
Targeting Glutaminolysis Shows Efficacy in Both Prednisolone-Sensitive and in Metabolically Rewired Prednisolone-Resistant B-Cell Childhood Acute Lymphoblastic Leukaemia Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
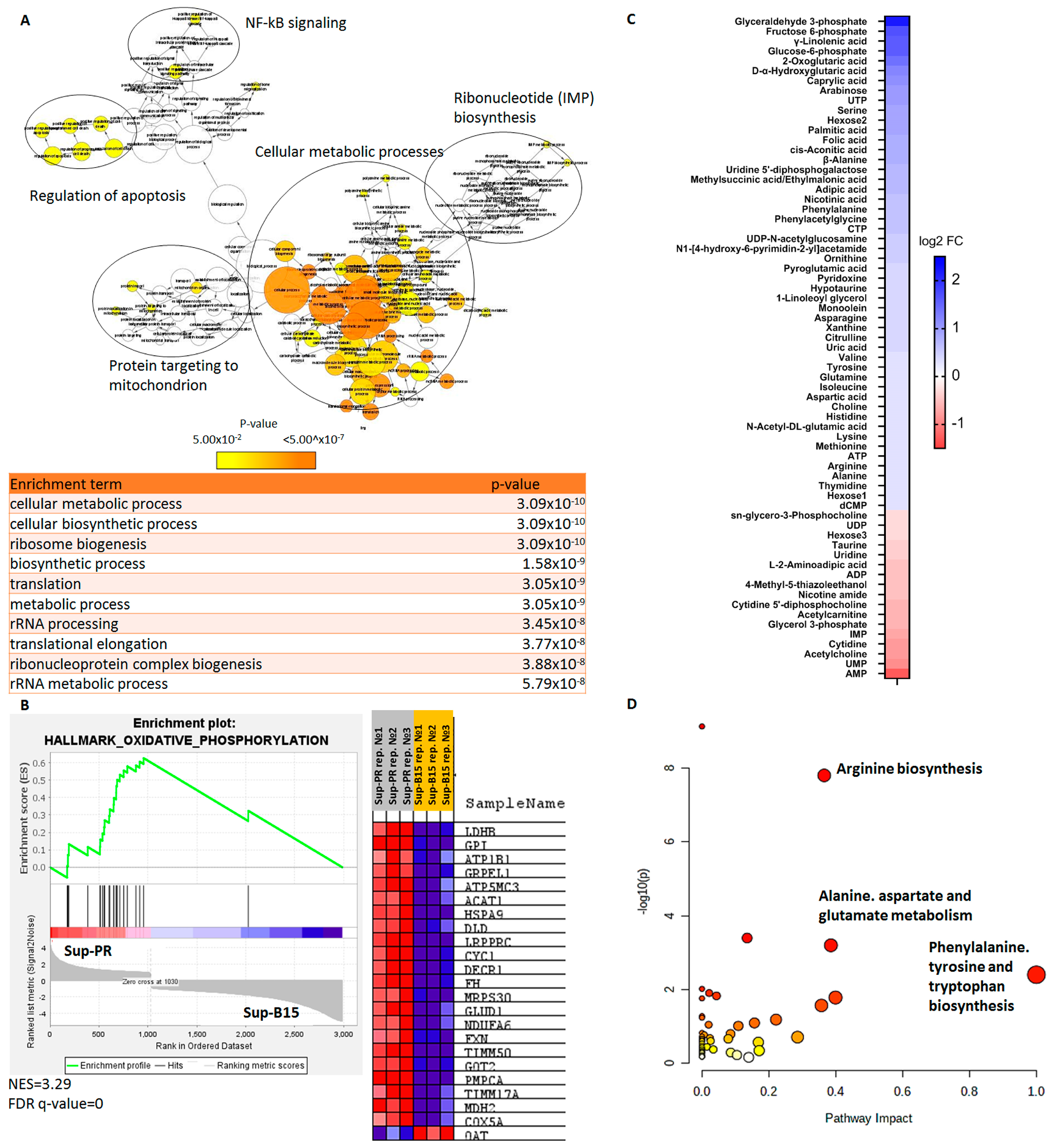
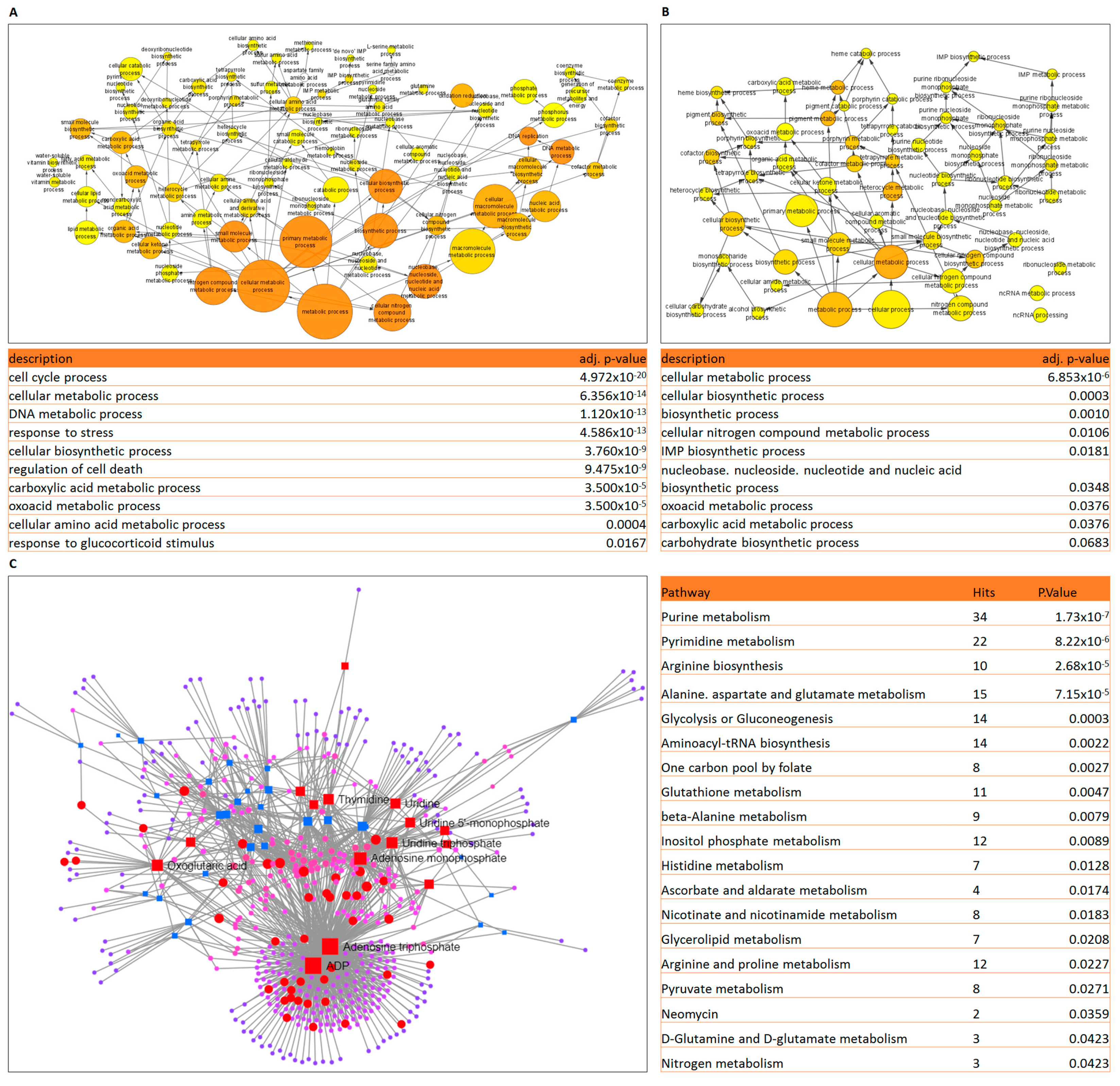
2.1. Gene Expression and Metabolomics Analyses of Prednisolone-Resistant Cells Suggest Dysregulated Glycolysis, Amino Acid and Nucleotide Metabolism
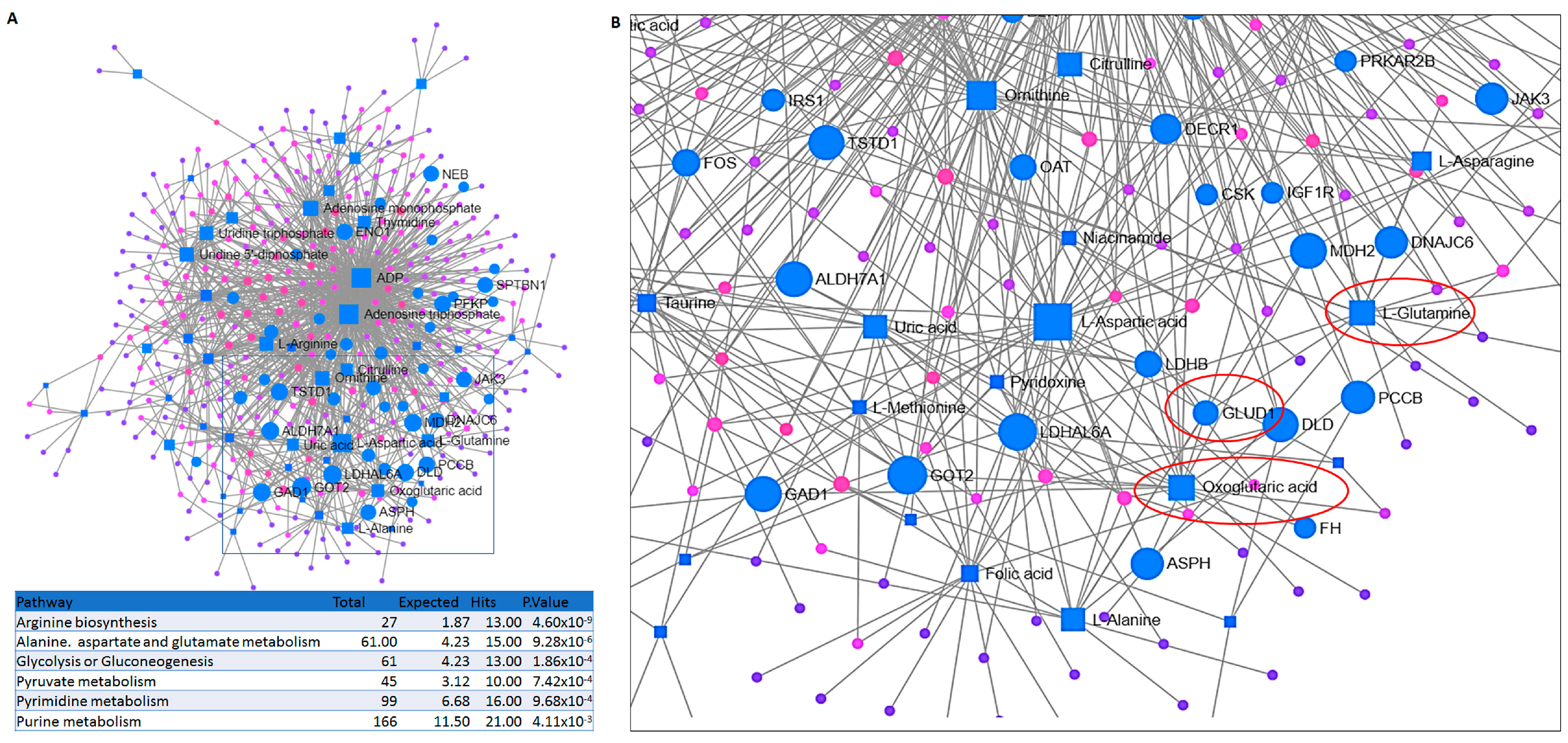
2.2. Integrated Transcriptomic and Metabolomic Analysis Reveals the Importance of Glutamine Metabolism in Prednisolone-Resistant Cells
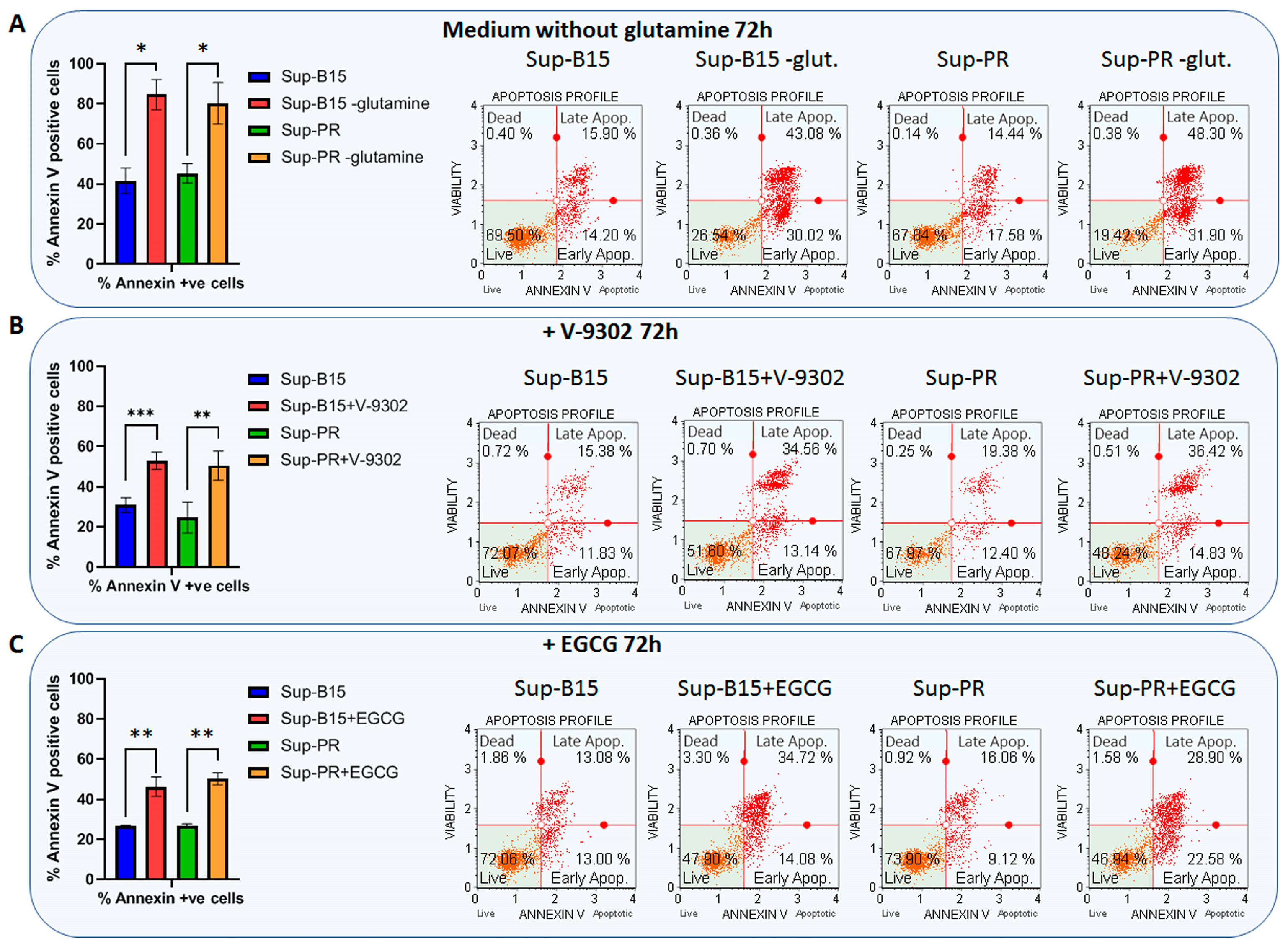
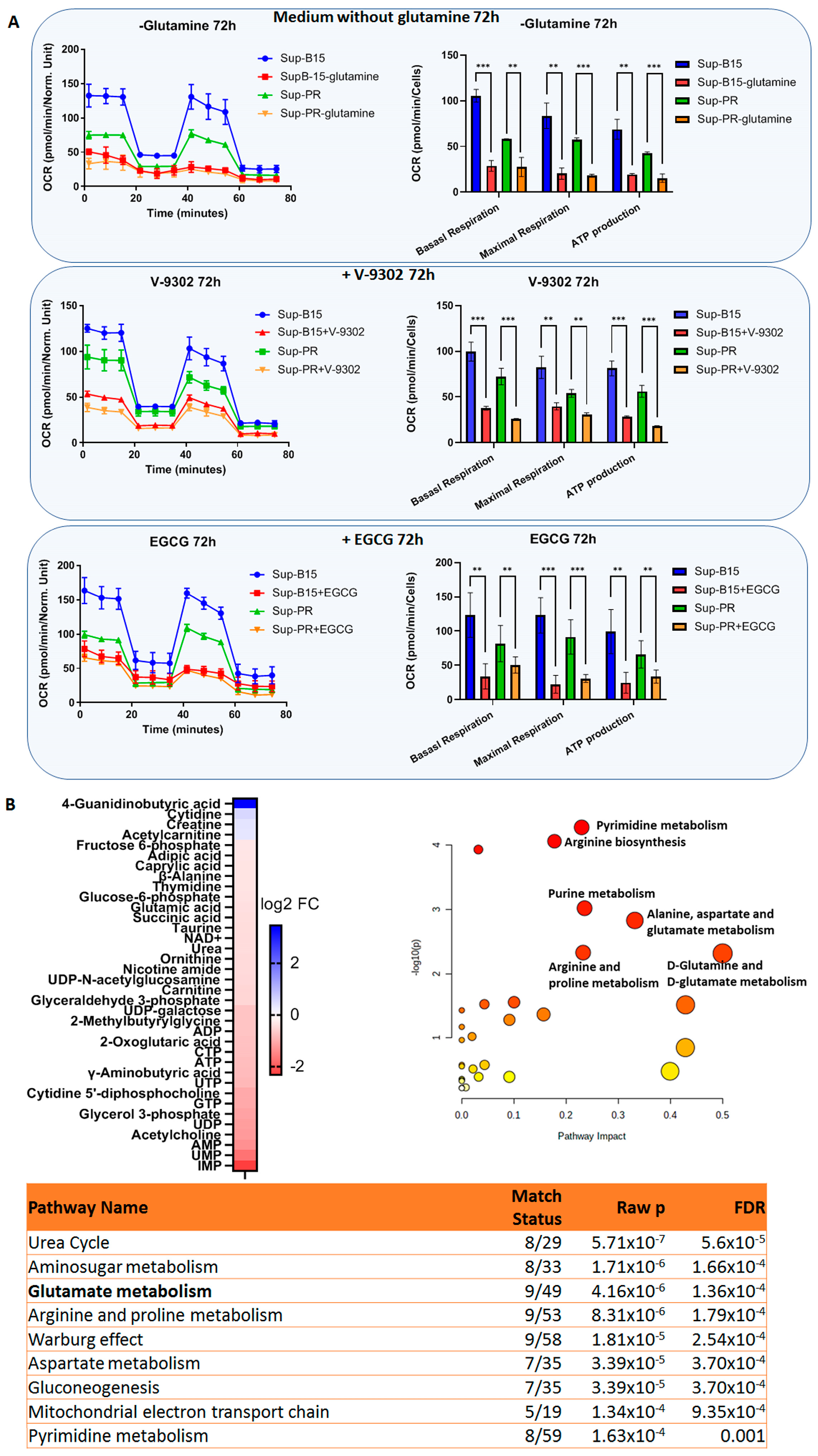
2.3. Targeting Glutamine Metabolism in cALL Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Drug Titrations, Cell Proliferation and Apoptosis Assays
4.2. RNA Sequencing and Data Analysis
4.3. Metabolomic Analysis
4.4. Seahorse Mitochondrial Function Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malczewska, M.; Kosmider, K.; Bednarz, K.; Ostapinska, K.; Lejman, M.; Zawitkowska, J. Recent Advances in Treatment Options for Childhood Acute Lymphoblastic Leukemia. Cancers 2022, 14, 2021. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Bhojwani, D.; Pui, C.H. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013, 14, e205–e217. [Google Scholar] [CrossRef] [PubMed]
- Goto, H. Childhood relapsed acute lymphoblastic leukemia: Biology and recent treatment progress. Pediatr. Int. 2015, 57, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- Sbirkov, Y.; Burnusuzov, H.; Sarafian, V. Metabolic reprogramming in childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2020, 67, e28255. [Google Scholar] [CrossRef]
- Staal, F.J.; de Ridder, D.; Szczepanski, T.; Schonewille, T.; van der Linden, E.C.; van Wering, E.R.; van der Velden, V.H.; van Dongen, J.J. Genome-wide expression analysis of paired diagnosis-relapse samples in ALL indicates involvement of pathways related to DNA replication, cell cycle and DNA repair, independent of immune phenotype. Leukemia 2010, 24, 491–499. [Google Scholar] [CrossRef]
- Szczepanek, J.; Styczynski, J.; Haus, O.; Tretyn, A.; Wysocki, M. Relapse of acute lymphoblastic leukemia in children in the context of microarray analyses. Arch. Immunol. Ther. Exp. 2011, 59, 61–68. [Google Scholar] [CrossRef]
- Beesley, A.H.; Cummings, A.J.; Freitas, J.R.; Hoffmann, K.; Firth, M.J.; Ford, J.; de Klerk, N.H.; Kees, U.R. The gene expression signature of relapse in paediatric acute lymphoblastic leukaemia: Implications for mechanisms of therapy failure. Br. J. Haematol. 2005, 131, 447–456. [Google Scholar] [CrossRef]
- Kirschner-Schwabe, R.; Lottaz, C.; Todling, J.; Rhein, P.; Karawajew, L.; Eckert, C.; von Stackelberg, A.; Ungethum, U.; Kostka, D.; Kulozik, A.E.; et al. Expression of late cell cycle genes and an increased proliferative capacity characterize very early relapse of childhood acute lymphoblastic leukemia. Clin. Cancer Res. 2006, 12, 4553–4561. [Google Scholar] [CrossRef]
- Chow, Y.P.; Alias, H.; Jamal, R. Meta-analysis of gene expression in relapsed childhood B-acute lymphoblastic leukemia. BMC Cancer 2017, 17, 120. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.N.; Chen, Z.; Braas, D.; Lee, J.W.; Xiao, G.; Geng, H.; Cosgun, K.N.; Hurtz, C.; Shojaee, S.; Cazzaniga, V.; et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 2017, 542, 479–483. [Google Scholar] [CrossRef]
- Sbirkov, Y.; Ivanova, T.; Burnusuzov, H.; Gercheva, K.; Petrie, K.; Schenk, T.; Sarafian, V. The Protozoan Inhibitor Atovaquone Affects Mitochondrial Respiration and Shows In Vitro Efficacy Against Glucocorticoid-Resistant Cells in Childhood B-Cell Acute Lymphoblastic Leukaemia. Front. Oncol. 2021, 11, 632181. [Google Scholar] [CrossRef]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C.; et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef]
- Edwards, D.N.; Ngwa, V.M.; Raybuck, A.L.; Wang, S.; Hwang, Y.; Kim, L.C.; Cho, S.H.; Paik, Y.; Wang, Q.; Zhang, S.; et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J. Clin. Investig. 2021, 131, e140100. [Google Scholar] [CrossRef]
- Jin, H.; Wang, S.; Zaal, E.A.; Wang, C.; Wu, H.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.; Lieftink, C.; et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. eLife 2020, 9, e56749. [Google Scholar] [CrossRef]
- Pocock, C.F.; Malone, M.; Booth, M.; Evans, M.; Morgan, G.; Greil, J.; Cotter, F.E. BCL-2 expression by leukaemic blasts in a SCID mouse model of biphenotypic leukaemia associated with the t(4;11)(q21;q23) translocation. Br. J. Haematol. 1995, 90, 855–867. [Google Scholar] [CrossRef]
- Stam, R.W.; Den Boer, M.L.; Schneider, P.; de Boer, J.; Hagelstein, J.; Valsecchi, M.G.; de Lorenzo, P.; Sallan, S.E.; Brady, H.J.; Armstrong, S.A.; et al. Association of high-level MCL-1 expression with in vitro and in vivo prednisone resistance in MLL-rearranged infant acute lymphoblastic leukemia. Blood 2010, 115, 1018–1025. [Google Scholar] [CrossRef]
- Sugimoto, K.; Suzuki, H.I.; Fujimura, T.; Ono, A.; Kaga, N.; Isobe, Y.; Sasaki, M.; Taka, H.; Miyazono, K.; Komatsu, N. A clinically attainable dose of L-asparaginase targets glutamine addiction in lymphoid cell lines. Cancer Sci. 2015, 106, 1534–1543. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Nokin, M.J.; Teres, S.; Tome, M.; Bodineau, C.; Galmar, O.; Pasquet, J.M.; Rousseau, B.; van Liempd, S.; Falcon-Perez, J.M.; et al. Downregulation of Glutamine Synthetase, not glutaminolysis, is responsible for glutamine addiction in Notch1-driven acute lymphoblastic leukemia. Mol. Oncol. 2021, 15, 1412–1431. [Google Scholar] [CrossRef]
- Schulte, M.L.; Khodadadi, A.B.; Cuthbertson, M.L.; Smith, J.A.; Manning, H.C. 2-Amino-4-bis(aryloxybenzyl)aminobutanoic acids: A novel scaffold for inhibition of ASCT2-mediated glutamine transport. Bioorg. Med. Chem. Lett. 2016, 26, 1044–1047. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Xu, J.; Sun, J.; Huang, H.; Zhang, Z.; Ma, W.; Wan, Z.; Liu, Y.; Pardeshi, A.; Li, S. Co-delivery of 2-Deoxyglucose and a glutamine metabolism inhibitor V9302 via a prodrug micellar formulation for synergistic targeting of metabolism in cancer. Acta Biomater. 2020, 105, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Broer, A.; Fairweather, S.; Broer, S. Disruption of Amino Acid Homeostasis by Novel ASCT2 Inhibitors Involves Multiple Targets. Front. Pharmacol. 2018, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Allen, A.; Kwagh, J.; Doliba, N.M.; Qin, W.; Najafi, H.; Collins, H.W.; Matschinsky, F.M.; Stanley, C.A.; Smith, T.J. Green tea polyphenols modulate insulin secretion by inhibiting glutamate dehydrogenase. J. Biol. Chem. 2006, 281, 10214–10221. [Google Scholar] [CrossRef] [PubMed]
- Peeters, T.H.; Lenting, K.; Breukels, V.; van Lith, S.A.M.; van den Heuvel, C.; Molenaar, R.; van Rooij, A.; Wevers, R.; Span, P.N.; Heerschap, A.; et al. Isocitrate dehydrogenase 1-mutated cancers are sensitive to the green tea polyphenol epigallocatechin-3-gallate. Cancer Metab. 2019, 7, 4. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, G.; Mao, Q.; Li, S.; Xiong, W.; Lin, Y.; Ge, J. Glutamate dehydrogenase (GDH) regulates bioenergetics and redox homeostasis in human glioma. Oncotarget 2014, 5. Available online: https://www.oncotarget.com/article/7657/text/ (accessed on 21 January 2022). [CrossRef]
- Jin, L.; Li, D.; Alesi, G.N.; Fan, J.; Kang, H.B.; Lu, Z.; Boggon, T.J.; Jin, P.; Yi, H.; Wright, E.R.; et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 2015, 27, 257–270. [Google Scholar] [CrossRef]
- Shin, E.S.; Park, J.; Shin, J.M.; Cho, D.; Cho, S.Y.; Shin, D.W.; Ham, M.; Kim, J.B.; Lee, T.R. Catechin gallates are NADP+-competitive inhibitors of glucose-6-phosphate dehydrogenase and other enzymes that employ NADP+ as a coenzyme. Bioorg. Med. Chem. 2008, 16, 3580–3586. [Google Scholar] [CrossRef]
- Whitelaw, B.S.; Robinson, M.B. Inhibitors of glutamate dehydrogenase block sodium-dependent glutamate uptake in rat brain membranes. Front. Endocrinol. 2013, 4, 123. [Google Scholar] [CrossRef]
- Halama, A.; Suhre, K. Advancing Cancer Treatment by Targeting Glutamine Metabolism-A Roadmap. Cancers 2022, 14, 553. [Google Scholar] [CrossRef]
- Ni, F.; Yu, W.M.; Li, Z.; Graham, D.K.; Jin, L.; Kang, S.; Rossi, M.R.; Li, S.; Broxmeyer, H.E.; Qu, C.K. Critical role of ASCT2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat. Metab. 2019, 1, 390–403. [Google Scholar] [CrossRef]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef]
- Raivio, K.O.; Andersson, L.C. Glutamine requirements for purine metabolism in leukemic lymphoblasts. Leuk. Res. 1982, 6, 111–115. [Google Scholar] [CrossRef]
- Cory, J.G.; Cory, A.H. Critical roles of glutamine as nitrogen donors in purine and pyrimidine nucleotide synthesis: Asparaginase treatment in childhood acute lymphoblastic leukemia. In Vivo 2006, 20, 587–589. [Google Scholar]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef]
- de la Cruz Lopez, K.G.; Toledo Guzman, M.E.; Sanchez, E.O.; Garcia Carranca, A. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373. [Google Scholar] [CrossRef]
- Stanulović, V.S.; Reed, M.A.C.; Patani, H.; Potluri, S.; Georgiadou, E.; Roberts, J.; Sarkar, S.; Pratt, G.; Jones, A.M.; Günther, U.; et al. Proliferation and Survival of T-cell Acute Lymphoblastic Leukaemia Depends on mTOR-regulated Glutamine Uptake and EAAT1-dependent Conversion of Glutamine to Aspartate and Nucleotides. biorXiv 2020. [Google Scholar] [CrossRef]
- Bodineau, C.; Tome, M.; Murdoch, P.D.S.; Duran, R.V. Glutamine, MTOR and autophagy: A multiconnection relationship. Autophagy 2022, 18, 2749–2750. [Google Scholar] [CrossRef]
- Simioni, C.; Martelli, A.M.; Zauli, G.; Melloni, E.; Neri, L.M. Targeting mTOR in Acute Lymphoblastic Leukemia. Cells 2019, 8, 190. [Google Scholar] [CrossRef] [Green Version]
- Ge, Z.; Song, C.; Ding, Y.; Tan, B.H.; Desai, D.; Sharma, A.; Gowda, R.; Yue, F.; Huang, S.; Spiegelman, V.; et al. Dual targeting of MTOR as a novel therapeutic approach for high-risk B-cell acute lymphoblastic leukemia. Leukemia 2021, 35, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Rashkovan, M.; Ferrando, A. Metabolic dependencies and vulnerabilities in leukemia. Genes Dev. 2019, 33, 1460–1474. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer Res. 2010, 70, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.; Wang, D.; Li, Z.; Marion, W.; Hughes, T.; Sousa, P.; Harada, T.; Sui, S.H.; Naumenko, S.; Kalfon, J.; et al. Hypoxic, glycolytic metabolism is a vulnerability of B-acute lymphoblastic leukemia-initiating cells. Cell Rep. 2022, 39, 110752. [Google Scholar] [CrossRef]
- Kim, G.; Jang, S.K.; Kim, Y.J.; Jin, H.O.; Bae, S.; Hong, J.; Park, I.C.; Lee, J.H. Inhibition of Glutamine Uptake Resensitizes Paclitaxel Resistance in SKOV3-TR Ovarian Cancer Cell via mTORC1/S6K Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 8761. [Google Scholar] [CrossRef]
- Burger, J.A. Bruton Tyrosine Kinase Inhibitors: Present and Future. Cancer J. 2019, 25, 386–393. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Han, L.; Zhou, Y.; Sun, S. Green tea polyphenol EGCG reverse cisplatin resistance of A549/DDP cell line through candidate genes demethylation. Biomed. Pharmacother. 2015, 69, 285–290. [Google Scholar] [CrossRef]
- Meng, J.; Chang, C.; Chen, Y.; Bi, F.; Ji, C.; Liu, W. EGCG overcomes gefitinib resistance by inhibiting autophagy and augmenting cell death through targeting ERK phosphorylation in NSCLC. OncoTargets Ther. 2019, 12, 6033–6043. [Google Scholar] [CrossRef]
- Tang, H.; Zeng, L.; Wang, J.; Zhang, X.; Ruan, Q.; Wang, J.; Cui, S.; Yang, D. Reversal of 5-fluorouracil resistance by EGCG is mediate by inactivation of TFAP2A/VEGF signaling pathway and down-regulation of MDR-1 and P-gp expression in gastric cancer. Oncotarget 2017, 8, 82842–82853. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Nastasi, C.; Willerlev-Olsen, A.; Dalhoff, K.; Ford, S.L.; Gadsboll, A.O.; Buus, T.B.; Gluud, M.; Danielsen, M.; Litman, T.; Bonefeld, C.M.; et al. Publisher Correction: Inhibition of succinate dehydrogenase activity impairs human T cell activation and function. Sci. Rep. 2021, 11, 8815. [Google Scholar] [CrossRef]
- Sun, Y.; Zou, H.; Li, X.; Xu, S.; Liu, C. Plasma Metabolomics Reveals Metabolic Profiling For Diabetic Retinopathy and Disease Progression. Front. Endocrinol. 2021, 12, 757088. [Google Scholar] [CrossRef]
- Feng, Q.; Liu, Z.; Zhong, S.; Li, R.; Xia, H.; Jie, Z.; Wen, B.; Chen, X.; Yan, W.; Fan, Y.; et al. Integrated metabolomics and metagenomics analysis of plasma and urine identified microbial metabolites associated with coronary heart disease. Sci. Rep. 2016, 6, 22525. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef]
- Jewison, T.; Su, Y.; Disfany, F.M.; Liang, Y.; Knox, C.; Maciejewski, A.; Poelzer, J.; Huynh, J.; Zhou, Y.; Arndt, D.; et al. SMPDB 2.0: Big improvements to the Small Molecule Pathway Database. Nucleic Acids Res. 2014, 42, D478–D484. [Google Scholar] [CrossRef]
- Karnovsky, A.; Weymouth, T.; Hull, T.; Tarcea, V.G.; Scardoni, G.; Laudanna, C.; Sartor, M.A.; Stringer, K.A.; Jagadish, H.V.; Burant, C.; et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 2012, 28, 373–380. [Google Scholar] [CrossRef] [Green Version]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sbirkov, Y.; Vergov, B.; Dzharov, V.; Schenk, T.; Petrie, K.; Sarafian, V. Targeting Glutaminolysis Shows Efficacy in Both Prednisolone-Sensitive and in Metabolically Rewired Prednisolone-Resistant B-Cell Childhood Acute Lymphoblastic Leukaemia Cells. Int. J. Mol. Sci. 2023, 24, 3378. https://doi.org/10.3390/ijms24043378
Sbirkov Y, Vergov B, Dzharov V, Schenk T, Petrie K, Sarafian V. Targeting Glutaminolysis Shows Efficacy in Both Prednisolone-Sensitive and in Metabolically Rewired Prednisolone-Resistant B-Cell Childhood Acute Lymphoblastic Leukaemia Cells. International Journal of Molecular Sciences. 2023; 24(4):3378. https://doi.org/10.3390/ijms24043378
Chicago/Turabian StyleSbirkov, Yordan, Bozhidar Vergov, Vasil Dzharov, Tino Schenk, Kevin Petrie, and Victoria Sarafian. 2023. "Targeting Glutaminolysis Shows Efficacy in Both Prednisolone-Sensitive and in Metabolically Rewired Prednisolone-Resistant B-Cell Childhood Acute Lymphoblastic Leukaemia Cells" International Journal of Molecular Sciences 24, no. 4: 3378. https://doi.org/10.3390/ijms24043378