
Pan-Plastome of Greater Yam (Dioscorea alata) in China: Intraspecific Genetic Variation, Comparative Genomics, and Phylogenetic Analyses

 , ,
, ,
Abstract
:1. Introduction
2. Results
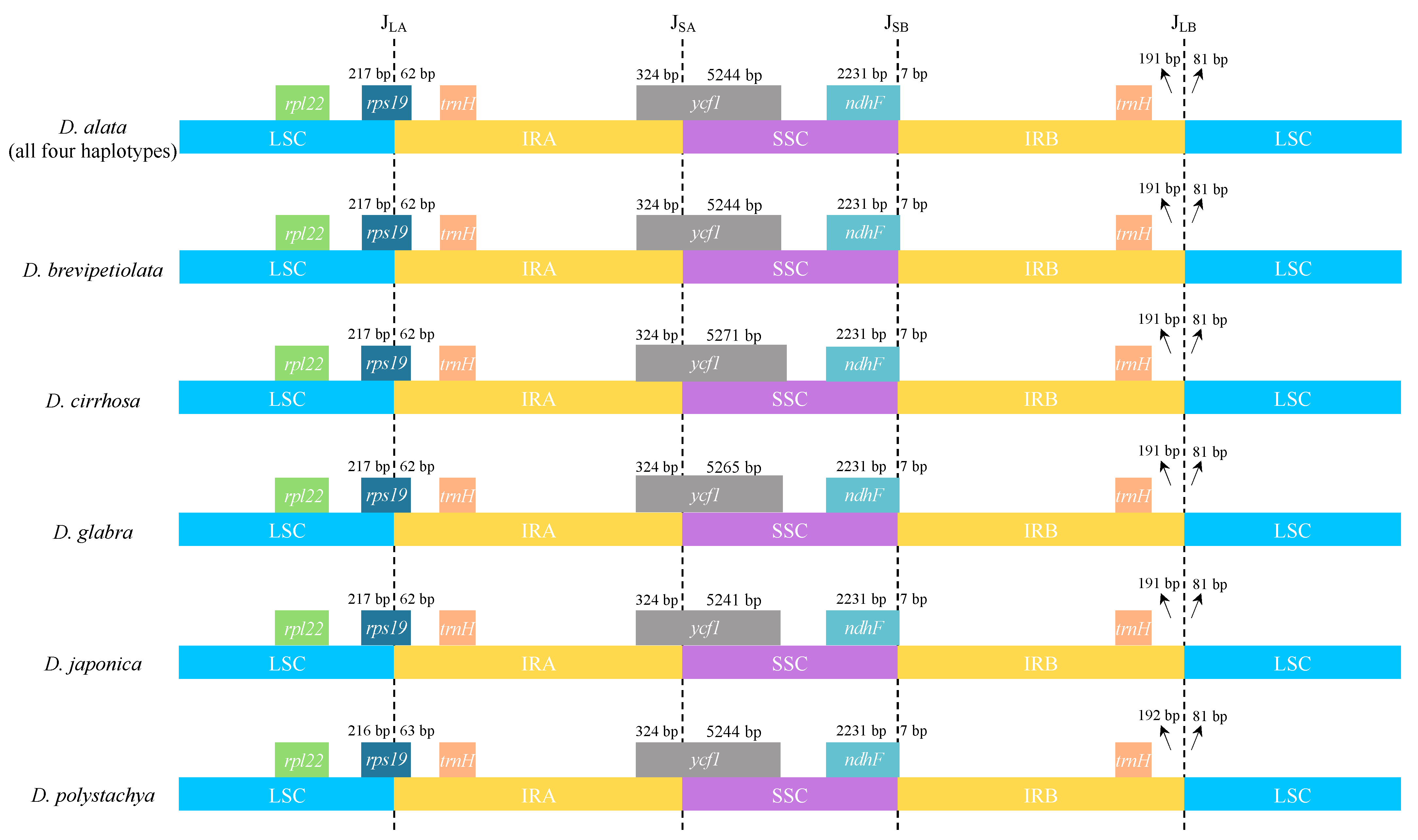
2.1. Plastome Structure and Organization of D. alata
2.2. Plastome Polymorphisms in D. alata
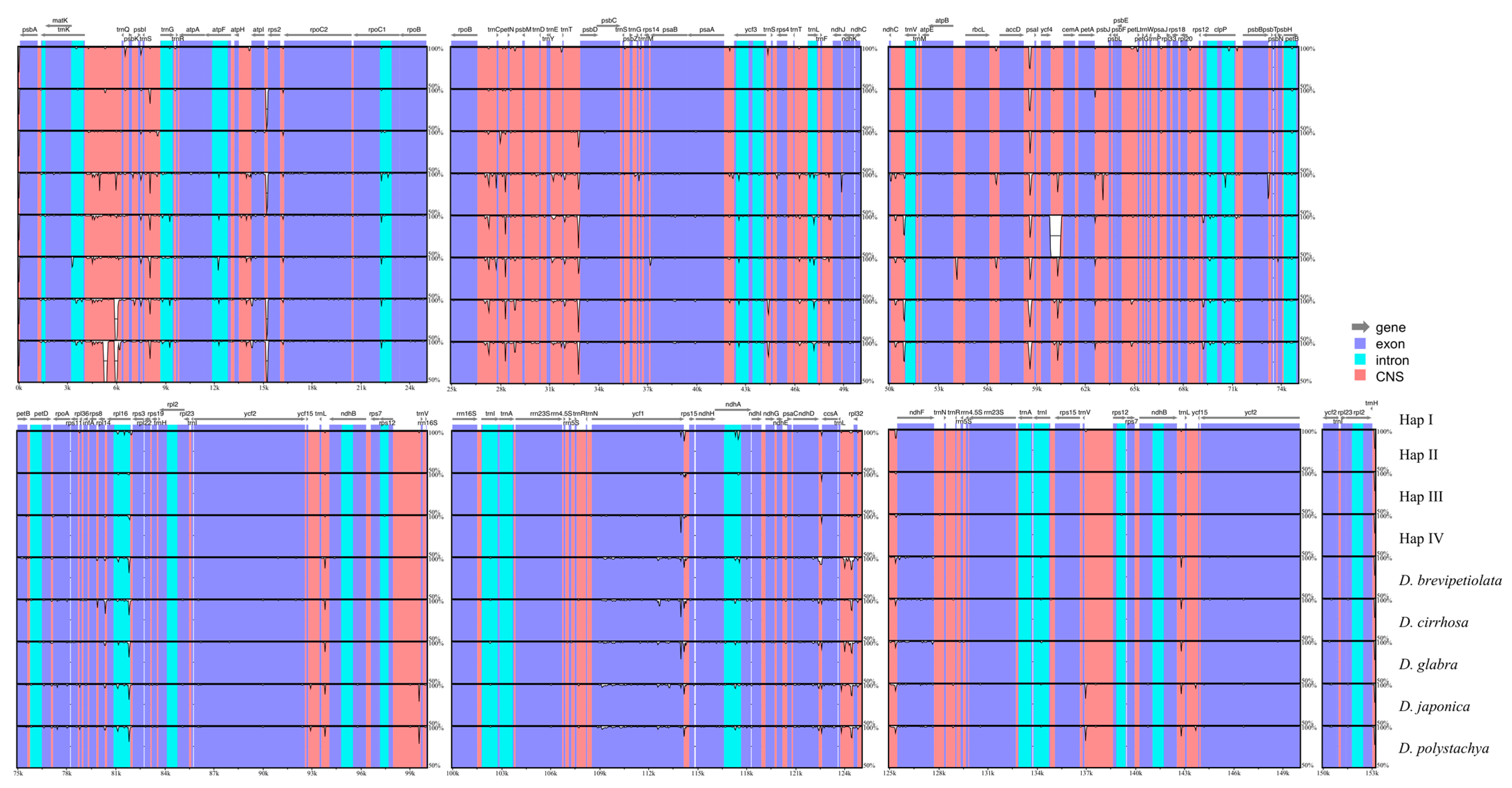
2.3. Whole Plastome Comparison at Intra- and Inter-Specific Levels
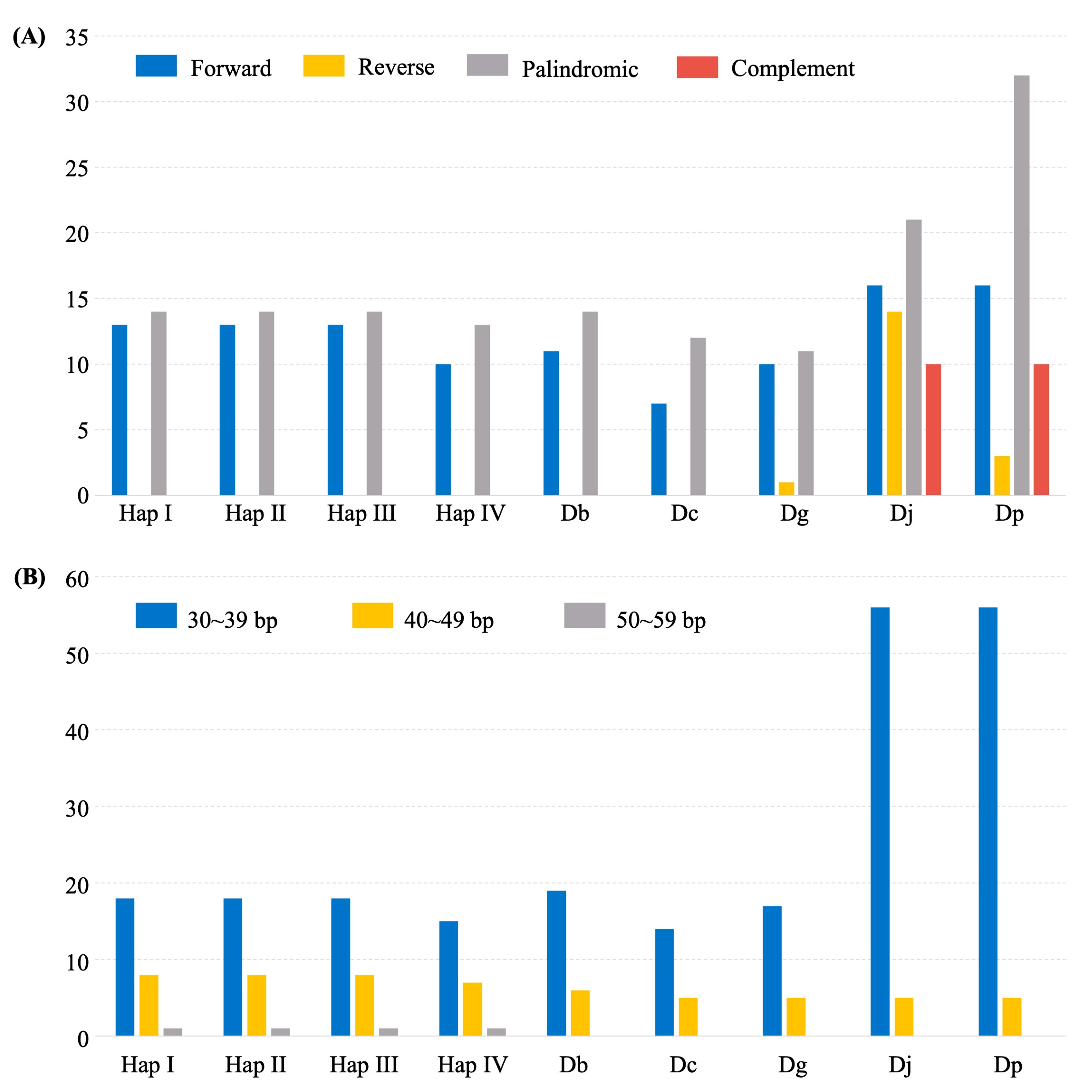
2.4. Dispersed Repeats and SSRs
2.5. Plastome-Divergent Hotspots in the Enantiophyllum
2.6. Phylogenetic Relationships within D. alata and among Members of Enantiophyllum
3. Discussion
3.1. Plastome Evolution in D. alata and Its Closely Related Species
3.2. Plastome-Derived Markers for Species/Cultivar Delimitation in Enantiophyllum
3.3. Phylogenetic Relationships within D. alata and among Species of the Enantiophyllum Section
4. Materials and Methods
4.1. Plant Samples, DNA Extraction, and Genomic Data Acquisition
4.2. Plastome Assembly and Annotation
4.3. Plastome Polymorphism Analysis
4.4. Comparative Plastome Genomics within D. alata and among Closely Related Species
4.5. Characterization of Dispersed Repeats and SSRs
4.6. Identification of Divergent Hotspots
4.7. Phylogenetic Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malapa, R.; Arnau, G.; Noyer, J.; Lebot, V. Genetic Diversity of the Greater Yam (Dioscorea alata L.) and Relatedness to D. nummularia Lam. and D. transversa Br. as Revealed with AFLP Markers. Genet. Resour. Crop Evol. 2005, 52, 919–929. [Google Scholar] [CrossRef]
- Cormier, F.; Lawac, F.; Maledon, E.; Gravillon, M.-C.; Nudol, E.; Mournet, P.; Vignes, H.; Chaïr, H.; Arnau, G. A reference high-density genetic map of greater yam (Dioscorea alata L.). Theor. Appl. Genet. 2019, 132, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Sharif, B.M.; Burgarella, C.; Cormier, F.; Mournet, P.; Causse, S.; Van, K.N.; Kaoh, J.; Rajaonah, M.T.; Lakshan, S.R.; Waki, J.; et al. Genome-wide genotyping elucidates the geographical diversification and dispersal of the polyploid and clonally propagated yam (Dioscorea alata). Ann. Bot. 2020, 126, 1029–1038. [Google Scholar] [CrossRef]
- Muzac-Tucker, I.; Asemota, H.N.; Ahmad, M.H. Biochemical composition and storage of Jamaican yams (Dioscorea spp). J. Sci. Food Agric. 1993, 62, 219–224. [Google Scholar] [CrossRef]
- Arnau, G.; Bhattacharjee, R.; Mn, S.; Chair, H.; Malapa, R.; Lebot, V.; Pavis, C. Understanding the genetic diversity and population structure of yam (Dioscorea alata L.) using microsatellite markers. PLoS ONE 2017, 12, e0174150. [Google Scholar] [CrossRef] [PubMed]
- Egesi, C.N.; Asiedu, R.; Ude, G.; Ogunyemi, S.; Egunjobi, J.K. AFLP marker diversity in water yam (Dioscorea alata L.). Plant Genet. Resour. Charact. Util. 2006, 4, 181–187. [Google Scholar] [CrossRef]
- Sartie, A.; Asiedu, R. Segregation of vegetative and reproductive traits associated with tuber yield and quality in water yam (Dioscorea alata L.). Afric. J. Biotech. 2014, 13, 2807–2818. [Google Scholar]
- Neina, D. Ecological and Edaphic Drivers of Yam Production in West Africa. Appl. Environ. Soil Sci. 2021, 2021, 5019481. [Google Scholar] [CrossRef]
- Asiedu, R.; Sartie, A. Crops that feed the World 1. Yams. Food Secur. 2010, 2, 305–315. [Google Scholar] [CrossRef]
- Shiwachi, H.; Kikuno, H.; Ohata, J.; Kikuchi, Y.U.; Irie, K. Growth of water yam (Dioscorea alata L.) under alkaline soil conditions. Trop. Agr. Develop. 2015, 59, 76–82. [Google Scholar]
- Lebot, V.; Lawac, F.; Legendre, L. The greater yam (Dioscorea alata L.): A review of its phytochemical content and potential for processed products and biofortification. J. Food Compos. Anal. 2023, 115, 104987. [Google Scholar] [CrossRef]
- Wu, W.; Chen, C.; Zhang, Q.; Ahmed, J.Z.; Xu, Y.; Huang, X.; Xie, J.; Xia, W.; Huang, D. A comparative assessment of diversity of greater yam (Dioscorea alata) in China. Sci. Hortic. 2018, 243, 116–124. [Google Scholar] [CrossRef]
- Lebot, V.; Trilles, B.; Noyer, J.; Modesto, J. Genetic relationships between Dioscorea alata L. cultivars. Genet. Resour. Crop Evol. 1998, 45, 499–509. [Google Scholar] [CrossRef]
- Vandenbroucke, H.; Mournet, P.; Vignes, H.; Chaïr, H.; Malapa, R.; Duval, M.F.; Lebot, V. Somaclonal variants of taro (Colocasia esculenta Schott) and yam (Dioscorea alata L.) are incorporated into farmers’ varietal portfolios in Vanuatu. Genet. Resour. Crop Evol. 2015, 63, 495–511. [Google Scholar] [CrossRef]
- Kumar, S.R.; Arumugam, T.; Anandakumar, C.R.; Premalakshmi, V. Genetic variability for quantitative and qualitative characters in Brinjal (Solanum melongena L.). Afr. J. Agric. Res. 2013, 8, 4956–4959. [Google Scholar]
- Chen, X.; Sun, J.; Zhu, Q.; Xiao, Y.; Zhang, H.; Huang, Y.; Wang, P.; Cao, T.; Hu, R.; Xiang, Z.; et al. Characterizing diversity based on phenotypes and molecular marker analyses of purple yam (Dioscorea alata L.) germplasm in southern China. Genet. Resour. Crop Evol. 2022, 69, 2501–2513. [Google Scholar] [CrossRef]
- Lu, R.-S.; Li, P.; Qiu, Y.-X. The Complete Chloroplast Genomes of Three Cardiocrinum (Liliaceae) Species: Comparative Genomic and Phylogenetic Analyses. Front. Plant Sci. 2017, 7, 2054. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, H.E.; Emes, M.J. Nonphotosynthetic Metabolism in Plastids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Birky Jr, C.W. Heterozygosity, heteromorphy, and phylogenetic trees in asexual eukaryotes. Genetics 1996, 144, 427–437. [Google Scholar] [CrossRef]
- Lu, R.-S.; Yang, T.; Chen, Y.; Wang, S.-Y.; Cai, M.-Q.; Cameron, K.M.; Li, P.; Fu, C.-X. Comparative plastome genomics and phylogenetic analyses of Liliaceae. Bot. J. Linn. Soc. 2021, 196, 279–293. [Google Scholar] [CrossRef]
- Wu, J.; Liu, B.; Cheng, F.; Ramchiary, N.; Choi, S.R.; Lim, Y.P.; Wang, X.-W. Sequencing of Chloroplast Genome Using Whole Cellular DNA and Solexa Sequencing Technology. Front. Plant Sci. 2012, 3, 243. [Google Scholar] [CrossRef]
- Muraguri, S.; Xu, W.; Chapman, M.; Muchugi, A.; Oluwaniyi, A.; Oyebanji, O.; Liu, A. Intraspecific variation within Castor bean (Ricinus communis L.) based on chloroplast genomes. Ind. Crops Prod. 2020, 155, 112779. [Google Scholar] [CrossRef]
- Barchi, L.; Rabanus-Wallace, M.T.; Prohens, J.; Toppino, L.; Padmarasu, S.; Portis, E.; Rotino, G.L.; Stein, N.; Lanteri, S.; Giuliano, G.; et al. Improved genome assembly and pan-genome provide key insights into eggplant domestication and breeding. Plant J. 2021, 107, 579–596. [Google Scholar] [CrossRef]
- Magdy, M.; Ou, L.; Yu, H.; Chen, R.; Zhou, Y.; Hassan, H.; Feng, B.; Taitano, N.; van der Knaap, E.; Zou, X.; et al. Pan-plastome approach empowers the assessment of genetic variation in cultivated Capsicum species. Hortic. Res. 2019, 6, 108. [Google Scholar] [CrossRef]
- Wang, J.; Liao, X.; Gu, C.; Xiang, K.; Li, S.; Tembrock, L.R.; Wu, Z.; He, W. The Asian lotus (Nelumbo nucifera) pan-plastome: Diversity and divergence in a living fossil grown for seed, rhizome, and aesthetics. Ornam. Plant Res. 2022, 2, 2. [Google Scholar] [CrossRef]
- Bredeson, J.V.; Lyons, J.B.; Oniyinde, I.O.; Okereke, N.R.; Kolade, O.; Nnabue, I.; Nwadili, C.O.; Hřibová, E.; Parker, M.; Nwogha, J.; et al. Chromosome evolution and the genetic basis of agronomically important traits in greater yam. Nat. Commun. 2022, 13, 2001. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Chen, M.; Feng, Y.; Yuan, N.; Zhang, Y.; Cao, M.; Liu, J.; Wang, Y.; Hang, Y.; Sun, X. Comparative plastome analyses and genomic resource development in wild rice (Zizania spp., Poaceae) using genome skimming data. Ind. Crops Prod. 2022, 186, 115244. [Google Scholar] [CrossRef]
- Cao, J.; Jiang, D.; Zhao, Z.; Yuan, S.; Zhang, Y.; Zhang, T.; Zhong, W.; Yuan, Q.; Huang, L. Development of Chloroplast Genomic Resources in Chinese Yam (Dioscorea polystachya). BioMed Res. Int. 2018, 2018, 6293847. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, X.; Yu, Y.; Yuan, S.; Jiang, D.; Zhang, Y.; Zhang, T.; Zhong, W.; Yuan, Q.; Huang, L. Complete chloroplast genome sequences of Dioscorea: Characterization, genomic resources, and phylogenetic analyses. PeerJ 2018, 6, e6032. [Google Scholar] [CrossRef]
- Xia, W.; Zhang, B.; Xing, D.; Li, Y.; Wu, W.; Xiao, Y.; Sun, J.; Dou, Y.; Tang, W.; Zhang, J.; et al. Development of high-resolution DNA barcodes for Dioscorea species discrimination and phylogenetic analysis. Ecol. Evol. 2019, 9, 10843–10853. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-J.; Cheng, C.-L.; Chang, C.-C.; Wu, C.-L.; Su, T.-M.; Chaw, S.-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Guo, W.; Gupta, S.; Fan, W.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2015, 209, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.L.; Ruhlman, T.A.; Jansen, R.K. Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 2017, 214, 842–851. [Google Scholar] [CrossRef]
- Wilkin, P.; Schols, P.; Chase, M.W.; Chayamarit, K.; Furness, C.A.; Huysmans, S.; Rakotonasolo, F.; Smets, E.; Thapyai, C. A Plastid Gene Phylogeny Of the Yam Genus, Dioscorea: Roots, Fruits and Madagascar. Syst. Bot. 2005, 30, 736–749. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Wu, B.; Zhao, Y.; Chen, J.; Hang, Y. Phylogeny of Dioscorea sect. Stenophora based on chloroplast matK, rbcL and trnL-F sequences. J. Syst. Evol. 2008, 46, 315–321. [Google Scholar]
- Viruel, J.; Segarra-Moragues, J.G.; Raz, L.; Forest, F.; Wilkin, P.; Sanmartín, I.; Catalán, P. Late Cretaceous–early Eocene origin of yams (Dioscorea, Dioscoreaceae) in the Laurasian Palaearctic and their subsequent Oligocene–Miocene diversification. J. Biogeogr. 2016, 43, 750–762. [Google Scholar] [CrossRef]
- Couto, R.S.; Martins, A.C.; Bolson, M.; Lopes, R.C.; Smidt, E.C.; Braga, J.M.A. Time calibrated tree of Dioscorea (Dioscoreaceae) indicates four origins of yams in the Neotropics since the Eocene. Bot. J. Linn. Soc. 2018, 188, 144–160. [Google Scholar] [CrossRef]
- Noda, H.; Yamashita, J.; Fuse, S.; Pooma, R.; Poopath, M.; Tobe, H.; Tamura, M.N. A large-scale phylogenetic analysis of Dioscorea (Dioscoreaceae), with reference to character evolution and subgeneric recognition. Acta. Phytotax. Geobot. 2020, 71, 103–128. [Google Scholar]
- Doyle, J.J. Gene Trees and Species Trees: Molecular Systematics as One-Character Taxonomy. Syst. Bot. 1992, 17, 144. [Google Scholar] [CrossRef]
- Stull, G.W.; De Stefano, R.D.; Soltis, D.E.; Soltis, P.S. Data from: Resolving basal lamiid phylogeny and the circumscription of Icacinaceae with a plastome-scale data set. Am. J. Bot. 2015, 102, 1794–1813. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.-M.; Tsai, J.-L.; Chen, M.-Y.; Ku, H.-M.; Liu, S.-C. Molecular phylogeny of Dioscorea (Dioscoreaceae) in East and Southeast Asia. Blumea-Biodivers. Evol. Biogeogr. Plants 2013, 58, 21–27. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Depamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; NISC Comparative Sequencing Program; Green, E.D.; Sidow, A.; Batzoglou, S. LAGAN and Multi-LAGAN: Efficient Tools for Large-Scale Multiple Alignment of Genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Magwé-Tindo, J.; Wieringa, J.J.; Sonké, B.; Zapfack, L.; Vigouroux, Y.; Couvreur, T.L.; Scarcelli, N. Guinea yam (Dioscorea spp., Dioscoreaceae) wild relatives identified using whole plastome phylogenetic analyses. Taxon 2018, 67, 905–915. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Cultivar or Local Name | Cultivation Region | Haplotype |
|---|---|---|---|
| CDa001 | ‘Minghuai No. 1’ | Sanming, Fujian | I |
| CDa002 | ‘Quanhuai 1515’ | Quanzhou, Fujian | I |
| CDa003 | ‘Quanhuai 1815’ | Quanzhou, Fujian | I |
| CDa004 | ‘Shanghang dashu’ | Fuzhou, Fujian | I |
| CDa005 | ‘Taihuai No. 6’ | Quanzhou, Fujian | I |
| CDa006 | ‘Wuyishan zishanyao’ | Wuyishan, Fujian | I |
| CDa007 | ‘Zhenghe hongxinshu’ | Nanping, Fujian | I |
| CDa008 | ‘Zhouning zishu’ | Ningde, Fujian | I |
| CDa009 | ‘Ziyushanyao’ | Quanzhou, Fujian | I |
| CDa010 | ‘Anxi saobashu’ | Quanzhou, Fujian | III |
| CDa011 | ‘Dongyou kuaishu’ | Nanping, Fujian | III |
| CDa012 | ‘Minghuai No. 3′ | Sanming, Fujian | III |
| CDa013 | ‘Zhenghe jiaobanshu’ | Nanping, Fujian | III |
| CDa014 | ‘Ziyushenshu’ | Guangzhou, Guangdong | I |
| CDa015 | ‘Hainan danshu’ | Sanya, Hainan | I |
| CDa016 | ‘Hainan zazi’ | Qiongzhong, Hainan | I |
| CDa017 | ‘Hainan zabai’ | Wuzhishan, Hainan | II |
| CDa018 | Da90 | Lingao, Hainan | III |
| CDa019 | Da94 | Danzhou, Hainan | III |
| CDa020 | ‘Yangxi nuomishu’ | Loudi, Hunan | IV |
| CDa021 | ‘Suyu No. 2’ | Xuzhou, Jiangsu | I |
| CDa022 | ‘Suyu No. 4’ | Xuzhou, Jiangsu | I |
| CDa023 | ‘Suyu No. 6’ | Nanjing, Jiangsu | III |
| CDa024 | ‘Ganzhoushangyou baixinshu’ | Ganzhou, Jiangxi | I |
| CDa025 | ‘Ganzhoushangyou zixinshu’ | Ganzhou, Jiangxi | I |
| CDa026 | ‘Hongshuzi’ | Ganzhou, Jiangxi | I |
| CDa027 | ‘Jiaobanshu’ | Ganzhou, Jiangxi | I |
| CDa028 | ‘Wantian zishu’ | Ruijin, Jiangxi | I |
| CDa029 | ‘Zixin yuantiao’ | Shangrao, Jiangxi | I |
| CDa030 | ‘Baishu’ | Ganzhou, Jiangxi | III |
| CDa031 | ‘Jiaobanshu purple’ | Fuzhou, Jiangxi | III |
| CDa032 | ‘Jiaobanshu’ | Ji’an, Jiangxi | III |
| CDa033 | ‘Longnante zipishanyao’ | Ganzhou, Jiangxi | III |
| CDa034 | ‘Ximazhuang jiaobanshu’ | Ruichang, Jiangxi | III |
| CDa035 | ‘Weihai baishu’ | Weihai, Shandong | III |
| CDa036 | ‘Miyi shanyao’ | Paizhihua, Sichuan | I |
| CDa037 | ‘Honglong shanyao’ | Jiayi, Taiwan | I |
| CDa038 | ‘Yangmingshan shanyao’ | Taibei, Taiwan | I |
| CDa039 | ‘Chengtuo baishanyao’ | Wenshan, Yunnan | I |
| CDa040 | ‘Lijiang zishu’ | Lijiang, Yunnan | I |
| CDa041 | ‘Quzhou kuaigenbaishu’ | Quzhou, Zhejiang | III |
| CDa042 | ‘Ruian shanyao’ | Wenzhou, Zhejiang | III |
| CDa043 | ‘Taizhou yuanshanyao’ | Taizhou, Zhejiang | III |
| CDa044 | ‘Wencheng nuomishanyao’ | Wenzhou, Zhejiang | IV |
| ADa001 | Tda00/00005 | Africa | I |
| ADa002 | Tda01/00039 | Africa | I |
| ADa003 | Tda02/00012 | Africa | I |
| ADa004 | Tda05/00015 | Africa | I |
| ADa005 | Tda95-310 | Africa | I |
| ADa006 | Tda95/00328 | Africa | I |
| ADa007 | Tda99/00048 | Africa | I |
| ADa008 | Tda99/00240 | Africa | I |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, R.-S.; Hu, K.; Zhang, F.-J.; Sun, X.-Q.; Chen, M.; Zhang, Y.-M. Pan-Plastome of Greater Yam (Dioscorea alata) in China: Intraspecific Genetic Variation, Comparative Genomics, and Phylogenetic Analyses. Int. J. Mol. Sci. 2023, 24, 3341. https://doi.org/10.3390/ijms24043341
Lu R-S, Hu K, Zhang F-J, Sun X-Q, Chen M, Zhang Y-M. Pan-Plastome of Greater Yam (Dioscorea alata) in China: Intraspecific Genetic Variation, Comparative Genomics, and Phylogenetic Analyses. International Journal of Molecular Sciences. 2023; 24(4):3341. https://doi.org/10.3390/ijms24043341
Chicago/Turabian StyleLu, Rui-Sen, Ke Hu, Feng-Jiao Zhang, Xiao-Qin Sun, Min Chen, and Yan-Mei Zhang. 2023. "Pan-Plastome of Greater Yam (Dioscorea alata) in China: Intraspecific Genetic Variation, Comparative Genomics, and Phylogenetic Analyses" International Journal of Molecular Sciences 24, no. 4: 3341. https://doi.org/10.3390/ijms24043341
APA StyleLu, R.-S., Hu, K., Zhang, F.-J., Sun, X.-Q., Chen, M., & Zhang, Y.-M. (2023). Pan-Plastome of Greater Yam (Dioscorea alata) in China: Intraspecific Genetic Variation, Comparative Genomics, and Phylogenetic Analyses. International Journal of Molecular Sciences, 24(4), 3341. https://doi.org/10.3390/ijms24043341