptability and Management in Pediatric Heart Transplantation. J. Hear. lung Transplant. Off. Publ. Int. Soc. Hear. Transplant. 2020, 39, 331–341. [Google Scholar] [CrossRef]
- Schlagenhauf, A.; Kalbhenn, J.; Geisen, U.; Beyersdorf, F.; Zieger, B. Acquired von Willebrand Syndrome and Platelet Function Defects during Extracorporeal Life Support (Mechanical Circulatory Support). Hamostaseologie 2020, 40, 221–225. [Google Scholar] [CrossRef]
- Lukito, P.; Wong, A.; Jing, J.; Arthur, J.F.; Marasco, S.F.; Murphy, D.A.; Bergin, P.J.; Shaw, J.A.; Collecutt, M.; Andrews, R.K.; et al. Mechanical Circulatory Support Is Associated with Loss of Platelet Receptors Glycoprotein Ibα and Glycoprotein VI. J. Thromb. Haemost. 2016, 14, 2253–2260. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Ohtsuki, S.; Eitoku, T.; Ousaka, D.; Kondo, M.; Kurita, Y.; Hirai, K.; Fukushima, Y.; Baba, K.; Goto, T.; et al. Intracoronary Cardiac Progenitor Cells in Single Ventricle Physiology: The PERSEUS (Cardiac Progenitor Cell Infusion to Treat Univentricular Heart Disease) Randomized Phase 2 Trial. Circ. Res. 2017, 120, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Makkar, R.R.; Kereiakes, D.J.; Aguirre, F.; Kowalchuk, G.; Chakravarty, T.; Malliaras, K.; Francis, G.S.; Povsic, T.J.; Schatz, R.; Traverse, J.H.; et al. Intracoronary ALLogeneic Heart STem Cells to Achieve Myocardial Regeneration (ALLSTAR): A Randomized, Placebo-Controlled, Double-Blinded Trial. Eur. Heart J. 2020, 41, 3451–3458. [Google Scholar] [CrossRef]
- Zhu, J.; Ide, H.; Fu, Y.Y.; Teichert, A.-M.; Kato, H.; Weisel, R.D.; Maynes, J.T.; Coles, J.G.; Caldarone, C.A. Losartan Ameliorates “Upstream” Pulmonary Vein Vasculopathy in a Piglet Model of Pulmonary Vein Stenosis. J. Thorac. Cardiovasc. Surg. 2014, 148, 2550–2557. [Google Scholar] [CrossRef]
- Ponzoni, M.; Frigo, A.C.; Castaldi, B.; Cerutti, A.; Di Salvo, G.; Vida, V.L.; Padalino, M.A. Surgical Strategies for the Management of End-Stage Heart Failure in Infants and Children: A 15-Year Experience with a Patient-Tailored Approach. Artif. Organs 2021, 45, 1543–1553. [Google Scholar] [CrossRef]
- Ponzoni, M.; Castaldi, B.; Padalino, M.A. Pulmonary Artery Banding for Dilated Cardiomyopathy in Children: Returning to the Bench from Bedside. Children 2022, 9, 1392. [Google Scholar] [CrossRef] [PubMed]
- Traister, A.; Patel, R.; Huang, A.; Patel, S.; Plakhotnik, J.; Lee, J.E.; Medina, M.G.; Welsh, C.; Ruparel, P.; Zhang, L.; et al. Cardiac Regenerative Capacity Is Age- and Disease-Dependent in Childhood Heart Disease. PLoS ONE 2018, 13, e0200342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yeganeh, A.; Bartkevics, M.; Perri, A.; Brown, C.; Coles, J.; Maynes, J.T. Mesenchymal Stromal Cells Isolated From Patients With Congenital Heart Disease Reveal an Age-Dependent Proinflammatory Phenotype. JACC Adv. 2022, 1, 100050. [Google Scholar] [CrossRef]
- Wang, P.; Li, H.; Zhu, M.; Han, R.Y.; Guo, S.; Han, R. Correction of DMD in Human IPSC-Derived Cardiomyocytes by Base-Editing-Induced Exon Skipping. Mol. Ther.—Methods Clin. Dev. 2023, 28, 40–50. [Google Scholar] [CrossRef]
- Nishiyama, T.; Zhang, Y.; Cui, M.; Li, H.; Sanchez-Ortiz, E.; McAnally, J.R.; Tan, W.; Kim, J.; Chen, K.; Xu, L.; et al. Precise Genomic Editing of Pathogenic Mutations in RBM20 Rescues Dilated Cardiomyopathy. Sci. Transl. Med. 2022, 14. [Google Scholar] [CrossRef]
- Patten, R.D.; Hall-Porter, M.R. Small Animal Models of Heart Failure: Development of Novel Therapies, Past and Present. Circ. Heart Fail. 2009, 2, 138–144. [Google Scholar] [CrossRef]
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.D.; Sussman, M.A.; et al. Animal Models of Heart Failure: A Scientific Statement from the American Heart Association. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef]
- Riehle, C.; Bauersachs, J. Small Animal Models of Heart Failure. Cardiovasc. Res. 2019, 115, 1838–1849. [Google Scholar] [CrossRef]
- Holtze, S.; Gorshkova, E.; Braude, S.; Cellerino, A.; Dammann, P.; Hildebrandt, T.B.; Hoeflich, A.; Hoffmann, S.; Koch, P.; Terzibasi Tozzini, E.; et al. Alternative Animal Models of Aging Research. Front. Mol. Biosci. 2021, 8, 660959. [Google Scholar] [CrossRef]
- Makowski, M.R.; Wiethoff, A.J.; Jansen, C.H.P.; Botnar, R.M. Cardiovascular MRI in Small Animals. Expert Rev. Cardiovasc. Ther. 2010, 8, 35–47. [Google Scholar] [CrossRef]
- Fu, X.; Segiser, A.; Carrel, T.P.; Tevaearai Stahel, H.T.; Most, H. Rat Heterotopic Heart Transplantation Model to Investigate Unloading-Induced Myocardial Remodeling. Front. Cardiovasc. Med. 2016, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Dobson, G.P. On Being the Right Size: Heart Design, Mitochondrial Efficiency and Lifespan Potential. Clin. Exp. Pharmacol. Physiol. 2003, 30, 590–597. [Google Scholar] [CrossRef]
- Zaragoza, C.; Gomez-Guerrero, C.; Martin-Ventura, J.L.; Blanco-Colio, L.; Lavin, B.; Mallavia, B.; Tarin, C.; Mas, S.; Ortiz, A.; Egido, J. Animal Models of Cardiovascular Diseases. J. Biomed. Biotechnol. 2011, 2011, 497841. [Google Scholar] [CrossRef]
- Nolan, S.E.; Mannisi, J.A.; Bush, D.E.; Healy, B.; Weisman, H.F. Increased Afterload Aggravates Infarct Expansion after Acute Myocardial Infarction. J. Am. Coll. Cardiol. 1988, 12, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.J. Dilated Cardiomyopathy: Concepts Derived from Gene Deficient and Transgenic Animal Models. Circ. J. 2002, 66, 219–224. [Google Scholar] [CrossRef]
- Willott, R.H.; Gomes, A.V.; Chang, A.N.; Parvatiyar, M.S.; Pinto, J.R.; Potter, J.D. Mutations in Troponin That Cause HCM, DCM AND RCM: What Can We Learn about Thin Filament Function? J. Mol. Cell. Cardiol. 2010, 48, 882–892. [Google Scholar] [CrossRef]
- Lee, Y.-K.; Jiang, Y.; Ran, X.-R.; Lau, Y.-M.; Ng, K.-M.; Lai, W.-H.K.; Siu, C.-W.; Tse, H.-F. Recent Advances in Animal and Human Pluripotent Stem Cell Modeling of Cardiac Laminopathy. Stem Cell Res. Ther. 2016, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Sitbon, Y.H.; Kazmierczak, K.; Szczesna-Cordary, D. Hereditary Heart Disease: Pathophysiology, Clinical Presentation, and Animal Models of HCM, RCM, and DCM Associated with Mutations in Cardiac Myosin Light Chains. Pflug. Arch. 2019, 471, 683–699. [Google Scholar] [CrossRef]
- Schultheiss, H.-P.; Fairweather, D.; Caforio, A.L.P.; Escher, F.; Hershberger, R.E.; Lipshultz, S.E.; Liu, P.P.; Matsumori, A.; Mazzanti, A.; McMurray, J.; et al. Dilated Cardiomyopathy. Nat. Rev. Dis. Prim. 2019, 5, 32. [Google Scholar] [CrossRef]
- Milani-Nejad, N.; Janssen, P.M.L. Small and Large Animal Models in Cardiac Contraction Research: Advantages and Disadvantages. Pharmacol. Ther. 2014, 141, 235–249. [Google Scholar] [CrossRef]
- Bozkurt, B.; Colvin, M.; Cook, J.; Cooper, L.T.; Deswal, A.; Fonarow, G.C.; Francis, G.S.; Lenihan, D.; Lewis, E.F.; McNamara, D.M.; et al. Current Diagnostic and Treatment Strategies for Specific Dilated Cardiomyopathies: A Scientific Statement From the American Heart Association. Circulation 2016, 134. [Google Scholar] [CrossRef]
- Faggiano, A.; Avallone, C.; Gentile, D.; Provenzale, G.; Toriello, F.; Merlo, M.; Sinagra, G.; Carugo, S. Echocardiographic Advances in Dilated Cardiomyopathy. J. Clin. Med. 2021, 10, 5518. [Google Scholar] [CrossRef]
- 2014 ESC Guidelines on Diagnosis and Management of Hypertrophic Cardiomyopathy. Eur. Heart J. 2014, 35, 2733–2779. [CrossRef]
- Rapezzi, C.; Aimo, A.; Barison, A.; Emdin, M.; Porcari, A.; Linhart, A.; Keren, A.; Merlo, M.; Sinagra, G. Restrictive Cardiomyopathy: Definition and Diagnosis. Eur. Heart J. 2022, 43, 4679–4693. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS Expert Consensus Statement on Evaluation, Risk Stratification, and Management of Arrhythmogenic Cardiomyopathy. Hear. Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef] [PubMed]
- Wijnker, P.J.M.; van der Velden, J. Mutation-Specific Pathology and Treatment of Hypertrophic Cardiomyopathy in Patients, Mouse Models and Human Engineered Heart Tissue. Biochim. Biophys. Acta—Mol. Basis Dis. 2020, 1866, 165774. [Google Scholar] [CrossRef]
- Gannon, M.P.; Link, M.S. Phenotypic Variation and Targeted Therapy of Hypertrophic Cardiomyopathy Using Genetic Animal Models. Trends Cardiovasc. Med. 2021, 31, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, L.; Pacciulli, D.; Zhao, J.; Nan, C.; Shen, W.; Quan, J.; Tian, J.; Huang, X. Restrictive Cardiomyopathy Caused by Troponin Mutations: Application of Disease Animal Models in Translational Studies. Front. Physiol. 2016, 7. [Google Scholar] [CrossRef]
- Chintanaphol, M.; Orgil, B.-O.; Alberson, N.R.; Towbin, J.A.; Purevjav, E. Restrictive Cardiomyopathy: From Genetics and Clinical Overview to Animal Modeling. Rev. Cardiovasc. Med. 2022, 23, 0108. [Google Scholar] [CrossRef]
- Gerull, B.; Brodehl, A. Genetic Animal Models for Arrhythmogenic Cardiomyopathy. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Padrón-Barthe, L.; Domínguez, F.; Garcia-Pavia, P.; Lara-Pezzi, E. Animal Models of Arrhythmogenic Right Ventricular Cardiomyopathy: What Have We Learned and Where Do We Go? Insight for Therapeutics. Basic Res. Cardiol. 2017, 112, 50. [Google Scholar] [CrossRef] [PubMed]
- Pilati, M.; Rebonato, M.; Formigari, R.; Butera, G. Endomyocardial Biopsy in Pediatric Myocarditis and Dilated Cardiomyopathy: A Tool in Search for a Role. J. Cardiovasc. Dev. Dis. 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Ponzoni, M.; Azzolina, D.; Vedovelli, L.; Gregori, D.; Di Salvo, G.; D’Udekem, Y.; Vida, V.; Padalino, M.A. Ventricular Morphology of Single Ventricle Hearts Has a Significant Impact on Outcomes after Fontan Palliation. A Meta-Analysis. Eur. J. Cardio-Thorac. Surg. 2022. [Google Scholar] [CrossRef] [PubMed]
- Sugino, H.; Shimada, H. Effect of Isoproterenol on Renal Uric Acid Excretion in Rats. Jpn. J. Pharmacol. 1987, 45, 343–348. [Google Scholar] [CrossRef]
- RONA, G.; CHAPPEL, C.I.; BALAZS, T.; GAUDRY, R. An Infarct-like Myocardial Lesion and Other Toxic Manifestations Produced by Isoproterenol in the Rat. AMA. Arch. Pathol. 1959, 67, 443–455. [Google Scholar] [PubMed]
- Adler, N.; Camin, L.L.; Shulkin, P. Rat Model for Acute Myocardial Infarction: Application to Technetium-Labeled Glucoheptonate, Tetracycline, and Polyphosphate. J. Nucl. Med. 1976, 17, 203–207. [Google Scholar] [PubMed]
- Ryu, J.H.; Kim, I.-K.; Cho, S.-W.; Cho, M.-C.; Hwang, K.-K.; Piao, H.; Piao, S.; Lim, S.H.; Hong, Y.S.; Choi, C.Y.; et al. Implantation of Bone Marrow Mononuclear Cells Using Injectable Fibrin Matrix Enhances Neovascularization in Infarcted Myocardium. Biomaterials 2005, 26, 319–326. [Google Scholar] [CrossRef]
- Yang, S.M.; Liu, J.; Li, C.X. Intermedin Protects against Myocardial Ischemia-Reperfusion Injury in Hyperlipidemia Rats. Genet. Mol. Res. 2014, 13, 8309–8319. [Google Scholar] [CrossRef] [PubMed]
- Kolk, M.V.V.; Meyberg, D.; Deuse, T.; Tang-Quan, K.R.; Robbins, R.C.; Reichenspurner, H.; Schrepfer, S. LAD-Ligation: A Murine Model of Myocardial Infarction. J. Vis. Exp. 2009. [Google Scholar] [CrossRef]
- Reichert, K.; Colantuono, B.; McCormack, I.; Rodrigues, F.; Pavlov, V.; Abid, M.R. Murine Left Anterior Descending (LAD) Coronary Artery Ligation: An Improved and Simplified Model for Myocardial Infarction. J. Vis. Exp. 2017. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Pfeffer, J.M.; Fishbein, M.C.; Fletcher, P.J.; Spadaro, J.; Kloner, R.A.; Braunwald, E. Myocardial Infarct Size and Ventricular Function in Rats. Circ. Res. 1979, 44, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Kainuma, S.; Miyagawa, S.; Fukushima, S.; Tsuchimochi, H.; Sonobe, T.; Fujii, Y.; Pearson, J.T.; Saito, A.; Harada, A.; Toda, K.; et al. Influence of Coronary Architecture on the Variability in Myocardial Infarction Induced by Coronary Ligation in Rats. PLoS ONE 2017, 12, e0183323. [Google Scholar] [CrossRef]
- Michael, L.H.; Entman, M.L.; Hartley, C.J.; Youker, K.A.; Zhu, J.; Hall, S.R.; Hawkins, H.K.; Berens, K.; Ballantyne, C.M. Myocardial Ischemia and Reperfusion: A Murine Model. Am. J. Physiol. 1995, 269, H2147–H2154. [Google Scholar] [CrossRef]
- Szabó, P.L.; Dostal, C.; Pilz, P.M.; Hamza, O.; Acar, E.; Watzinger, S.; Mathew, S.; Kager, G.; Hallström, S.; Podesser, B.K.; et al. Remote Ischemic Perconditioning Ameliorates Myocardial Ischemia and Reperfusion-Induced Coronary Endothelial Dysfunction and Aortic Stiffness in Rats. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 702–713. [Google Scholar] [CrossRef]
- Peterss, S.; Guenther, S.; Kellermann, K.; Jungwirth, B.; Lichtinghagen, R.; Haverich, A.; Hagl, C.; Khaladj, N. An Experimental Model of Myocardial Infarction and Controlled Reperfusion Using a Miniaturized Cardiopulmonary Bypass in Rats. Interact. Cardiovasc. Thorac. Surg. 2014, 19, 561–566. [Google Scholar] [CrossRef]
- Rockman, H.A.; Wachhorst, S.P.; Mao, L.; Ross, J.J. ANG II Receptor Blockade Prevents Ventricular Hypertrophy and ANF Gene Expression with Pressure Overload in Mice. Am. J. Physiol. 1994, 266, H2468–H2475. [Google Scholar] [CrossRef]
- Nakao, Y.; Aono, J.; Hamaguchi, M.; Takahashi, K.; Sakaue, T.; Inoue, K.; Ikeda, S.; Yamaguchi, O. O-Ring-Induced Transverse Aortic Constriction (OTAC) Is a New Simple Method to Develop Cardiac Hypertrophy and Heart Failure in Mice. Sci. Rep. 2022, 12, 85. [Google Scholar] [CrossRef]
- Feldman, A.M.; Weinberg, E.O.; Ray, P.E.; Lorell, B.H. Selective Changes in Cardiac Gene Expression during Compensated Hypertrophy and the Transition to Cardiac Decompensation in Rats with Chronic Aortic Banding. Circ. Res. 1993, 73, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Silva, D.; Gonçalves-Rodrigues, P.; Almeida-Coelho, J.; Hamdani, N.; Lima, T.; Conceição, G.; Sousa-Mendes, C.; Cláudia-Moura; González, A.; Díez, J.; et al. Characterization of Biventricular Alterations in Myocardial (Reverse) Remodelling in Aortic Banding-Induced Chronic Pressure Overload. Sci. Rep. 2019, 9, 2956. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Oydanich, M.; Zhang, J.; Babici, D.; Fraidenraich, D.; Vatner, D.E.; Vatner, S.F. Rats Are Protected from the Stress of Chronic Pressure Overload Compared with Mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 318, R894–R900. [Google Scholar] [CrossRef] [PubMed]
- Litwin, S.E.; Katz, S.E.; Weinberg, E.O.; Lorell, B.H.; Aurigemma, G.P.; Douglas, P.S. Serial Echocardiographic-Doppler Assessment of Left Ventricular Geometry and Function in Rats with Pressure-Overload Hypertrophy. Chronic Angiotensin-Converting Enzyme Inhibition Attenuates the Transition to Heart Failure. Circulation 1995, 91, 2642–2654. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chemaly, E.R.; Liang, L.F.; LaRocca, T.J.; Yaniz-Galende, E.; Hajjar, R.J. A New Model of Congestive Heart Failure in Rats. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H994–H1003. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.S.; Lee, J.; Park, K.C.; Yang, K.-J.; Cho, E.J. The Relationship between MiRNA-26b and Connective Tissue Growth Factor in Rat Models of Aortic Banding and Debanding. Korean J. Intern. Med. 2021, 36, 596–607. [Google Scholar] [CrossRef]
- Gao, J.P.; Chen, C.X.; Wu, Q.; Gu, W.L.; Li, X. Effect of Sodium Houttuyfonate on Inhibiting Ventricular Remodeling Induced by Abdominal Aortic Banding in Rats. Can. J. Physiol. Pharmacol. 2010, 88, 693–701. [Google Scholar] [CrossRef]
- Hirata, M.; Ousaka, D.; Arai, S.; Okuyama, M.; Tarui, S.; Kobayashi, J.; Kasahara, S.; Sano, S. Novel Model of Pulmonary Artery Banding Leading to Right Heart Failure in Rats. Biomed Res. Int. 2015, 2015, 753210. [Google Scholar] [CrossRef]
- Andersen, S.; Schultz, J.G.; Holmboe, S.; Axelsen, J.B.; Hansen, M.S.; Lyhne, M.D.; Nielsen-Kudsk, J.E.; Andersen, A. A Pulmonary Trunk Banding Model of Pressure Overload Induced Right Ventricular Hypertrophy and Failure. J. Vis. Exp. 2018. [Google Scholar] [CrossRef]
- Bossers, G.P.L.; Hagdorn, Q.A.J.; Koop, A.-M.C.; van der Feen, D.E.; Bartelds, B.; van Leusden, T.; De Boer, R.A.; Silljé, H.H.W.; Berger, R.M.F. Female Rats Are Less Prone to Clinical Heart Failure than Male Rats in a Juvenile Rat Model of Right Ventricular Pressure Load. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H994–H1002. [Google Scholar] [CrossRef]
- Mendes-Ferreira, P.; Santos-Ribeiro, D.; Adão, R.; Maia-Rocha, C.; Mendes-Ferreira, M.; Sousa-Mendes, C.; Leite-Moreira, A.F.; Brás-Silva, C. Distinct Right Ventricle Remodeling in Response to Pressure Overload in the Rat. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H85–H95. [Google Scholar] [CrossRef]
- Ruppert, M.; Barta, B.A.; Korkmaz-Icöz, S.; Loganathan, S.; Oláh, A.; Sayour, A.A.; Benke, K.; Nagy, D.; Bálint, T.; Karck, M.; et al. Sex Similarities and Differences in the Reverse and Anti-Remodeling Effect of Pressure Unloading Therapy in a Rat Model of Aortic Banding and Debanding. Am. J. Physiol. Heart Circ. Physiol. 2022, 323, H204–H222. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Silva, D.G.; Rodrigues, P.; Alves, E.; Rizo, D.; Fonseca, A.C.R.G.; Lima, T.; Baganha, F.; Conceição, G.; Sousa, C.; Gonçalves, A.; et al. Mitochondrial Reversible Changes Determine Diastolic Function Adaptations During Myocardial (Reverse) Remodeling. Circ. Heart Fail. 2020, 13, e006170. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Xie, Q.; Wang, L.; Lu, Y.; Liu, P.; Yang, P.; Chen, R.; Shao, C.; Qiao, C.; Wang, Z.; et al. The TIR/BB-Loop Mimetic AS-1 Prevents Ang II-Induced Hypertensive Cardiac Hypertrophy via NF-ΚB Dependent Downregulation of MiRNA-143. Sci. Rep. 2019, 9, 6354. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, S.; Nishina, T.; Nishimura, K.; Miwa, S.; Ikeda, T.; Hanyu, M.; Fujioka, Y.; Kihara, Y.; Sasayama, S.; Komeda, M. A Rat Model of Dilated Cardiomyopathy to Investigate Partial Left Ventriculectomy. J. Card. Surg. 2001, 16, 40–47. [Google Scholar] [CrossRef][Green Version]
- OKAMOTO, K.; AOKI, K. Development of a Strain of Spontaneously Hypertensive Rats. Jpn. Circ. J. 1963, 27, 282–293. [Google Scholar] [CrossRef]
- Heyen, J.R.R.; Blasi, E.R.; Nikula, K.; Rocha, R.; Daust, H.A.; Frierdich, G.; Van Vleet, J.F.; De Ciechi, P.; McMahon, E.G.; Rudolph, A.E. Structural, Functional, and Molecular Characterization of the SHHF Model of Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1775–H1784. [Google Scholar] [CrossRef]
- Andersen, A.; van der Feen, D.E.; Andersen, S.; Schultz, J.G.; Hansmann, G.; Bogaard, H.J. Animal Models of Right Heart Failure. Cardiovasc. Diagn. Ther. 2020, 10, 1561–1579. [Google Scholar] [CrossRef]
- Bogaard, H.J.; Natarajan, R.; Henderson, S.C.; Long, C.S.; Kraskauskas, D.; Smithson, L.; Ockaili, R.; McCord, J.M.; Voelkel, N.F. Chronic Pulmonary Artery Pressure Elevation Is Insufficient to Explain Right Heart Failure. Circulation 2009, 120, 1951–1960. [Google Scholar] [CrossRef]
- Yamazaki, H.; Tajima, H.; Yamamoto, Y.; Munesue, S.; Okazaki, M.; Ohbatake, Y.; Nakanuma, S.; Makino, I.; Miyashita, T.; Takamura, H.; et al. Thrombopoietin Accumulation in Hepatocytes Induces a Decrease in Its Serum Levels in a Sinusoidal Obstruction Syndrome Model. Mol. Med. Rep. 2022, 25. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, J.; Cheng, Y.; Li, X.; He, M.; Zhu, J.; Han, H.; Wei, G.; Kong, H.; Xie, W.; et al. Glucagon-Like Peptide-1 Mediates the Protective Effect of the Dipeptidyl Peptidase IV Inhibitor on Renal Fibrosis via Reducing the Phenotypic Conversion of Renal Microvascular Cells in Monocrotaline-Treated Rats. Biomed Res. Int. 2018, 2018, 1864107. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, B.; Chen, X.; Hirenallur-Shanthappa, D.; Zhao, Y.; Stansfield, J.C.; Zhang, B.B.; Sheikh, A.; Wu, Z. Neutralization of GDF15 Prevents Anorexia and Weight Loss in the Monocrotaline-Induced Cardiac Cachexia Rat Model. Cells 2022, 11, 1073. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Gomez-Arroyo, J. The Role of Vascular Endothelial Growth Factor in Pulmonary Arterial Hypertension. The Angiogenesis Paradox. Am. J. Respir. Cell Mol. Biol. 2014, 51, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Jayasekera, G.; Wilson, K.S.; Buist, H.; Woodward, R.; Uckan, A.; Hughes, C.; Nilsen, M.; Church, A.C.; Johnson, M.K.; Gallagher, L.; et al. Understanding Longitudinal Biventricular Structural and Functional Changes in a Pulmonary Hypertension Sugen–Hypoxia Rat Model by Cardiac Magnetic Resonance Imaging. Pulm. Circ. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zelt, J.G.E.; Cadete, V.; Deng, Y.; Godoy, R.; Cuillerier, A.; Rowe, K.; Abdul-Ghani, M.; Megeney, L.; Burelle, Y.; Giulivi, A.; et al. Right Ventricular Maladaptation to Pressure Overload in Fischer Rats Is Associated With Profound Deficiency in Adenylate Kinase 1 and Impaired Ventricular Energetics. Hypertension 2022, 79, 2774–2786. [Google Scholar] [CrossRef]
- Havlenova, T.; Skaroupkova, P.; Miklovic, M.; Behounek, M.; Chmel, M.; Jarkovska, D.; Sviglerova, J.; Stengl, M.; Kolar, M.; Novotny, J.; et al. Right versus Left Ventricular Remodeling in Heart Failure Due to Chronic Volume Overload. Sci. Rep. 2021, 11, 17136. [Google Scholar] [CrossRef]
- Brower, G.L.; Janicki, J.S. Contribution of Ventricular Remodeling to Pathogenesis of Heart Failure in Rats. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H674–H683. [Google Scholar] [CrossRef]
- Aimoto, M.; Yagi, K.; Ezawa, A.; Tsuneoka, Y.; Kumada, K.; Hasegawa, T.; Kuze, T.; Chiba, T.; Nagasawa, Y.; Tanaka, H.; et al. Chronic Volume Overload Caused by Abdominal Aorto-Venocaval Shunt Provides Arrhythmogenic Substrates in the Rat Atrium. Biol. Pharm. Bull. 2022, 45, 635–642. [Google Scholar] [CrossRef]
- Oliver-Dussault, C.; Ascah, A.; Marcil, M.; Matas, J.; Picard, S.; Pibarot, P.; Burelle, Y.; Deschepper, C.F. Early Predictors of Cardiac Decompensation in Experimental Volume Overload. Mol. Cell. Biochem. 2010, 338, 271–282. [Google Scholar] [CrossRef]
- Tung, Y.-C.; Cheng, M.-L.; Wu, L.-S.; Tang, H.-Y.; Huang, C.-Y.; Chang, G.-J.; Chang, C.-J. Derangements and Reversibility of Energy Metabolism in Failing Hearts Resulting from Volume Overload: Transcriptomics and Metabolomics Analyses. Int. J. Mol. Sci. 2022, 23, 6809. [Google Scholar] [CrossRef]
- Corporan, D.; Onohara, D.; Amedi, A.; Saadeh, M.; Guyton, R.A.; Kumar, S.; Padala, M. Hemodynamic and Transcriptomic Studies Suggest Early Left Ventricular Dysfunction in a Preclinical Model of Severe Mitral Regurgitation. J. Thorac. Cardiovasc. Surg. 2021, 161, 961–976.e22. [Google Scholar] [CrossRef]
- Corporan, D.; Segura, A.; Padala, M. Ultrastructural Adaptation of the Cardiomyocyte to Chronic Mitral Regurgitation. Front. Cardiovasc. Med. 2021, 8, 714774. [Google Scholar] [CrossRef] [PubMed]
- Onohara, D.; Corporan, D.; Hernandez-Merlo, R.; Guyton, R.A.; Padala, M. Mitral Regurgitation Worsens Cardiac Remodeling in Ischemic Cardiomyopathy in an Experimental Model. J. Thorac. Cardiovasc. Surg. 2020, 160, e107–e125. [Google Scholar] [CrossRef] [PubMed]
- Kono, T.; Onohara, D.; Amedi, A.; Corporan, D.; Padala, M. Effect of Early versus Late Onset Mitral Regurgitation on Left Ventricular Remodeling in Ischemic Cardiomyopathy in an Animal Model. J. Thorac. Cardiovasc. Surg. 2021. [Google Scholar] [CrossRef]
- Arsenault, M.; Plante, E.; Drolet, M.-C.; Couet, J. Experimental Aortic Regurgitation in Rats under Echocardiographic Guidance. J. Heart Valve Dis. 2002, 11, 128–134. [Google Scholar]
- Bussoni, M.; Okoshi, M.P.; Matsubara, L.S.; Polegato, B.F.; Roscani, M.G.; Pereira, E.J.; de Paiva, S.A.R.; Zornoff, L.A.M.; Okoshi, K.; Minicucci, M.F.; et al. The Role of Extracellular Matrix in the Experimental Acute Aortic Regurgitation Model in Rats. Heart. Lung Circ. 2022, 31, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Walsh-Wilkinson, E.; Drolet, M.-C.; Arsenault, M.; Couet, J. Sex Differences in the Evolution of Left Ventricle Remodeling in Rats with Severe Volume Overload. BMC Cardiovasc. Disord. 2020, 20, 51. [Google Scholar] [CrossRef]
- Akazawa, Y.; Fujioka, T.; Ide, H.; Yazaki, K.; Honjo, O.; Sun, M.; Friedberg, M.K. Impaired Right and Left Ventricular Function and Relaxation Induced by Pulmonary Regurgitation Are Not Reversed by Tardive Antifibrosis Treatment. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H38–H51. [Google Scholar] [CrossRef]
- Droogmans, S.; Franken, P.R.; Garbar, C.; Weytjens, C.; Cosyns, B.; Lahoutte, T.; Caveliers, V.; Pipeleers-Marichal, M.; Bossuyt, A.; Schoors, D.; et al. In Vivo Model of Drug-Induced Valvular Heart Disease in Rats: Pergolide-Induced Valvular Heart Disease Demonstrated with Echocardiography and Correlation with Pathology. Eur. Heart J. 2007, 28, 2156–2162. [Google Scholar] [CrossRef][Green Version]
- Khosroshahi, A.J.; Mokhtari, B.; Badalzadeh, R. Combination of Nicotinamide Mononucleotide and Troxerutin Induces Full Protection against Doxorubicin-Induced Cardiotoxicity by Modulating Mitochondrial Biogenesis and Inflammatory Response. Mol. Biol. Rep. 2022. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.L.; Romano, M.M.D.; Campos Pulici, E.C.; Carvalho, E.E.V.; de Souza, F.R.; Tanaka, D.M.; Maciel, B.C.; Salgado, H.C.; Fazan-Júnior, R.; Rossi, M.A.; et al. Short-Term and Long-Term Models of Doxorubicin-Induced Cardiomyopathy in Rats: A Comparison of Functional and Histopathological Changes. Exp. Toxicol. Pathol. Off. J. Ges. Fur Toxikol. Pathol. 2017, 69, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Chakouri, N.; Farah, C.; Matecki, S.; Amedro, P.; Vincenti, M.; Saumet, L.; Vergely, L.; Sirvent, N.; Lacampagne, A.; Cazorla, O. Screening for In-Vivo Regional Contractile Defaults to Predict the Delayed Doxorubicin Cardiotoxicity in Juvenile Rat. Theranostics 2020, 10, 8130–8142. [Google Scholar] [CrossRef] [PubMed]
- Rahmanifard, M.; Vessal, M.; Noorafshan, A.; Karbalay-Doust, S.; Naseh, M. The Protective Effects of Coenzyme Q10 and Lisinopril Against Doxorubicin-Induced Cardiotoxicity in Rats: A Stereological and Electrocardiogram Study. Cardiovasc. Toxicol. 2021, 21, 936–946. [Google Scholar] [CrossRef]
- Saleh, D.O.; Mahmoud, S.S.; Hassan, A.; Sanad, E.F. Doxorubicin-Induced Hepatic Toxicity in Rats: Mechanistic Protective Role of Omega-3 Fatty Acids through Nrf2/HO-1 Activation and PI3K/Akt/GSK-3β Axis Modulation. Saudi J. Biol. Sci. 2022, 29, 103308. [Google Scholar] [CrossRef]
- Mahzari, S.; Hosseinian, S.; Hadjzadeh, M.-A.-R.; Mohebbati, R.; Noshahr, Z.S.; Rad, A.K. Kidney Dysfunction and Oxidative Stress in Doxorubicin-Induced Nephrotic Rat: Protective Role of Sesame Oil. Saudi J. Kidney Dis. Transplant. Off. Publ. Saudi Cent. Organ Transplant. Saudi Arab. 2021, 32, 1243–1252. [Google Scholar] [CrossRef]
- Shaldoum, F.; El-Kott, A.F.; Ouda, M.M.A.; Abd-Ella, E.M. Immunomodulatory Effects of Bee Pollen on Doxorubicin-Induced Bone Marrow/Spleen Immunosuppression in Rat. J. Food Biochem. 2021, 45, e13747. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Joseph, L.; Shekhawat, N.S.; Devi, S.; Wang, J.; Melchert, R.B.; Hauer-Jensen, M.; Kennedy, R.H. Hyperhomocysteinemia Leads to Pathological Ventricular Hypertrophy in Normotensive Rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H679–H686. [Google Scholar] [CrossRef]
- Liu, B.; Ma, S.; Wang, T.; Zhao, C.; Li, Y.; Yin, J.; Liu, C.; Gao, C.; Sun, L.; Yue, W.; et al. A Novel Rat Model of Heart Failure Induced by High Methionine Diet Showing Evidence of Association between Hyperhomocysteinemia and Activation of NF-KappaB. Am. J. Transl. Res. 2016, 8, 117–124. [Google Scholar]
- Capasso, J.M.; Li, P.; Guideri, G.; Malhotra, A.; Cortese, R.; Anversa, P. Myocardial Mechanical, Biochemical, and Structural Alterations Induced by Chronic Ethanol Ingestion in Rats. Circ. Res. 1992, 71, 346–356. [Google Scholar] [CrossRef]
- Capasso, J.M.; Li, P.; Guideri, G.; Anversa, P. Left Ventricular Dysfunction Induced by Chronic Alcohol Ingestion in Rats. Am. J. Physiol. 1991, 261, H212–H219. [Google Scholar] [CrossRef]
- Marchini, G.S.; Cestari, I.N.; Salemi, V.M.C.; Irigoyen, M.C.; Arnold, A.; Kakoi, A.; Rocon, C.; Aiello, V.D.; Cestari, I.A. Early Changes in Myocyte Contractility and Cardiac Function in Streptozotocin-Induced Type 1 Diabetes in Rats. PLoS ONE 2020, 15, e0237305. [Google Scholar] [CrossRef]
- Lakomkin, V.L.; Abramov, A.A.; Lukoshkova, E.V.; Prosvirnin, A.V.; Kapelko, V.I. Systolic Dysfunction of the Heart in Type 1 Diabetes Mellitus. Bull. Exp. Biol. Med. 2021, 172, 14–17. [Google Scholar] [CrossRef]
- Sklifasovskaya, A.P.; Blagonravov, M.L.; Ryabinina, A.Y.; Azova, M.M.; Goryachev, V.A. Expression of Bax and Bcl-2 Proteins in Left-Ventricular Cardiomyocytes in Wistar-Kyoto and SHR Rats with Insulin-Dependent Diabetes Mellitus. Bull. Exp. Biol. Med. 2021, 171, 576–581. [Google Scholar] [CrossRef]
- López y López, G.; Tepox Galicia, A.Y.; Atonal Flores, F.; Flores Hernández, J.; Pérez Vizcaino, F.; Villa Mancera, A.E.; Miguél, G.G.; Reynoso Palomar, A. Echocardiographic Follow-up to Right Ventricular Modifications in Secondary Pulmonary Hypertension to Diabetes in Rats. Clin. Exp. Hypertens. 2021, 43, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Mátyás, C.; Kovács, A.; Németh, B.T.; Oláh, A.; Braun, S.; Tokodi, M.; Barta, B.A.; Benke, K.; Ruppert, M.; Lakatos, B.K.; et al. Comparison of Speckle-Tracking Echocardiography with Invasive Hemodynamics for the Detection of Characteristic Cardiac Dysfunction in Type-1 and Type-2 Diabetic Rat Models. Cardiovasc. Diabetol. 2018, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Nana-Leventaki, E.; Nana, M.; Poulianitis, N.; Sampaziotis, D.; Perrea, D.; Sanoudou, D.; Rontogianni, D.; Malliaras, K. Cardiosphere-Derived Cells Attenuate Inflammation, Preserve Systolic Function, and Prevent Adverse Remodeling in Rat Hearts With Experimental Autoimmune Myocarditis. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 70–77. [Google Scholar] [CrossRef]
- Wu, Y.; Zheng, Z.; Cao, X.; Yang, Q.; Norton, V.; Adini, A.; Maiti, A.K.; Adini, I.; Wu, H. RIP1/RIP3/MLKL Mediates Myocardial Function Through Necroptosis in Experimental Autoimmune Myocarditis. Front. Cardiovasc. Med. 2021, 8, 696362. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, Y.; Xu, Z.; Wang, S.; Wang, L.; Liu, X.; Gao, F. Non-Invasive Assessment of Early and Acute Myocarditis in a Rat Model Using Cardiac Magnetic Resonance Tissue Tracking Analysis of Myocardial Strain. Quant. Imaging Med. Surg. 2020, 10, 2157–2167. [Google Scholar] [CrossRef]
- Korkusuz, H.; Esters, P.; Naguib, N.; Nour Eldin, N.-E.; Lindemayr, S.; Huebner, F.; Koujan, A.; Bug, R.; Ackermann, H.; Vogl, T.J. Acute Myocarditis in a Rat Model: Late Gadolinium Enhancement with Histopathological Correlation. Eur. Radiol. 2009, 19, 2672–2678. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, M.; Gonano, L.A.; Back, T.G.; Chen, S.R.W.; Vila Petroff, M. Role of CaMKII and ROS in Rapid Pacing-Induced Apoptosis. J. Mol. Cell. Cardiol. 2013, 63, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Fu, W.-D.; Chen, L. MiRNA-182 Regulates the Cardiomyocyte Apoptosis in Heart Failure. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4917–4923. [Google Scholar] [CrossRef] [PubMed]
- Aistrup, G.L.; Kelly, J.E.; Kapur, S.; Kowalczyk, M.; Sysman-Wolpin, I.; Kadish, A.H.; Wasserstrom, J.A. Pacing-Induced Heterogeneities in Intracellular Ca2+ Signaling, Cardiac Alternans, and Ventricular Arrhythmias in Intact Rat Heart. Circ. Res. 2006, 99, e65–e73. [Google Scholar] [CrossRef]
- Greaser, M.L.; Warren, C.M.; Esbona, K.; Guo, W.; Duan, Y.; Parrish, A.M.; Krzesinski, P.R.; Norman, H.S.; Dunning, S.; Fitzsimons, D.P.; et al. Mutation That Dramatically Alters Rat Titin Isoform Expression and Cardiomyocyte Passive Tension. J. Mol. Cell. Cardiol. 2008, 44, 983–991. [Google Scholar] [CrossRef]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a Gene for Hereditary Cardiomyopathy, Regulates Titin Splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhu, C.; Yin, Z.; Zhang, Y.; Wang, C.; Walk, A.S.; Lin, Y.-H.; McKinsey, T.A.; Woulfe, K.C.; Ren, J.; et al. The Ryanodine Receptor Stabilizer S107 Ameliorates Contractility of Adult Rbm20 Knockout Rat Cardiomyocytes. Physiol. Rep. 2021, 9, e15011. [Google Scholar] [CrossRef]
- Sugihara, H.; Kimura, K.; Yamanouchi, K.; Teramoto, N.; Okano, T.; Daimon, M.; Morita, H.; Takenaka, K.; Shiga, T.; Tanihata, J.; et al. Age-Dependent Echocardiographic and Pathologic Findings in a Rat Model with Duchenne Muscular Dystrophy Generated by CRISPR/Cas9 Genome Editing. Int. Heart J. 2020, 61, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Fujikura, Y.; Kimura, K.; Yamanouchi, K.; Sugihara, H.; Hatakeyama, M.; Zhuang, H.; Abe, T.; Daimon, M.; Morita, H.; Komuro, I.; et al. A Medium-Chain Triglyceride Containing Ketogenic Diet Exacerbates Cardiomyopathy in a CRISPR/Cas9 Gene-Edited Rat Model with Duchenne Muscular Dystrophy. Sci. Rep. 2022, 12, 11580. [Google Scholar] [CrossRef]
- Ling, Y.; Ma, J.; Qi, X.; Zhang, X.; Kong, Q.; Guan, F.; Dong, W.; Chen, W.; Gao, S.; Gao, X.; et al. Novel Rat Model of Multiple Mitochondrial Dysfunction Syndromes (MMDS) Complicated with Cardiomyopathy. Anim. Model. Exp. Med. 2021, 4, 381–390. [Google Scholar] [CrossRef]
- Ling, Y.; Yang, X.; Zhang, X.; Guan, F.; Qi, X.; Dong, W.; Liu, M.; Ma, J.; Jiang, X.; Gao, K.; et al. Myocardium-Specific Isca1 Knockout Causes Iron Metabolism Disorder and Myocardial Oncosis in Rat. Life Sci. 2022, 297, 120485. [Google Scholar] [CrossRef]
- Steinhorn, B.; Sorrentino, A.; Badole, S.; Bogdanova, Y.; Belousov, V.; Michel, T. Chemogenetic Generation of Hydrogen Peroxide in the Heart Induces Severe Cardiac Dysfunction. Nat. Commun. 2018, 9, 4044. [Google Scholar] [CrossRef] [PubMed]
- Spyropoulos, F.; Sorrentino, A.; van der Reest, J.; Yang, P.; Waldeck-Weiermair, M.; Steinhorn, B.; Eroglu, E.; Saeedi Saravi, S.S.; Yu, P.; Haigis, M.; et al. Metabolomic and Transcriptomic Signatures of Chemogenetic Heart Failure. Am. J. Physiol. Heart Circ. Physiol. 2022, 322, H451–H465. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, A.; Steinhorn, B.; Troncone, L.; Saravi, S.S.S.; Badole, S.; Eroglu, E.; Kijewski, M.F.; Divakaran, S.; Di Carli, M.; Michel, T. Reversal of Heart Failure in a Chemogenetic Model of Persistent Cardiac Redox Stress. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H617–H626. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Johnson, B.A.; Grinsfelder, D.; Canseco, D.; Mammen, P.P.; Rothermel, B.A.; Olson, E.N.; Sadek, H.A. Regulation of Neonatal and Adult Mammalian Heart Regeneration by the MiR-15 Family. Proc. Natl. Acad. Sci. USA 2013, 110, 187–192. [Google Scholar] [CrossRef]
- Kalra, S.; Bhatt, H.; Kirtane, A.J. Stenting in Primary Percutaneous Coronary Intervention for Acute ST-Segment Elevation Myocardial Infarction. Methodist Debakey Cardiovasc. J. 2018, 14, 14–22. [Google Scholar] [CrossRef]
- Vidal-Calés, P.; Cepas-Guillén, P.L.; Brugaletta, S.; Sabaté, M. New Interventional Therapies beyond Stenting to Treat ST-Segment Elevation Acute Myocardial Infarction. J. Cardiovasc. Dev. Dis. 2021, 8, 100. [Google Scholar] [CrossRef]
- Kato, H.; Fu, Y.Y.; Zhu, J.; Wang, L.; Aafaqi, S.; Rahkonen, O.; Slorach, C.; Traister, A.; Leung, C.H.; Chiasson, D.; et al. Pulmonary Vein Stenosis and the Pathophysiology of “Upstream” Pulmonary Veins. J. Thorac. Cardiovasc. Surg. 2014, 148, 245–253. [Google Scholar] [CrossRef]
- Ryan, J.J.; Marsboom, G.; Archer, S.L. Rodent Models of Group 1 Pulmonary Hypertension. In Pharmacotherapy of Pulmonary Hypertension; Springer: Berlin/Heidelberg, Germany, 2013; pp. 105–149. [Google Scholar]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. Pharmacol. 2015, 70, 5.47.1–5.47.20. [Google Scholar] [CrossRef]
- Traister, A.; Li, M.; Aafaqi, S.; Lu, M.; Arab, S.; Radisic, M.; Gross, G.; Guido, F.; Sherret, J.; Verma, S.; et al. Integrin-Linked Kinase Mediates Force Transduction in Cardiomyocytes by Modulating SERCA2a/PLN Function. Nat. Commun. 2014, 5, 4533. [Google Scholar] [CrossRef]
- Boczar, K.E.; Aseyev, O.; Sulpher, J.; Johnson, C.; Burwash, I.G.; Turek, M.; Dent, S.; Dwivedi, G. Right Heart Function Deteriorates in Breast Cancer Patients Undergoing Anthracycline-Based Chemotherapy. Echo Res. Pract. 2016, 3, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Abdel-Rahman, A.A. Tetrahydrobiopterin Paradoxically Mediates Cardiac Oxidative Stress and Mitigates Ethanol-Evoked Cardiac Dysfunction in Conscious Female Rats. Eur. J. Pharmacol. 2021, 909, 174406. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Dvorakova, M.C. Future Perspective of Diabetic Animal Models. Endocr. Metab. Immune Disord.—Drug Targets 2020, 20, 25–38. [Google Scholar] [CrossRef]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Nagasaka, A.; Terawaki, T.; Noda, M.; Takashima, M.; Fujino, M.; Yamauchi, Y.; Arawaka, S.; Kato, T.; Nakaya, M. GRK5 -mediated Inflammation and Fibrosis Exert Cardioprotective Effects during the Acute Phase of Myocardial Infarction. FEBS Open Bio 2023. [Google Scholar] [CrossRef] [PubMed]
- Chalise, U.; Becirovic-Agic, M.; Lindsey, M.L. The Cardiac Wound Healing Response to Myocardial Infarction. WIREs Mech. Dis. 2023, 15. [Google Scholar] [CrossRef]
- Del Re, D.P. Hippo-Yap Signaling in Cardiac and Fibrotic Remodeling. Curr. Opin. Physiol. 2022, 26, 100492. [Google Scholar] [CrossRef]
- Sarohi, V.; Chakraborty, S.; Basak, T. Exploring the Cardiac ECM during Fibrosis: A New Era with next-Gen Proteomics. Front. Mol. Biosci. 2022, 9. [Google Scholar] [CrossRef]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and Inflammatory Cardiomyopathy: Current Evidence and Future Directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef]
- Farrag, N.A.; Thornhill, R.E.; Prato, F.S.; Skanes, A.C.; Sullivan, R.; Sebben, D.; Butler, J.; Sykes, J.; Wilk, B.; Ukwatta, E. Assessment of Left Atrial Fibrosis Progression in Canines Following Rapid Ventricular Pacing Using 3D Late Gadolinium Enhanced CMR Images. PLoS ONE 2022, 17, e0269592. [Google Scholar] [CrossRef]
- Schiabor Barrett, K.M.; Cirulli, E.T.; Bolze, A.; Rowan, C.; Elhanan, G.; Grzymski, J.J.; Lee, W.; Washington, N.L. Cardiomyopathy Prevalence Exceeds 30% in Individuals with TTN Variants and Early Atrial Fibrillation. Genet. Med. 2023, 100012. [Google Scholar] [CrossRef]
- Marcello, M.; Cetrangolo, V.; Savarese, M.; Udd, B. Use of Animal Models to Understand Titin Physiology and Pathology. J. Cell. Mol. Med. 2022, 26, 5103–5112. [Google Scholar] [CrossRef]
- Schultz, T.I.; Raucci, F.J.; Salloum, F.N. Cardiovascular Disease in Duchenne Muscular Dystrophy. JACC Basic Transl. Sci. 2022, 7, 608–625. [Google Scholar] [CrossRef] [PubMed]
- Fujikura, Y.; Sugihara, H.; Hatakeyama, M.; Oishi, K.; Yamanouchi, K. Ketogenic Diet with Medium-chain Triglycerides Restores Skeletal Muscle Function and Pathology in a Rat Model of Duchenne Muscular Dystrophy. FASEB J. 2021, 35. [Google Scholar] [CrossRef] [PubMed]
- Ruppert, T.; Heckmann, M.B.; Rapti, K.; Schultheis, D.; Jungmann, A.; Katus, H.A.; Winter, L.; Frey, N.; Clemen, C.S.; Schröder, R.; et al. AAV-Mediated Cardiac Gene Transfer of Wild-Type Desmin in Mouse Models for Recessive Desminopathies. Gene Ther. 2020, 27, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Caravia, X.M.; Ramirez-Martinez, A.; Gan, P.; Wang, F.; McAnally, J.R.; Xu, L.; Bassel-Duby, R.; Liu, N.; Olson, E.N. Loss of Function of the Nuclear Envelope Protein LEMD2 Causes DNA Damage–Dependent Cardiomyopathy. J. Clin. Invest. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Belke, D.D.; Garnett, L.; Martens, K.; Abdelfatah, N.; Rodriguez, M.; Diao, C.; Chen, Y.-X.; Gordon, P.M.K.; Nygren, A.; et al. Transgenic Mice Overexpressing Desmocollin-2 (DSC2) Develop Cardiomyopathy Associated with Myocardial Inflammation and Fibrotic Remodeling. PLoS ONE 2017, 12, e0174019. [Google Scholar] [CrossRef]
- Kloos, W.; Katus, H.A.; Meder, B. Genetic Cardiomyopathies. Herz 2012, 37, 612–618. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).