

Extracellular Histone-Induced Protein Kinase C Alpha Activation and Troponin Phosphorylation Is a Potential Mechanism of Cardiac Contractility Depression in Sepsis
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
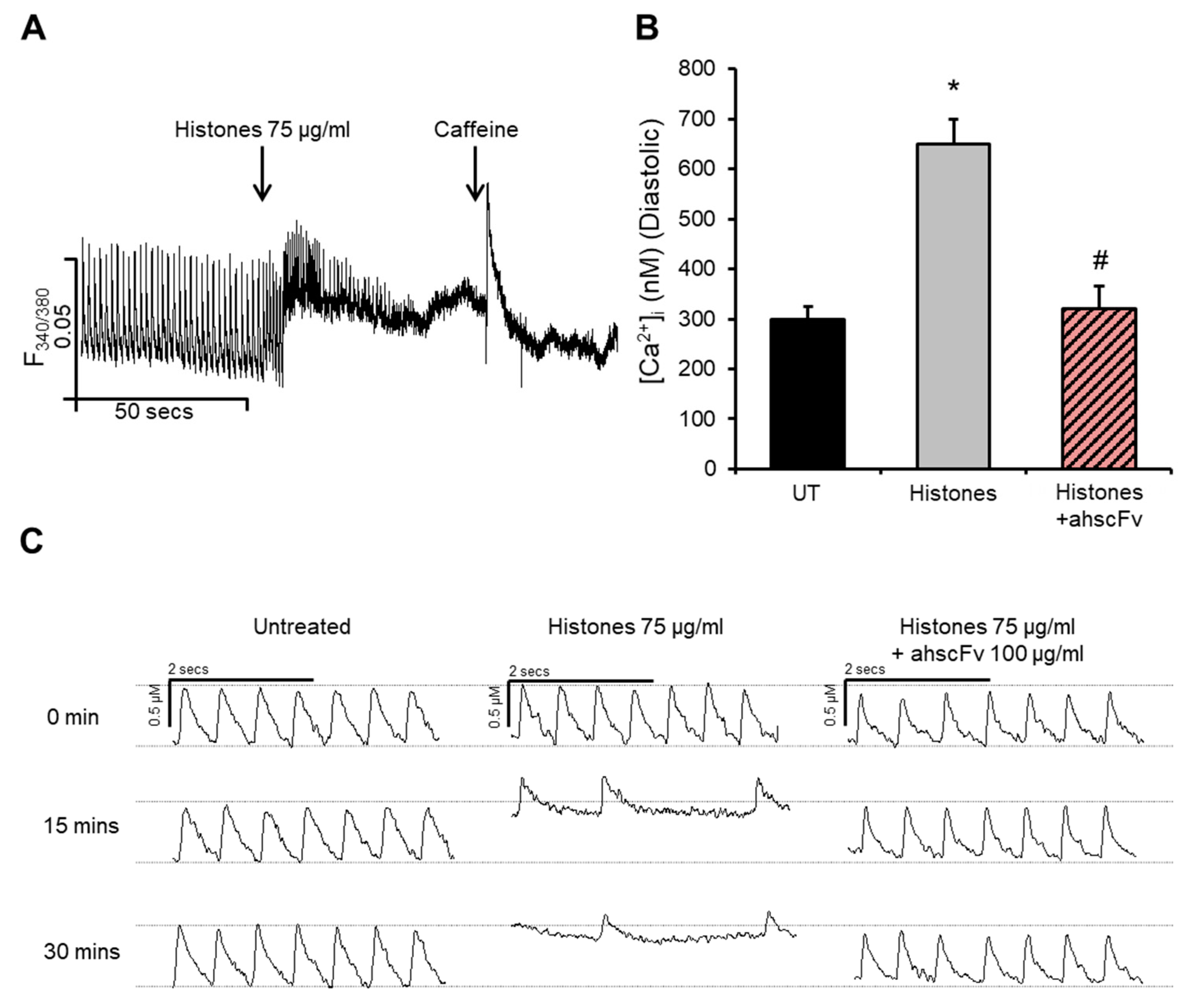
2.1. Histones Disturb Intracellular Calcium Dynamics
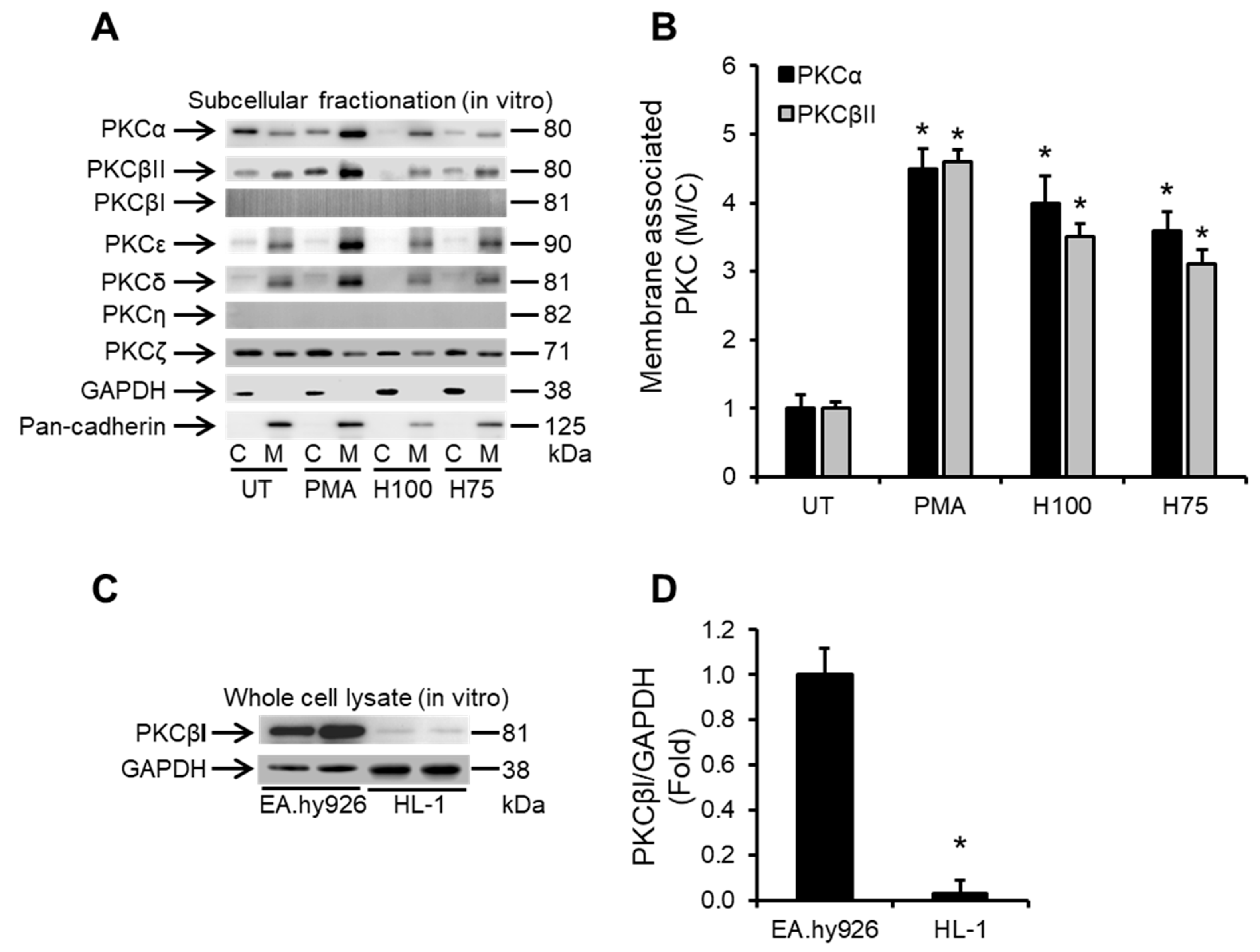
2.2. Histones Induce Ca2+-Dependent PKC Isoform Activation in Cardiomyocytes
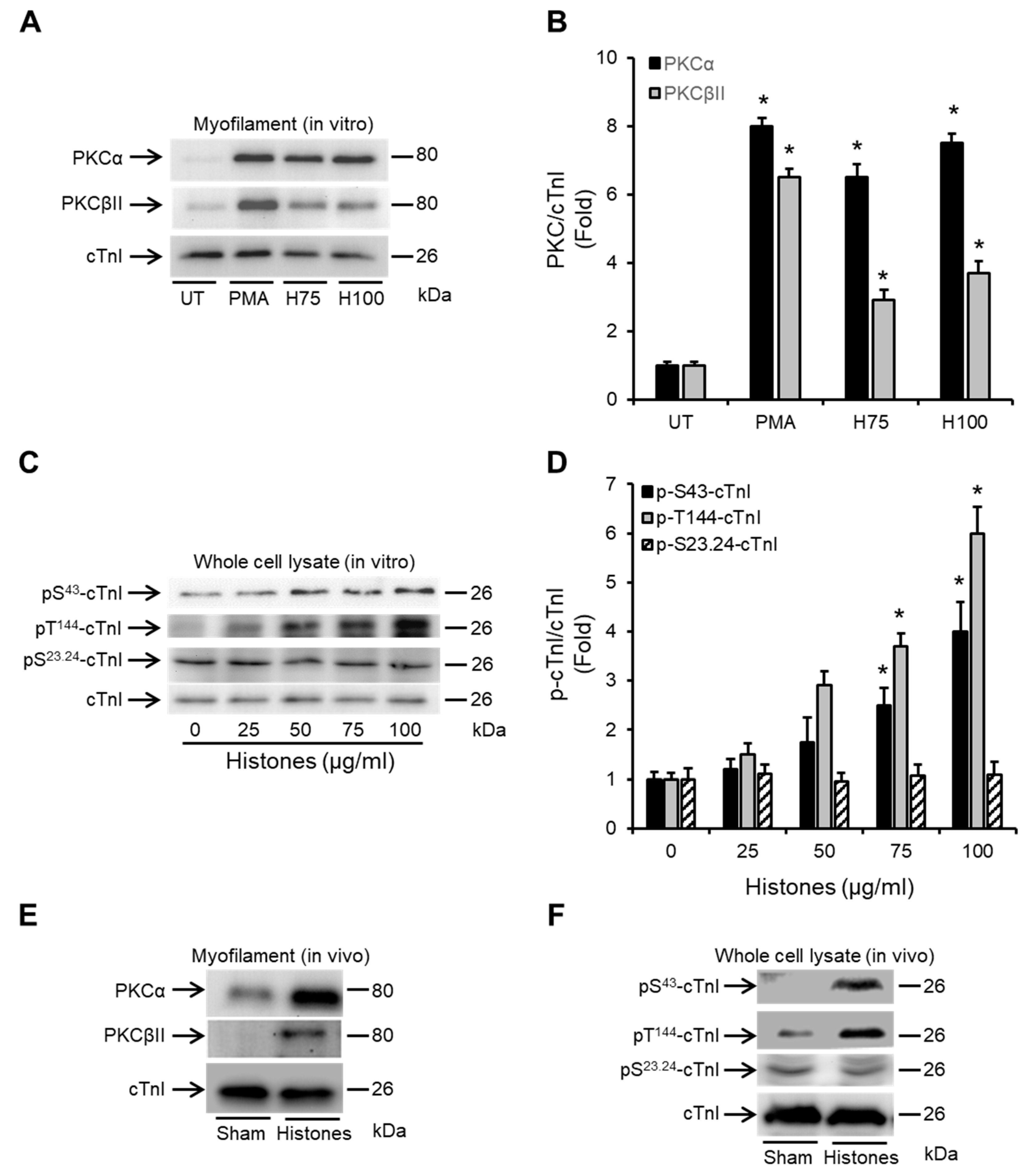
2.3. Histones Enhance the Association of PKCα and PKCβII with Cardiac Myofilaments and Induce cTnI Phosphorylation
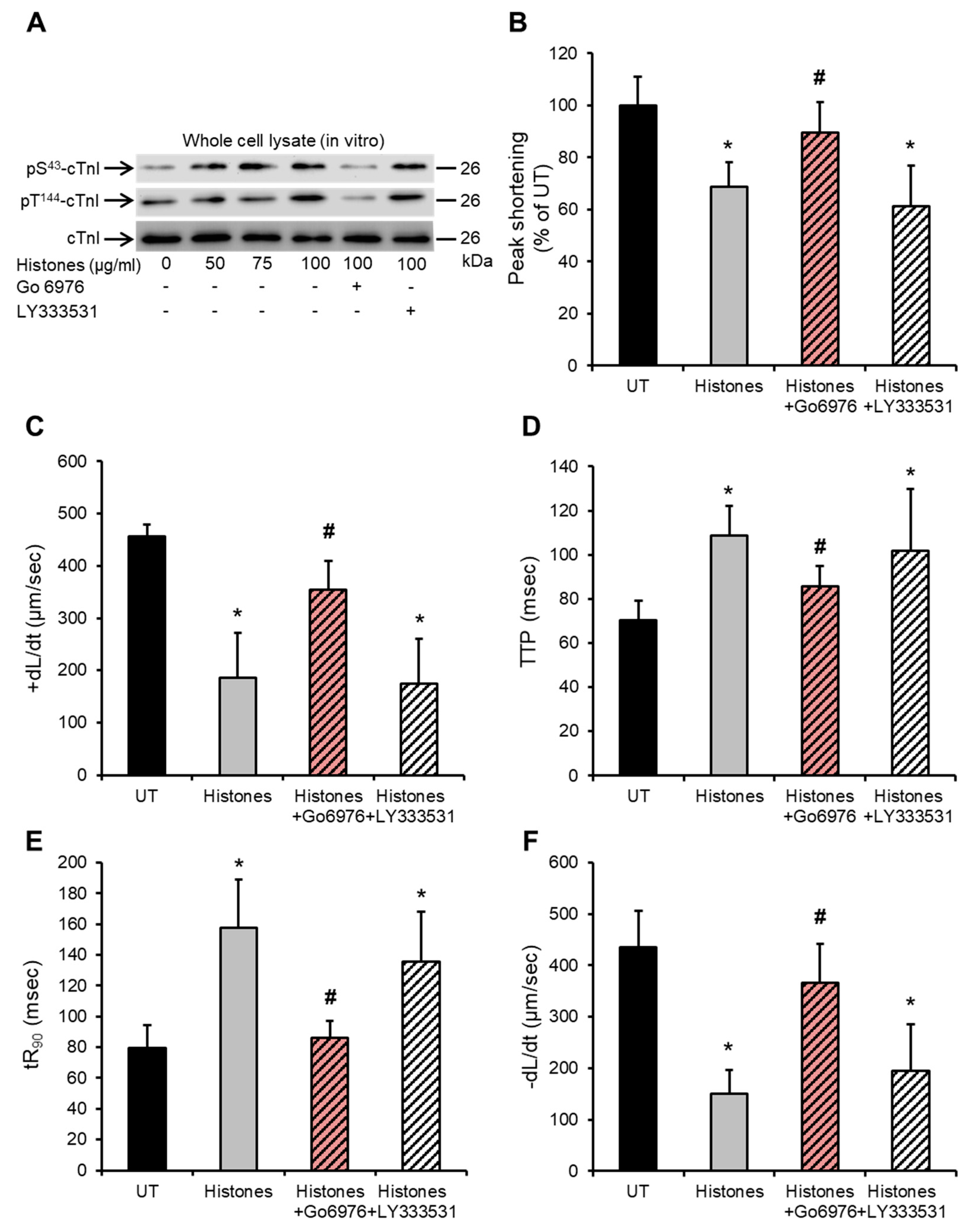
2.4. Histone-Induced cTnI Phosphorylation Is Mediated by PKCα and Contributes to Depressed Cardiomyocyte Contractility
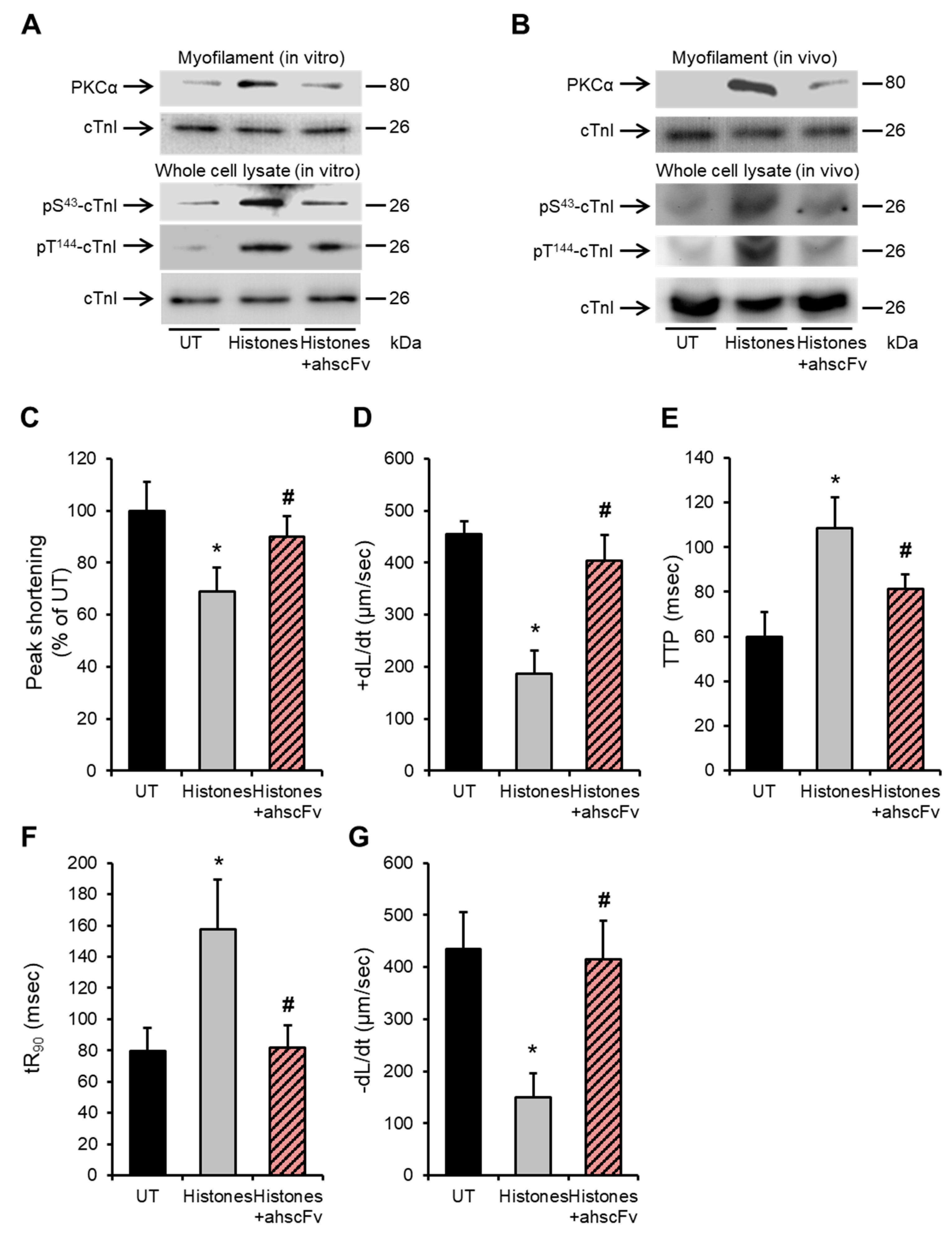
2.5. Anti-Histone Antibody Abrogates Histone-Induced Activation of PKCα-cTnI Pathway to Rescue Cardiomyocyte Contractility
3. Discussion
4. Materials and Methods
4.1. HL-1 Cardiomyocyte Culture
4.2. Animal Experiments
4.3. Ca2+ Flux Measurement in HL-1 Cardiomyocytes
4.4. Measurement of HL-1 Cardiomyocytes Contractility
4.5. Whole Cell Lysates Preparation of HL-1 and Mouse Cardiomyocytes
4.6. Subcellular Fractionation of HL-1 and Mouse Cardiomyocytes
4.7. Western Blotting
4.8. Development of Anti-Histone Single Chain Variable Fragment Anti-Body (ahscFv)
4.9. Culture of Endothelial Cells
4.10. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fleischmann, C.; Scherag, A.; Adhikari, N.K.; Hartog, C.S.; Tsaganos, T.; Schlattmann, P.; Angus, D.C.; Reinhart, K.; International Forum of Acute Care Trialists. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Prest, J.; Sathananthan, M.; Jeganathan, N. Current Trends in Sepsis-Related Mortality in the United States. Crit Care Med. 2021, 49, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Kakihana, Y.; Ito, T.; Nakahara, M.; Yamaguchi, K.; Yasuda, T. Sepsis-induced myocardial dysfunction: Pathophysiology and management. J. Intensive Care 2016, 4, 22. [Google Scholar] [CrossRef]
- Walley, K.R. Sepsis-induced myocardial dysfunction. Curr. Opin. Crit. Care 2018, 24, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Habimana, R.; Choi, I.; Cho, H.J.; Kim, D.; Lee, K.; Jeong, I. Sepsis-induced cardiac dysfunction: A review of pathophysiology. Acute Crit. Care 2020, 35, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Gentile, L.F.; Moldawer, L.L. DAMPs, PAMPs, and the origins of SIRS in bacterial sepsis. Shock 2013, 39, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Raymond, S.L.; Holden, D.C.; Mira, J.C.; Stortz, J.A.; Loftus, T.J.; Mohr, A.M.; Moldawer, L.L.; Moore, F.A.; Larson, S.D.; Efron, P.A. Microbial recognition and danger signals in sepsis and trauma. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2564–2573. [Google Scholar] [CrossRef] [PubMed]
- Alhamdi, Y.; Neill, D.R.; Abrams, S.T.; Malak, H.A.; Yahya, R.; Barrett-Jolley, R.; Wang, G.; Kadioglu, A.; Toh, C.H. Circulating Pneumolysin Is a Potent Inducer of Cardiac Injury during Pneumococcal Infection. PLoS Pathog. 2015, 11, e1004836. [Google Scholar] [CrossRef] [PubMed]
- Denning, N.L.; Aziz, M.; Gurien, S.D.; Wang, P. DAMPs and NETs in Sepsis. Front. Immunol. 2019, 10, 2536. [Google Scholar] [CrossRef]
- Alhamdi, Y.; Abrams, S.T.; Cheng, Z.; Jing, S.; Su, D.; Liu, Z.; Lane, S.; Welters, I.; Wang, G.; Toh, C.H. Circulating Histones Are Major Mediators of Cardiac Injury in Patients With Sepsis. Crit. Care Med. 2015, 43, 2094–2103. [Google Scholar] [CrossRef]
- Pathan, N.; Hemingway, C.A.; Alizadeh, A.A.; Stephens, A.C.; Boldrick, J.C.; Oragui, E.E.; McCabe, C.; Welch, S.B.; Whitney, A.; O’Gara, P.; et al. Role of interleukin 6 in myocardial dysfunction of meningococcal septic shock. Lancet 2004, 363, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Morin, E.E.; Guo, L.; Schwendeman, A.; Li, X.A. HDL in sepsis—Risk factor and therapeutic approach. Front. Pharmacol. 2015, 6, 244. [Google Scholar] [CrossRef] [PubMed]
- De Geest, B.; Mishra, M. Role of high-density lipoproteins in cardioprotection and in reverse remodeling: Therapeutic implications. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 159022. [Google Scholar] [CrossRef] [PubMed]
- De Geest, B.; Mishra, M. Impact of High-Density Lipoproteins on Sepsis. Int. J. Mol. Sci. 2022, 23, 12965. [Google Scholar] [CrossRef]
- Shah, M.; He, Z.; Rauf, A.; Beikoghli Kalkhoran, S.; Heiestad, C.M.; Stenslokken, K.O.; Parish, C.R.; Soehnlein, O.; Arjun, S.; Davidson, S.M.; et al. Extracellular histones are a target in myocardial ischaemia-reperfusion injury. Cardiovasc. Res. 2022, 118, 1115–1125. [Google Scholar] [CrossRef]
- Cheng, Z.; Abrams, S.T.; Alhamdi, Y.; Toh, J.; Yu, W.; Toh, C.; Wang, G. Circulating histones are major mediators of multiple organ dysfunction syndrome in acute critical illnesses. Crit. Care Med. 2019, 47, e677–e684. [Google Scholar] [CrossRef]
- Kalbitz, M.; Grailer, J.J.; Fattahi, F.; Jajou, L.; Herron, T.J.; Campbell, K.F.; Zetoune, F.S.; Bosmann, M.; Sarma, J.V.; Huber-Lang, M.; et al. Role of extracellular histones in the cardiomyopathy of sepsis. FASEB J. 2015, 29, 2185–2193. [Google Scholar] [CrossRef]
- Ramakrishnan, V. Histone structure and the organization of the nucleosome. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 83–112. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef]
- Ekaney, M.L.; Otto, G.P.; Sossdorf, M.; Sponholz, C.; Boehringer, M.; Loesche, W.; Rittirsch, D.; Wilharm, A.; Kurzai, O.; Bauer, M.; et al. Impact of plasma histones in human sepsis and their contribution to cellular injury and inflammation. Crit. Care 2014, 18, 543. [Google Scholar] [CrossRef]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating histones are mediators of trauma-associated lung injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef]
- Kutcher, M.E.; Xu, J.; Vilardi, R.F.; Ho, C.; Esmon, C.T.; Cohen, M.J. Extracellular histone release in response to traumatic injury: Implications for a compensatory role of activated protein C. J. Trauma Acute Care Surg. 2012, 73, 1389–1394. [Google Scholar] [CrossRef]
- Ou, X.; Cheng, Z.; Liu, T.; Tang, Z.; Huang, W.; Szatmary, P.; Zheng, S.; Sutton, R.; Toh, C.H.; Zhang, N.; et al. Circulating Histone Levels Reflect Disease Severity in Animal Models of Acute Pancreatitis. Pancreas 2015, 44, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Allam, R.; Kumar, S.V.; Darisipudi, M.N.; Anders, H.J. Extracellular histones in tissue injury and inflammation. J. Mol. Med. 2014, 92, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Alhamdi, Y.; Zi, M.; Abrams, S.T.; Liu, T.; Su, D.; Welters, I.; Dutt, T.; Cartwright, E.J.; Wang, G.; Toh, C.H. Circulating Histone Concentrations Differentially Affect the Predominance of Left or Right Ventricular Dysfunction in Critical Illness. Crit. Care Med. 2016, 44, e278–e288. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Nasu, M. A review of sepsis-induced cardiomyopathy. J. Intensive Care 2015, 3, 48. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium fluxes involved in control of cardiac myocyte contraction. Circ. Res. 2000, 87, 275–281. [Google Scholar] [CrossRef]
- Marks, A.R. Calcium and the heart: A question of life and death. J. Clin. Investig. 2003, 111, 597–600. [Google Scholar] [CrossRef]
- Vassalle, M.; Lin, C.I. Calcium overload and cardiac function. J. Biomed. Sci. 2004, 11, 542–565. [Google Scholar] [CrossRef]
- Braz, J.C.; Gregory, K.; Pathak, A.; Zhao, W.; Sahin, B.; Klevitsky, R.; Kimball, T.F.; Lorenz, J.N.; Nairn, A.C.; Liggett, S.B.; et al. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat. Med. 2004, 10, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Parmacek, M.S.; Solaro, R.J. Biology of the troponin complex in cardiac myocytes. Prog. Cardiovasc. Dis. 2004, 47, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Solaro, R.J. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu. Rev. Physiol. 2005, 67, 39–67. [Google Scholar] [CrossRef] [PubMed]
- Layland, J.; Solaro, R.J.; Shah, A.M. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc. Res. 2005, 66, 12–21. [Google Scholar] [CrossRef]
- Metzger, J.M.; Westfall, M.V. Covalent and noncovalent modification of thin filament action: The essential role of troponin in cardiac muscle regulation. Circ. Res. 2004, 94, 146–158. [Google Scholar] [CrossRef]
- Sumandea, M.P.; Burkart, E.M.; Kobayashi, T.; De Tombe, P.P.; Solaro, R.J. Molecular and integrated biology of thin filament protein phosphorylation in heart muscle. Ann. NY Acad. Sci. 2004, 1015, 39–52. [Google Scholar] [CrossRef]
- Kachooei, E.; Cordina, N.M.; Potluri, P.R.; Guse, J.A.; McCamey, D.; Brown, L.J. Phosphorylation of Troponin I finely controls the positioning of Troponin for the optimal regulation of cardiac muscle contraction. J. Mol. Cell Cardiol. 2021, 150, 44–53. [Google Scholar] [CrossRef]
- Pavadai, E.; Rynkiewicz, M.J.; Yang, Z.; Gould, I.R.; Marston, S.B.; Lehman, W. Modulation of cardiac thin filament structure by phosphorylated troponin-I analyzed by protein-protein docking and molecular dynamics simulation. Arch. Biochem. Biophys. 2022, 725, 109282. [Google Scholar] [CrossRef]
- Qvit, N.; Lin, A.J.; Elezaby, A.; Ostberg, N.P.; Campos, J.C.; Ferreira, J.C.B.; Mochly-Rosen, D. A Selective Inhibitor of Cardiac Troponin I Phosphorylation by Delta Protein Kinase C (deltaPKC) as a Treatment for Ischemia-Reperfusion Injury. Pharmaceuticals 2022, 15, 271. [Google Scholar] [CrossRef]
- Wu, L.L.; Tang, C.; Liu, M.S. Altered phosphorylation and calcium sensitivity of cardiac myofibrillar proteins during sepsis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R408–R416. [Google Scholar] [CrossRef]
- Kooij, V.; Zhang, P.; Piersma, S.R.; Sequeira, V.; Boontje, N.M.; Wijnker, P.J.; Jimenez, C.R.; Jaquet, K.E.; dos Remedios, C.; Murphy, A.M.; et al. PKCalpha-specific phosphorylation of the troponin complex in human myocardium: A functional and proteomics analysis. PLoS ONE 2013, 8, e74847. [Google Scholar] [CrossRef] [PubMed]
- Tavernier, B.; Li, J.M.; El-Omar, M.M.; Lanone, S.; Yang, Z.K.; Trayer, I.P.; Mebazaa, A.; Shah, A.M. Cardiac contractile impairment associated with increased phosphorylation of troponin I in endotoxemic rats. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Tveita, T.; Arteaga, G.M.; Han, Y.S.; Sieck, G.C. Cardiac troponin-I phosphorylation underlies myocardial contractile dysfunction induced by hypothermia rewarming. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H726–H731. [Google Scholar] [CrossRef] [PubMed]
- Bodor, G.S.; Oakeley, A.E.; Allen, P.D.; Crimmins, D.L.; Ladenson, J.H.; Anderson, P.A. Troponin I phosphorylation in the normal and failing adult human heart. Circulation 1997, 96, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Belmonte, S.L.; Blaxall, B.C. PKC-ing is believing: Targeting protein kinase C in heart failure. Circ. Res. 2011, 109, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef]
- Wolf, M.; Cuatrecasas, P.; Sahyoun, N. Interaction of protein kinase C with membranes is regulated by Ca2+, phorbol esters, and ATP. J. Biol. Chem. 1985, 260, 15718–15722. [Google Scholar] [CrossRef]
- Way, K.J.; Chou, E.; King, G.L. Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol. Sci. 2000, 21, 181–187. [Google Scholar] [CrossRef]
- Steinberg, S.F. Cardiac actions of protein kinase C isoforms. Physiology 2012, 27, 130–139. [Google Scholar] [CrossRef]
- Takeishi, Y.; Chu, G.; Kirkpatrick, D.M.; Li, Z.; Wakasaki, H.; Kranias, E.G.; King, G.L.; Walsh, R.A. In vivo phosphorylation of cardiac troponin I by protein kinase Cbeta2 decreases cardiomyocyte calcium responsiveness and contractility in transgenic mouse hearts. J. Clin. Investig. 1998, 102, 72–78. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, X.; Macdonnell, S.M.; Kranias, E.G.; Lorenz, J.N.; Leitges, M.; Houser, S.R.; Molkentin, J.D. Protein kinase C{alpha}, but not PKC{beta} or PKC{gamma}, regulates contractility and heart failure susceptibility: Implications for ruboxistaurin as a novel therapeutic approach. Circ. Res. 2009, 105, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Grant, J.E.; Doede, C.M.; Sadayappan, S.; Robbins, J.; Walker, J.W. PKC-betaII sensitizes cardiac myofilaments to Ca2+ by phosphorylating troponin I on threonine-144. J. Mol. Cell Cardiol. 2006, 41, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Molkentin, J.D. Protein kinase Calpha as a heart failure therapeutic target. J. Mol. Cell Cardiol. 2011, 51, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Biesiadecki, B.J.; Westfall, M.V. Troponin I modulation of cardiac performance: Plasticity in the survival switch. Arch. Biochem. Biophys. 2019, 664, 9–14. [Google Scholar] [CrossRef]
- Lang, S.E.; Stevenson, T.K.; Schatz, T.M.; Biesiadecki, B.J.; Westfall, M.V. Functional communication between PKC-targeted cardiac troponin I phosphorylation sites. Arch. Biochem. Biophys. 2017, 627, 1–9. [Google Scholar] [CrossRef]
- Takimoto, E.; Soergel, D.G.; Janssen, P.M.; Stull, L.B.; Kass, D.A.; Murphy, A.M. Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circ. Res. 2004, 94, 496–504. [Google Scholar] [CrossRef]
- Yasuda, S.; Coutu, P.; Sadayappan, S.; Robbins, J.; Metzger, J.M. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ. Res. 2007, 101, 377–386. [Google Scholar] [CrossRef]
- Lang, S.E.; Schwank, J.; Stevenson, T.K.; Jensen, M.A.; Westfall, M.V. Independent modulation of contractile performance by cardiac troponin I Ser43 and Ser45 in the dynamic sarcomere. J. Mol. Cell Cardiol. 2015, 79, 264–274. [Google Scholar] [CrossRef]
- Kirk, J.A.; MacGowan, G.A.; Evans, C.; Smith, S.H.; Warren, C.M.; Mamidi, R.; Chandra, M.; Stewart, A.F.; Solaro, R.J.; Shroff, S.G. Left ventricular and myocardial function in mice expressing constitutively pseudophosphorylated cardiac troponin I. Circ. Res. 2009, 105, 1232–1239. [Google Scholar] [CrossRef]
- Burkart, E.M.; Sumandea, M.P.; Kobayashi, T.; Nili, M.; Martin, A.F.; Homsher, E.; Solaro, R.J. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J. Biol. Chem. 2003, 278, 11265–11272. [Google Scholar] [CrossRef]
- Chaudary, N.; Naydenova, Z.; Shuralyova, I.; Coe, I.R. Hypoxia regulates the adenosine transporter, mENT1, in the murine cardiomyocyte cell line, HL-1. Cardiovasc. Res. 2004, 61, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.H.; Kan, B.; Roberts, H.; Wang, Y.; Walley, K.R. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ. Res. 2008, 102, 1239–1246. [Google Scholar] [CrossRef]
- Streng, A.S.; Jacobs, L.H.; Schwenk, R.W.; Cardinaels, E.P.; Meex, S.J.; Glatz, J.F.; Wodzig, W.K.; van Dieijen-Visser, M.P. Cardiac troponin in ischemic cardiomyocytes: Intracellular decrease before onset of cell death. Exp. Mol. Pathol. 2014, 96, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Andersson, H.; Kagedal, B.; Mandenius, C.F. Monitoring of troponin release from cardiomyocytes during exposure to toxic substances using surface plasmon resonance biosensing. Anal. Bioanal. Chem. 2010, 398, 1395–1402. [Google Scholar] [CrossRef]
- Steinberg, S.F. Decoding the Cardiac Actions of Protein Kinase D Isoforms. Mol. Pharmacol. 2021, 100, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Monestier, M.; Fasy, T.M.; Losman, M.J.; Novick, K.E.; Muller, S. Structure and binding properties of monoclonal antibodies to core histones from autoimmune mice. Mol. Immunol. 1993, 30, 1069–1075. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abrams, S.T.; Alhamdi, Y.; Zi, M.; Guo, F.; Du, M.; Wang, G.; Cartwright, E.J.; Toh, C.-H. Extracellular Histone-Induced Protein Kinase C Alpha Activation and Troponin Phosphorylation Is a Potential Mechanism of Cardiac Contractility Depression in Sepsis. Int. J. Mol. Sci. 2023, 24, 3225. https://doi.org/10.3390/ijms24043225
Abrams ST, Alhamdi Y, Zi M, Guo F, Du M, Wang G, Cartwright EJ, Toh C-H. Extracellular Histone-Induced Protein Kinase C Alpha Activation and Troponin Phosphorylation Is a Potential Mechanism of Cardiac Contractility Depression in Sepsis. International Journal of Molecular Sciences. 2023; 24(4):3225. https://doi.org/10.3390/ijms24043225
Chicago/Turabian StyleAbrams, Simon T., Yasir Alhamdi, Min Zi, Fengmei Guo, Min Du, Guozheng Wang, Elizabeth J. Cartwright, and Cheng-Hock Toh. 2023. "Extracellular Histone-Induced Protein Kinase C Alpha Activation and Troponin Phosphorylation Is a Potential Mechanism of Cardiac Contractility Depression in Sepsis" International Journal of Molecular Sciences 24, no. 4: 3225. https://doi.org/10.3390/ijms24043225