
Genetic Heterogeneity of Familial Hypercholesterolemia: Repercussions for Molecular Diagnosis


Abstract
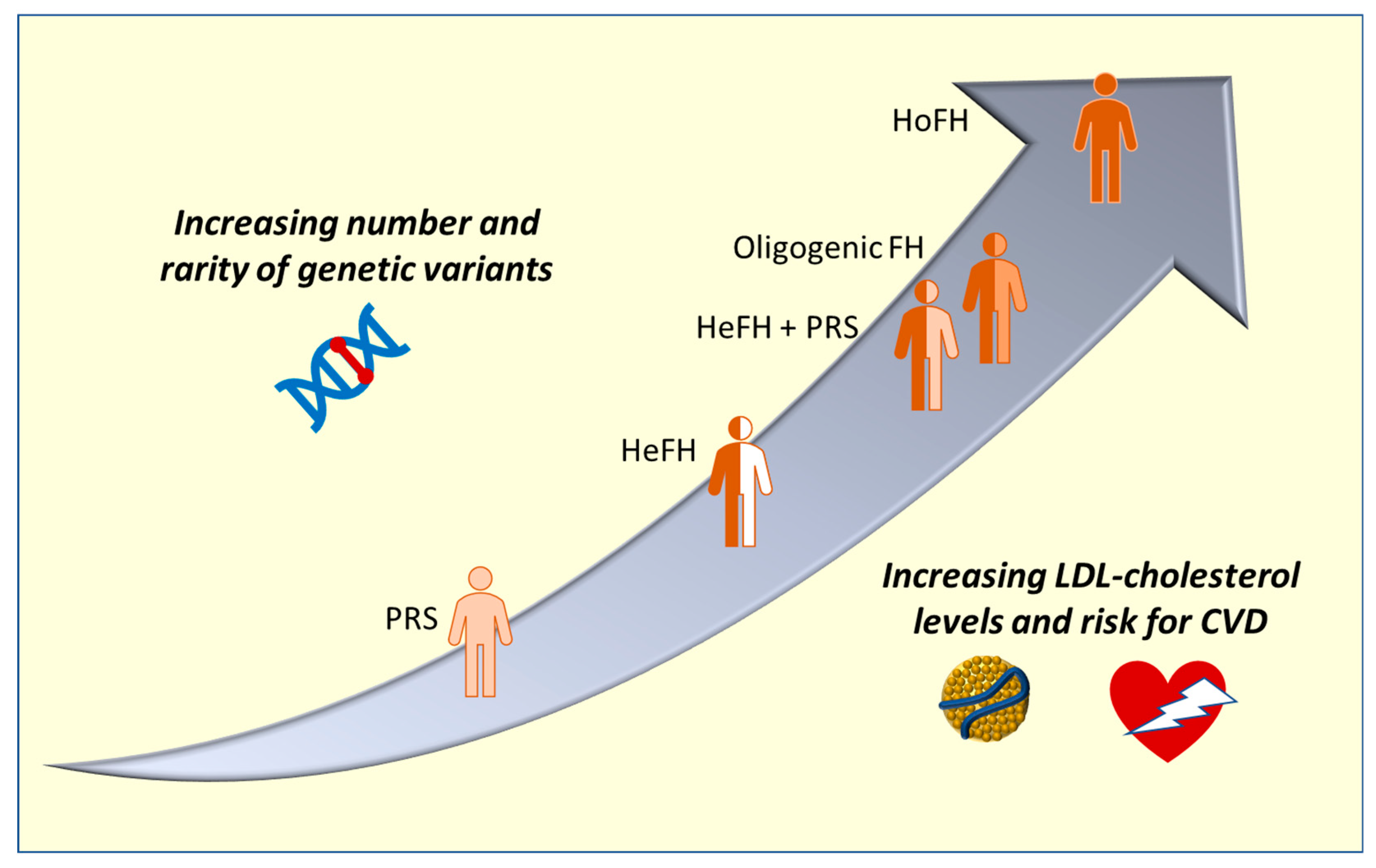
:1. Introduction
2. Genetics and Molecular Basis
2.1. Classical FH-Causative Genes
2.2. Heterozygous and Homozygous FH
2.3. Other Genes Involved in FH
2.4. FH Phenocopies
- Among these genes, the ABCG5 and ABCG8 encoding for the two subunits of the sterol transporter mediating efflux of plant sterols both in the enterocytes, immediately after their absorption or in the liver causing their excretion in the bile [58]. These genes are causative for sitosterolemia, an autosomal recessive disease leading to severe xanthomatosis and high cholesterol levels. Variants in the ABCG5 and ABCG8 genes were identified in 2.4% of Dutch FH patients [59].
- Cerebrotendinous xanthomatosis is another disease characterized by diffuse xanthomas and, in some cases, by high cholesterol levels that can mimic FH [60,61]. Variants in the causative gene, the CYP27A1, were identified in suspected FH patients without other pathogenic variants [62] or in presence of a FH-causative variant as worsening effects [63]. The molecular defect is due to the absence of functional sterol 27-hydroxylase, an enzyme essential for turning cholesterol in bile acids.
- The LIPA gene encodes for the lysosomal acid lipase, the enzyme essential for the degradation of cholesteryl esters that in absence of a functional enzyme accumulate within lysosomes giving rise to a severe disease characterized by high cholesterol levels that can be misdiagnosed as FH (Lysosomal Acid Lipase Deficiency—LALD) [64]. In Slovenia, during the universal screening for FH, 3 children suffering from LALD were identified among hypercholesterolemic children [65].
2.5. Oligogenic FH and Modifier Genes
2.6. Polygenic Risk Scores
3. Molecular Diagnosis
Pathogenicity Evaluation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial Hypercholesterolaemia Is Underdiagnosed and Undertreated in the General Population: Guidance for Clinicians to Prevent Coronary Heart Disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Cuchel, M.; Bruckert, E.; Ginsberg, H.N.; Raal, F.J.; Santos, R.D.; Hegele, R.A.; Kuivenhoven, J.A.; Nordestgaard, B.G.; Descamps, O.S.; Steinhagen-Thiessen, E.; et al. Homozygous Familial Hypercholesterolaemia: New Insights and Guidance for Clinicians to Improve Detection and Clinical Management. A Position Paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 2014, 35, 2146–2157. [Google Scholar] [CrossRef] [PubMed]
- Akioyamen, L.E.; Genest, J.; Shan, S.D.; Reel, R.L.; Albaum, J.M.; Chu, A.; Tu, J.V. Estimating the Prevalence of Heterozygous Familial Hypercholesterolaemia: A Systematic Review and Meta-Analysis. BMJ Open 2017, 7, e016461. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Dharmayat, K.I.; Stevens, C.A.T.; Sharabiani, M.T.A.; Jones, R.S.; Watts, G.F.; Genest, J.; Ray, K.K.; Vallejo-Vaz, A.J. Prevalence of Familial Hypercholesterolemia Among the General Population and Patients With Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-Analysis. Circulation 2020, 141, 1742–1759. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, S.O.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef] [PubMed]
- Toft-Nielsen, F.; Emanuelsson, F.; Benn, M. Familial Hypercholesterolemia Prevalence Among Ethnicities-Systematic Review and Meta-Analysis. Front. Genet. 2022, 13, 840797. [Google Scholar] [CrossRef]
- Laufs, U.; Dent, R.; Kostenuik, P.J.; Toth, P.P.; Catapano, A.L.; Chapman, M.J. Why Is Hypercholesterolaemia so Prevalent? A View from Evolutionary Medicine. Eur. Heart J. 2019, 40, 2825–2830. [Google Scholar] [CrossRef]
- Civeira, F.; Arca, M.; Cenarro, A.; Hegele, R.A. A Mechanism-Based Operational Definition and Classification of Hypercholesterolemia. J. Clin. Lipidol. 2022, 16, 813–821. [Google Scholar] [CrossRef]
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial Hypercholesterolaemia. Nat. Rev. Dis. Primers 2017, 3, 17093. [Google Scholar] [CrossRef]
- Feingold, K.R. Lipid and Lipoprotein Metabolism. Endocrinol. Metab. Clin. N. Am. 2022, 51, 437–458. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Familial Hypercholesterolemia: A Genetic Defect in the Low-Density Lipoprotein Receptor. N. Engl. J. Med. 1976, 294, 1386–1390. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Garcia, U.; Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Larrea-Sebal, A.; Alonso-Estrada, R.; Aguilo-Arce, J.; Ostolaza, H.; Palacios, L.; Martin, C. Mutation Type Classification and Pathogenicity Assignment of Sixteen Missense Variants Located in the EGF-Precursor Homology Domain of the LDLR. Sci. Rep. 2020, 10, 1727. [Google Scholar] [CrossRef] [PubMed]
- Futema, M.; Ramaswami, U.; Tichy, L.; Bogsrud, M.P.; Holven, K.B.; Roeters van Lennep, J.; Wiegman, A.; Descamps, O.S.; De Leener, A.; Fastre, E.; et al. Comparison of the Mutation Spectrum and Association with Pre and Post Treatment Lipid Measures of Children with Heterozygous Familial Hypercholesterolaemia (FH) from Eight European Countries. Atherosclerosis 2021, 319, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Rieck, L.; Bardey, F.; Grenkowitz, T.; Bertram, L.; Helmuth, J.; Mischung, C.; Spranger, J.; Steinhagen-Thiessen, E.; Bobbert, T.; Kassner, U.; et al. Mutation Spectrum and Polygenic Score in German Patients with Familial Hypercholesterolemia. Clin. Genet. 2020, 98, 457–467. [Google Scholar] [CrossRef]
- Pirillo, A.; Garlaschelli, K.; Arca, M.; Averna, M.; Bertolini, S.; Calandra, S.; Tarugi, P.; Catapano, A.L. LIPIGEN Group Spectrum of Mutations in Italian Patients with Familial Hypercholesterolemia: New Results from the LIPIGEN Study. Atheroscler. Suppl. 2017, 29, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bañares, V.G. Preliminary Spectrum of Genetic Variants in Familial Hypercholesterolemia in Argentina. J. Clin. Lipidol. 2017, 11, 8. [Google Scholar] [CrossRef]
- Sánchez-Hernández, R.M.; González-Lleó, A.M.; Tugores, A.; Brito-Casillas, Y.; Civeira, F.; Boronat, M.; Wägner, A. Familial Hypercholesterolemia in Gran Canaria: Founder Mutation Effect and High Frequency of Diabetes. Clin. Investig. Arterioscler 2021, 33, 247–253. [Google Scholar] [CrossRef]
- Tada, H.; Hori, M.; Nomura, A.; Hosomichi, K.; Nohara, A.; Kawashiri, M.-A.; Harada-Shiba, M. A Catalog of the Pathogenic Mutations of LDL Receptor Gene in Japanese Familial Hypercholesterolemia. J. Clin. Lipidol. 2020, 14, 346–351. [Google Scholar] [CrossRef]
- Burnett, J.R.; Hooper, A.J.; Hegele, R.A. APOB-Related Familial Hypobetalipoproteinemia. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Abifadel, M.; Boileau, C. Genetic and Molecular Architecture of Familial Hypercholesterolemia. J. Intern. Med. 2023, 193, 144–165. [Google Scholar] [CrossRef]
- Alves, A.C.; Benito-Vicente, A.; Medeiros, A.M.; Reeves, K.; Martin, C.; Bourbon, M. Further Evidence of Novel APOB Mutations as a Cause of Familial Hypercholesterolaemia. Atherosclerosis 2018, 277, 448–456. [Google Scholar] [CrossRef]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 Cause Autosomal Dominant Hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Alnouri, F.; Santos, R.D. New Trends and Therapies for Familial Hypercholesterolemia. J. Clin. Med. 2022, 11, 6638. [Google Scholar] [CrossRef] [PubMed]
- Dron, J.S.; Hegele, R.A. Complexity of Mechanisms among Human Proprotein Convertase Subtilisin-Kexin Type 9 Variants. Curr. Opin. Lipidol. 2017, 28, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Poirier, S.; Mayer, G.; Poupon, V.; McPherson, P.S.; Desjardins, R.; Ly, K.; Asselin, M.-C.; Day, R.; Duclos, F.J.; Witmer, M.; et al. Dissection of the Endogenous Cellular Pathways of PCSK9-Induced Low Density Lipoprotein Receptor Degradation: Evidence for an Intracellular Route. J. Biol. Chem. 2009, 284, 28856–28864. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Hernández, R.M.; Di Taranto, M.D.; Benito-Vicente, A.; Uribe, K.B.; Lamiquiz-Moneo, I.; Larrea-Sebal, A.; Jebari, S.; Galicia-Garcia, U.; Nóvoa, F.J.; Boronat, M.; et al. The Arg499His Gain-of-Function Mutation in the C-Terminal Domain of PCSK9. Atherosclerosis 2019, 289, 162–172. [Google Scholar] [CrossRef] [PubMed]
- D’Erasmo, L.; Di Costanzo, A.; Arca, M. Autosomal Recessive Hypercholesterolemia: Update for 2020. Curr. Opin. Lipidol. 2020, 31, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Won, H.-H.; Peloso, G.M.; Lawson, K.S.; Bartz, T.M.; Deng, X.; van Leeuwen, E.M.; Natarajan, P.; Emdin, C.A.; Bick, A.G.; et al. Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 2016, 67, 2578–2589. [Google Scholar] [CrossRef] [PubMed]
- Sturm, A.C.; Knowles, J.W.; Gidding, S.S.; Ahmad, Z.S.; Ahmed, C.D.; Ballantyne, C.M.; Baum, S.J.; Bourbon, M.; Carrié, A.; Cuchel, M.; et al. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J. Am. Coll. Cardiol. 2018, 72, 662–680. [Google Scholar] [CrossRef]
- Rubba, P.; Gentile, M.; Marotta, G.; Iannuzzi, A.; Sodano, M.; De Simone, B.; Jossa, F.; Iannuzzo, G.; Giacobbe, C.; Di Taranto, M.D.; et al. Causative Mutations and Premature Cardiovascular Disease in Patients with Heterozygous Familial Hypercholesterolaemia. Eur. J. Prev. Cardiol. 2017, 24, 1051–1059. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; Giacobbe, C.; Palma, D.; Iannuzzo, G.; Gentile, M.; Calcaterra, I.; Guardamagna, O.; Auricchio, R.; Di Minno, M.N.D.; Fortunato, G. Genetic Spectrum of Familial Hypercholesterolemia and Correlations with Clinical Expression: Implications for Diagnosis Improvement. Clin. Genet. 2021, 100, 529–541. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; de Falco, R.; Guardamagna, O.; Massini, G.; Giacobbe, C.; Auricchio, R.; Malamisura, B.; Proto, M.; Palma, D.; Greco, L.; et al. Lipid Profile and Genetic Status in a Familial Hypercholesterolemia Pediatric Population: Exploring the LDL/HDL Ratio. Clin. Chem. Lab. Med. 2019, 57, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzo, G.; Buonaiuto, A.; Calcaterra, I.; Gentile, M.; Forte, F.; Tripaldella, M.; Di Taranto, M.D.; Giacobbe, C.; Fortunato, G.; Rubba, P.O.; et al. Association between Causative Mutations and Response to PCSK9 Inhibitor Therapy in Subjects with Familial Hypercholesterolemia: A Single Center Real-World Study. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Di Minno, M.N.D.; Gentile, M.; Di Minno, A.; Iannuzzo, G.; Calcaterra, I.; Buonaiuto, A.; Di Taranto, M.D.; Giacobbe, C.; Fortunato, G.; Rubba, P.O.F. Changes in Carotid Stiffness in Patients with Familial Hypercholesterolemia Treated with Evolocumab®: A Prospective Cohort Study. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Tromp, T.R.; Hartgers, M.L.; Hovingh, G.K.; Vallejo-Vaz, A.J.; Ray, K.K.; Soran, H.; Freiberger, T.; Bertolini, S.; Harada-Shiba, M.; Blom, D.J.; et al. Worldwide Experience of Homozygous Familial Hypercholesterolaemia: Retrospective Cohort Study. Lancet 2022, 399, 719–728. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; Giacobbe, C.; Buonaiuto, A.; Calcaterra, I.; Palma, D.; Maione, G.; Iannuzzo, G.; Di Minno, M.N.D.; Rubba, P.; Fortunato, G. A Real-World Experience of Clinical, Biochemical and Genetic Assessment of Patients with Homozygous Familial Hypercholesterolemia. J. Clin. Med. 2020, 9, 219. [Google Scholar] [CrossRef]
- D’Erasmo, L.; Giammanco, A.; Suppressa, P.; Pavanello, C.; Iannuzzo, G.; Di Costanzo, A.; Tramontano, D.; Minicocci, I.; Bini, S.; Vogt, A.; et al. Efficacy of Long-Term Treatment of Autosomal Recessive Hypercholesterolemia With Lomitapide: A Subanalysis of the Pan-European Lomitapide Study. Front. Genet. 2022, 13, 937750. [Google Scholar] [CrossRef] [PubMed]
- Tromp, T.R.; Cuchel, M. New Algorithms for Treating Homozygous Familial Hypercholesterolemia. Curr. Opin. Lipidol. 2022, 33, 326–335. [Google Scholar] [CrossRef]
- Reeskamp, L.F.; Millar, J.S.; Wu, L.; Jansen, H.; van Harskamp, D.; Schierbeek, H.; Gipe, D.A.; Rader, D.J.; Dallinga-Thie, G.M.; Hovingh, G.K.; et al. ANGPTL3 Inhibition With Evinacumab Results in Faster Clearance of IDL and LDL ApoB in Patients With Homozygous Familial Hypercholesterolemia-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1753–1759. [Google Scholar] [CrossRef]
- Ruscica, M.; Sirtori, C.R.; Carugo, S.; Banach, M.; Corsini, A. Bempedoic Acid: For Whom and When. Curr. Atheroscler. Rep. 2022, 24, 791–801. [Google Scholar] [CrossRef]
- Averna, M.; Cefalù, A.B.; Casula, M.; Noto, D.; Arca, M.; Bertolini, S.; Calandra, S.; Catapano, A.L.; Tarugi, P. LIPIGEN Group Familial Hypercholesterolemia: The Italian Atherosclerosis Society Network (LIPIGEN). Atheroscler. Suppl. 2017, 29, 11–16. [Google Scholar] [CrossRef]
- Wang, J.; Dron, J.S.; Ban, M.R.; Robinson, J.F.; McIntyre, A.D.; Alazzam, M.; Zhao, P.J.; Dilliott, A.A.; Cao, H.; Huff, M.W.; et al. Polygenic Versus Monogenic Causes of Hypercholesterolemia Ascertained Clinically. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2439–2445. [Google Scholar] [CrossRef] [PubMed]
- Rios, J.; Stein, E.; Shendure, J.; Hobbs, H.H.; Cohen, J.C. Identification by Whole-Genome Resequencing of Gene Defect Responsible for Severe Hypercholesterolemia. Hum. Mol. Genet. 2010, 19, 4313–4318. [Google Scholar] [CrossRef] [PubMed]
- Futema, M.; Plagnol, V.; Li, K.; Whittall, R.A.; Neil, H.A.W.; Seed, M.; Simon Broome Consortium; Bertolini, S.; Calandra, S.; Descamps, O.S.; et al. Whole Exome Sequencing of Familial Hypercholesterolaemia Patients Negative for LDLR/APOB/PCSK9 Mutations. J. Med. Genet. 2014, 51, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Marques-Pinheiro, A.; Marduel, M.; Rabès, J.-P.; Devillers, M.; Villéger, L.; Allard, D.; Weissenbach, J.; Guerin, M.; Zair, Y.; Erlich, D.; et al. A Fourth Locus for Autosomal Dominant Hypercholesterolemia Maps at 16q22.1. Eur. J. Hum. Genet. 2010, 18, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Cenarro, A.; García-Otín, A.-L.; Tejedor, M.T.; Solanas, M.; Jarauta, E.; Junquera, C.; Ros, E.; Mozas, P.; Puzo, J.; Pocoví, M.; et al. A Presumptive New Locus for Autosomal Dominant Hypercholesterolemia Mapping to 8q24.22. Clin. Genet. 2011, 79, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, S.W.; Dallinga-Thie, G.M.; Meijers, J.C.M.; Zelcer, N.; Kastelein, J.J.P.; Defesche, J.C.; Hovingh, G.K. Mutations in STAP1 Are Associated with Autosomal Dominant Hypercholesterolemia. Circ. Res. 2014, 115, 552–555. [Google Scholar] [CrossRef]
- Marduel, M.; Ouguerram, K.; Serre, V.; Bonnefont-Rousselot, D.; Marques-Pinheiro, A.; Erik Berge, K.; Devillers, M.; Luc, G.; Lecerf, J.-M.; Tosolini, L.; et al. Description of a Large Family with Autosomal Dominant Hypercholesterolemia Associated with the APOE p.Leu167del Mutation. Hum. Mutat. 2013, 34, 83–87. [Google Scholar] [CrossRef]
- Wintjens, R.; Bozon, D.; Belabbas, K.; MBou, F.; Girardet, J.-P.; Tounian, P.; Jolly, M.; Boccara, F.; Cohen, A.; Karsenty, A.; et al. Global Molecular Analysis and APOE Mutations in a Cohort of Autosomal Dominant Hypercholesterolemia Patients in France. J. Lipid Res. 2016, 57, 482–491. [Google Scholar] [CrossRef]
- Sacks, F.M. The Crucial Roles of Apolipoproteins E and C-III in ApoB Lipoprotein Metabolism in Normolipidemia and Hypertriglyceridemia. Curr. Opin. Lipidol. 2015, 26, 56–63. [Google Scholar] [CrossRef]
- Khalil, Y.A.; Rabès, J.-P.; Boileau, C.; Varret, M. APOE Gene Variants in Primary Dyslipidemia. Atherosclerosis 2021, 328, 11–22. [Google Scholar] [CrossRef]
- Abou Khalil, Y.; Marmontel, O.; Ferrières, J.; Paillard, F.; Yelnik, C.; Carreau, V.; Charrière, S.; Bruckert, E.; Gallo, A.; Giral, P.; et al. APOE Molecular Spectrum in a French Cohort with Primary Dyslipidemia. Int. J. Mol. Sci. 2022, 23, 5792. [Google Scholar] [CrossRef] [PubMed]
- Civeira, F.; Jarauta, E.; Cenarro, A.; García-Otín, A.L.; Tejedor, D.; Zambón, D.; Mallen, M.; Ros, E.; Pocoví, M. Frequency of Low-Density Lipoprotein Receptor Gene Mutations in Patients with a Clinical Diagnosis of Familial Combined Hyperlipidemia in a Clinical Setting. J. Am. Coll. Cardiol. 2008, 52, 1546–1553. [Google Scholar] [CrossRef] [PubMed]
- Staiano, A.; di Taranto, M.D.; Bloise, E.; D’Agostino, M.N.; D’Angelo, A.; Marotta, G.; Gentile, M.; Jossa, F.; Iannuzzi, A.; Rubba, P.; et al. Investigation of Single Nucleotide Polymorphisms Associated to Familial Combined Hyperlipidemia with Random Forests. Smart Innov. Syst. Technol. 2013, 19, 169–178. [Google Scholar]
- Minicocci, I.; Prisco, C.; Montali, A.; Di Costanzo, A.; Ceci, F.; Pigna, G.; Arca, M. Contribution of Mutations in Low Density Lipoprotein Receptor (LDLR) and Lipoprotein Lipase (LPL) Genes to Familial Combined Hyperlipidemia (FCHL): A Reappraisal by Using a Resequencing Approach. Atherosclerosis 2015, 242, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Knowles, J.W.; Horton, J.D. Delisting STAP1: The Rise and Fall of a Putative Hypercholesterolemia Gene. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Vrablik, M.; Tichý, L.; Freiberger, T.; Blaha, V.; Satny, M.; Hubacek, J.A. Genetics of Familial Hypercholesterolemia: New Insights. Front. Genet. 2020, 11, 574474. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Liu, M.; Portincasa, P.; Wang, D.Q.-H. Recent Advances in the Critical Role of the Sterol Efflux Transporters ABCG5/G8 in Health and Disease. Adv. Exp. Med. Biol. 2020, 1276, 105–136. [Google Scholar] [CrossRef]
- Reeskamp, L.F.; Volta, A.; Zuurbier, L.; Defesche, J.C.; Hovingh, G.K.; Grefhorst, A. ABCG5 and ABCG8 Genetic Variants in Familial Hypercholesterolemia. J. Clin. Lipidol. 2020, 14, 207–217. [Google Scholar] [CrossRef]
- Stelten, B.M.L.; Raal, F.J.; Marais, A.D.; Riksen, N.P.; Roeters van Lennep, J.E.; Duell, P.B.; van der Graaf, M.; Kluijtmans, L.A.J.; Wevers, R.A.; Verrips, A. Cerebrotendinous Xanthomatosis without Neurological Involvement. J. Intern. Med. 2021, 290, 1039–1047. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; Gelzo, M.; Giacobbe, C.; Gentile, M.; Marotta, G.; Savastano, S.; Dello Russo, A.; Fortunato, G.; Corso, G. Cerebrotendinous Xanthomatosis, a Metabolic Disease with Different Neurological Signs: Two Case Reports. Metab. Brain Dis. 2016, 31, 1185–1188. [Google Scholar] [CrossRef]
- Corral, P.; Geller, A.S.; Polisecki, E.Y.; Lopez, G.I.; Bañares, V.G.; Cacciagiu, L.; Berg, G.; Hegele, R.A.; Schaefer, E.J.; Schreier, L.E. Unusual Genetic Variants Associated with Hypercholesterolemia in Argentina. Atherosclerosis 2018, 277, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Huijgen, R.; Stork, A.D.M.; Defesche, J.C.; Peter, J.; Alonso, R.; Cuevas, A.; Kastelein, J.J.P.; Duran, M.; Stroes, E.S.G. Extreme Xanthomatosis in Patients with Both Familial Hypercholesterolemia and Cerebrotendinous Xanthomatosis. Clin. Genet. 2012, 81, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Mashima, R.; Takada, S. Lysosomal Acid Lipase Deficiency: Genetics, Screening, and Preclinical Study. Int. J. Mol. Sci. 2022, 23, 15549. [Google Scholar] [CrossRef] [PubMed]
- Sustar, U.; Groselj, U.; Trebusak Podkrajsek, K.; Mlinaric, M.; Kovac, J.; Thaler, M.; Drole Torkar, A.; Skarlovnik, A.; Battelino, T.; Hovnik, T. Early Discovery of Children With Lysosomal Acid Lipase Deficiency With the Universal Familial Hypercholesterolemia Screening Program. Front. Genet. 2022, 13, 936121. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Kawashiri, M.-A.; Nomura, A.; Teramoto, R.; Hosomichi, K.; Nohara, A.; Inazu, A.; Mabuchi, H.; Tajima, A.; Yamagishi, M. Oligogenic Familial Hypercholesterolemia, LDL Cholesterol, and Coronary Artery Disease. J. Clin. Lipidol. 2018, 12, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Kawashiri, M.; Ohtani, R.; Noguchi, T.; Nakanishi, C.; Konno, T.; Hayashi, K.; Nohara, A.; Inazu, A.; Kobayashi, J.; et al. A Novel Type of Familial Hypercholesterolemia: Double Heterozygous Mutations in LDL Receptor and LDL Receptor Adaptor Protein 1 Gene. Atherosclerosis 2011, 219, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, Y.; Elbitar, S.; Philippi, A.; El Khoury, P.; Azar, Y.; Andrianirina, M.; Loste, A.; Abou-Khalil, Y.; Nicolas, G.; Le Borgne, M.; et al. Whole Exome/Genome Sequencing Joint Analysis of a Family with Oligogenic Familial Hypercholesterolemia. Metabolites 2022, 12, 262. [Google Scholar] [CrossRef]
- Nishikawa, R.; Furuhashi, M.; Hori, M.; Ogura, M.; Harada-Shiba, M.; Okada, T.; Koseki, M.; Kujiraoka, T.; Hattori, H.; Ito, R.; et al. A Resuscitated Case of Acute Myocardial Infarction with Both Familial Hypercholesterolemia Phenotype Caused by Possibly Oligogenic Variants of the PCSK9 and ABCG5 Genes and Type I CD36 Deficiency. J. Atheroscler. Thromb. 2022, 29, 551–557. [Google Scholar] [CrossRef]
- Huijgen, R.; Sjouke, B.; Vis, K.; de Randamie, J.S.E.; Defesche, J.C.; Kastelein, J.J.P.; Hovingh, G.K.; Fouchier, S.W. Genetic Variation in APOB, PCSK9, and ANGPTL3 in Carriers of Pathogenic Autosomal Dominant Hypercholesterolemic Mutations with Unexpected Low LDL-Cl Levels. Hum. Mutat. 2012, 33, 448–455. [Google Scholar] [CrossRef]
- Winther, M.; Shpitzen, S.; Yaacov, O.; Landau, J.; Oren, L.; Foroozan-Rosenberg, L.; Lev Cohain, N.; Schurr, D.; Meiner, V.; Szalat, A.; et al. In Search of a Genetic Explanation for LDLc Variability in an FH Family: Common SNPs and a Rare Mutation in MTTP Explain Only Part of LDL Variability in an FH Family. J. Lipid Res. 2019, 60, 1733–1740. [Google Scholar] [CrossRef]
- Sasaki, K.; Tada, H.; Kawashiri, M.-A.; Ito, T. Case Report: Unusual Coexistence between Familial Hypercholesterolemia and Familial Hypobetalipoproteinemia. Front. Cardiovasc. Med. 2022, 9, 942772. [Google Scholar] [CrossRef] [PubMed]
- Ellis, K.L.; Pang, J.; Chan, D.C.; Hooper, A.J.; Bell, D.A.; Burnett, J.R.; Watts, G.F. Familial Combined Hyperlipidemia and Hyperlipoprotein(a) as Phenotypic Mimics of Familial Hypercholesterolemia: Frequencies, Associations and Predictions. J. Clin. Lipidol. 2016, 10, 1329–1337.e3. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in Atherosclerotic Cardiovascular Disease and Aortic Stenosis: A European Atherosclerosis Society Consensus Statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef] [PubMed]
- Marco-Benedí, V.; Cenarro, A.; Laclaustra, M.; Larrea-Sebal, A.; Jarauta, E.; Lamiquiz-Moneo, I.; Calmarza, P.; Bea, A.M.; Plana, N.; Pintó, X.; et al. Lipoprotein(a) in Hereditary Hypercholesterolemia: Influence of the Genetic Cause, Defective Gene and Type of Mutation. Atherosclerosis 2022, 349, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Pederiva, C.; Capra, M.E.; Biasucci, G.; Banderali, G.; Fabrizi, E.; Gazzotti, M.; Casula, M.; Catapano, A.L.; LIPIGEN Paediatric Group; Members of the Lipigen Steering Committee; et al. Lipoprotein(a) and Family History for Cardiovascular Disease in Paediatric Patients: A New Frontier in Cardiovascular Risk Stratification. Data from the LIPIGEN Paediatric Group. Atherosclerosis 2022, 349, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Langsted, A.; Nordestgaard, B.G. Lipoprotein(a) as Part of the Diagnosis of Clinical Familial Hypercholesterolemia. Curr. Atheroscler. Rep. 2022, 24, 289–296. [Google Scholar] [CrossRef]
- Talmud, P.J.; Shah, S.; Whittall, R.; Futema, M.; Howard, P.; Cooper, J.A.; Harrison, S.C.; Li, K.; Drenos, F.; Karpe, F.; et al. Use of Low-Density Lipoprotein Cholesterol Gene Score to Distinguish Patients with Polygenic and Monogenic Familial Hypercholesterolaemia: A Case-Control Study. Lancet 2013, 381, 1293–1301. [Google Scholar] [CrossRef]
- Futema, M.; Shah, S.; Cooper, J.A.; Li, K.; Whittall, R.A.; Sharifi, M.; Goldberg, O.; Drogari, E.; Mollaki, V.; Wiegman, A.; et al. Refinement of Variant Selection for the LDL Cholesterol Genetic Risk Score in the Diagnosis of the Polygenic Form of Clinical Familial Hypercholesterolemia and Replication in Samples from 6 Countries. Clin. Chem. 2015, 61, 231–238. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.; Telkar, N.; Reiker, T.; Walters, R.G.; Lin, K.; Eriksson, A.; Gurdasani, D.; Gilly, A.; Southam, L.; Tsafantakis, E.; et al. The Transferability of Lipid Loci across African, Asian and European Cohorts. Nat. Commun. 2019, 10, 4330. [Google Scholar] [CrossRef]
- Vanhoye, X.; Bardel, C.; Rimbert, A.; Moulin, P.; Rollat-Farnier, P.-A.; Muntaner, M.; Marmontel, O.; Dumont, S.; Charrière, S.; Cornélis, F.; et al. A New 165-SNP Low-Density Lipoprotein Cholesterol Polygenic Risk Score Based on next Generation Sequencing Outperforms Previously Published Scores in Routine Diagnostics of Familial Hypercholesterolemia. Transl. Res. 2022, S1931-5244(22)00281-X. [Google Scholar] [CrossRef]
- Natarajan, P.; Peloso, G.M.; Zekavat, S.M.; Montasser, M.; Ganna, A.; Chaffin, M.; Khera, A.V.; Zhou, W.; Bloom, J.M.; Engreitz, J.M.; et al. Deep-Coverage Whole Genome Sequences and Blood Lipids among 16,324 Individuals. Nat. Commun. 2018, 9, 3391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinder, M.; Paquette, M.; Cermakova, L.; Ban, M.R.; Hegele, R.A.; Baass, A.; Brunham, L.R. Polygenic Contribution to Low-Density Lipoprotein Cholesterol Levels and Cardiovascular Risk in Monogenic Familial Hypercholesterolemia. Circ. Genom. Precis. Med. 2020, 13, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, L.; Pinkier, I.; Sałacińska, K.; Kępczyński, Ł.; Salachna, D.; Lewek, J.; Banach, M.; Matusik, P.; Starostecka, E.; Lewiński, A.; et al. Identification of New Copy Number Variation and the Evaluation of a CNV Detection Tool for NGS Panel Data in Polish Familial Hypercholesterolemia Patients. Genes 2022, 13, 1424. [Google Scholar] [CrossRef] [PubMed]
- Iacocca, M.A.; Wang, J.; Sarkar, S.; Dron, J.S.; Lagace, T.; McIntyre, A.D.; Lau, P.; Robinson, J.F.; Yang, P.; Knoll, J.H.; et al. Whole-Gene Duplication of PCSK9 as a Novel Genetic Mechanism for Severe Familial Hypercholesterolemia. Can. J. Cardiol. 2018, 34, 1316–1324. [Google Scholar] [CrossRef]
- Selvaraj, M.S.; Li, X.; Li, Z.; Pampana, A.; Zhang, D.Y.; Park, J.; Aslibekyan, S.; Bis, J.C.; Brody, J.A.; Cade, B.E.; et al. Whole Genome Sequence Analysis of Blood Lipid Levels in >66,000 Individuals. Nat. Commun. 2022, 13, 5995. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Chora, J.R.; Medeiros, A.M.; Alves, A.C.; Bourbon, M. Analysis of Publicly Available LDLR, APOB, and PCSK9 Variants Associated with Familial Hypercholesterolemia: Application of ACMG Guidelines and Implications for Familial Hypercholesterolemia Diagnosis. Genet. Med. 2018, 20, 591–598. [Google Scholar] [CrossRef]
- Chora, J.R.; Iacocca, M.A.; Tichý, L.; Wand, H.; Kurtz, C.L.; Zimmermann, H.; Leon, A.; Williams, M.; Humphries, S.E.; Hooper, A.J.; et al. The Clinical Genome Resource (ClinGen) Familial Hypercholesterolemia Variant Curation Expert Panel Consensus Guidelines for LDLR Variant Classification. Genet. Med. 2022, 24, 293–306. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; D’Agostino, M.N.; Fortunato, G. Functional Characterization of Mutant Genes Associated with Autosomal Dominant Familial Hypercholesterolemia: Integration and Evolution of Genetic Diagnosis. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 979–987. [Google Scholar] [CrossRef]
- Iacocca, M.A.; Chora, J.R.; Carrié, A.; Freiberger, T.; Leigh, S.E.; Defesche, J.C.; Kurtz, C.L.; DiStefano, M.T.; Santos, R.D.; Humphries, S.E.; et al. ClinVar Database of Global Familial Hypercholesterolemia-Associated DNA Variants. Hum. Mutat. 2018, 39, 1631–1640. [Google Scholar] [CrossRef]
- Barbosa, T.K.A.; Hirata, R.D.C.; Ferreira, G.M.; Borges, J.B.; Oliveira, V.F.; Gorjão, R.; da Silva Marçal, E.R.; Gonçalves, R.M.; Faludi, A.A.; Freitas, R.C.C.; et al. LDLR Missense Variants Disturb Structural Conformation and LDLR Activity in T-Lymphocytes of Familial Hypercholesterolemia Patients. Gene 2022, 853, 147084. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Di Taranto, M.D.; Mirabelli, P.; D’Agostino, M.N.; Iannuzzi, A.; Marotta, G.; Gentile, M.; Raia, M.; Di Noto, R.; Del Vecchio, L.; et al. An Improved Method on Stimulated T-Lymphocytes to Functionally Characterize Novel and Known LDLR Mutations. J. Lipid Res. 2011, 52, 2095–2100. [Google Scholar] [CrossRef] [PubMed]
- Di Taranto, M.D.; Benito-Vicente, A.; Giacobbe, C.; Uribe, K.B.; Rubba, P.; Etxebarria, A.; Guardamagna, O.; Gentile, M.; Martín, C.; Fortunato, G. Identification and in Vitro Characterization of Two New PCSK9 Gain of Function Variants Found in Patients with Familial Hypercholesterolemia. Sci. Rep. 2017, 7, 15282. [Google Scholar] [CrossRef] [PubMed]
- Ruotolo, A.; Di Taranto, M.D.; D’Agostino, M.N.; Marotta, G.; Gentile, M.; Nunziata, M.; Sodano, M.; Di Noto, R.; Del Vecchio, L.; Rubba, P.; et al. The Novel Variant p.Ser465Leu in the PCSK9 Gene Does Not Account for the Decreased LDLR Activity in Members of a FH Family. Clin. Chem. Lab. Med. 2014, 52, e175–e178. [Google Scholar] [CrossRef] [PubMed]
- Pfisterer, S.G.; Brock, I.; Kanerva, K.; Hlushchenko, I.; Paavolainen, L.; Ripatti, P.; Islam, M.M.; Kyttälä, A.; Di Taranto, M.D.; Scotto di Frega, A.; et al. Multiparametric Platform for Profiling Lipid Trafficking in Human Leukocytes. Cell Rep. Methods 2022, 2, 100166. [Google Scholar] [CrossRef]
- Larrea-Sebal, A.; Benito-Vicente, A.; Fernandez-Higuero, J.A.; Jebari-Benslaiman, S.; Galicia-Garcia, U.; Uribe, K.B.; Cenarro, A.; Ostolaza, H.; Civeira, F.; Arrasate, S.; et al. MLb-LDLr: A Machine Learning Model for Predicting the Pathogenicity of LDLr Missense Variants. JACC Basic Transl. Sci. 2021, 6, 815–827. [Google Scholar] [CrossRef]
- Camastra, F.; Di Taranto, M.D.; Staiano, A. Statistical and Computational Methods for Genetic Diseases: An Overview. Comput. Math. Methods Med. 2015, 2015, 954598. [Google Scholar] [CrossRef]

{kind=link}
| Hypercholesterolemia Form | Genes | Genetic Status |
|---|---|---|
| Homozygous FH | LDLR, APOB, PCSK9, LDLRAP1 | at homozygous status |
| at compound heterozygous status | ||
| at double heterozygous status | ||
| Oligogenic FH | LDLR, APOB, PCSK9 | at heterozygous status |
| and | ||
| ABCG5, ABCG8 or other modifier genes | at heterozygous status | |
| Heterozygous FH | LDLR, APOB, PCSK9, APOE genes | heterozygous status |
| Polygenic hypercholesterolemia | Multiple | combination of heterozygous and homozygous variant according to determined score attribution |
| Resource | Available Information | Website Link |
|---|---|---|
| ClinVar | Previous identification of variants and related condition; number of patients with the variant; pathogenicity evaluation and related evidence; literature references | https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 29 December 2022) |
| ClinGen curated variants in ClinVar | FH-related variants curated by the ClinGen Familial Hypercholesterolemia Variant Curation Expert Panel | https://www.ncbi.nlm.nih.gov/clinvar/submitters/508055/ (accessed on 29 December 2022) |
| ClinGen Familial Hypercholesterolemia Variant Curation Expert Panel | Revised pathogenicity criteria for variants in FH-causative variants; Evidence Repository about analyzed variants | https://www.clinicalgenome.org/affiliation/50004 (accessed on 29 December 2022) |
| Human Gene Mutation Database (HGMD) | Database of variants associated with different diseases; literature references; bioinformatic predictions | https://www.hgmd.cf.ac.uk/ac/index.php (accessed on 29 December 2022) |
| LOVD 3.0 | Database of variants associated with different diseases; literature references; functional data; bioinformatic predictions | https://www.lovd.nl/3.0/home (accessed on 29 December 2022) |
| LitVar | Search instrument to retrieve information from scientific literature | https://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/LitVar/#!?query= (accessed on 29 December 2022) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Taranto, M.D.; Fortunato, G. Genetic Heterogeneity of Familial Hypercholesterolemia: Repercussions for Molecular Diagnosis. Int. J. Mol. Sci. 2023, 24, 3224. https://doi.org/10.3390/ijms24043224
Di Taranto MD, Fortunato G. Genetic Heterogeneity of Familial Hypercholesterolemia: Repercussions for Molecular Diagnosis. International Journal of Molecular Sciences. 2023; 24(4):3224. https://doi.org/10.3390/ijms24043224
Chicago/Turabian StyleDi Taranto, Maria Donata, and Giuliana Fortunato. 2023. "Genetic Heterogeneity of Familial Hypercholesterolemia: Repercussions for Molecular Diagnosis" International Journal of Molecular Sciences 24, no. 4: 3224. https://doi.org/10.3390/ijms24043224
APA StyleDi Taranto, M. D., & Fortunato, G. (2023). Genetic Heterogeneity of Familial Hypercholesterolemia: Repercussions for Molecular Diagnosis. International Journal of Molecular Sciences, 24(4), 3224. https://doi.org/10.3390/ijms24043224