
The Dynamic Interactions of a Multitargeting Domain in Ameloblastin Protein with Amelogenin and Membrane

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Characterization of Spin-Labeled Ambn Peptides
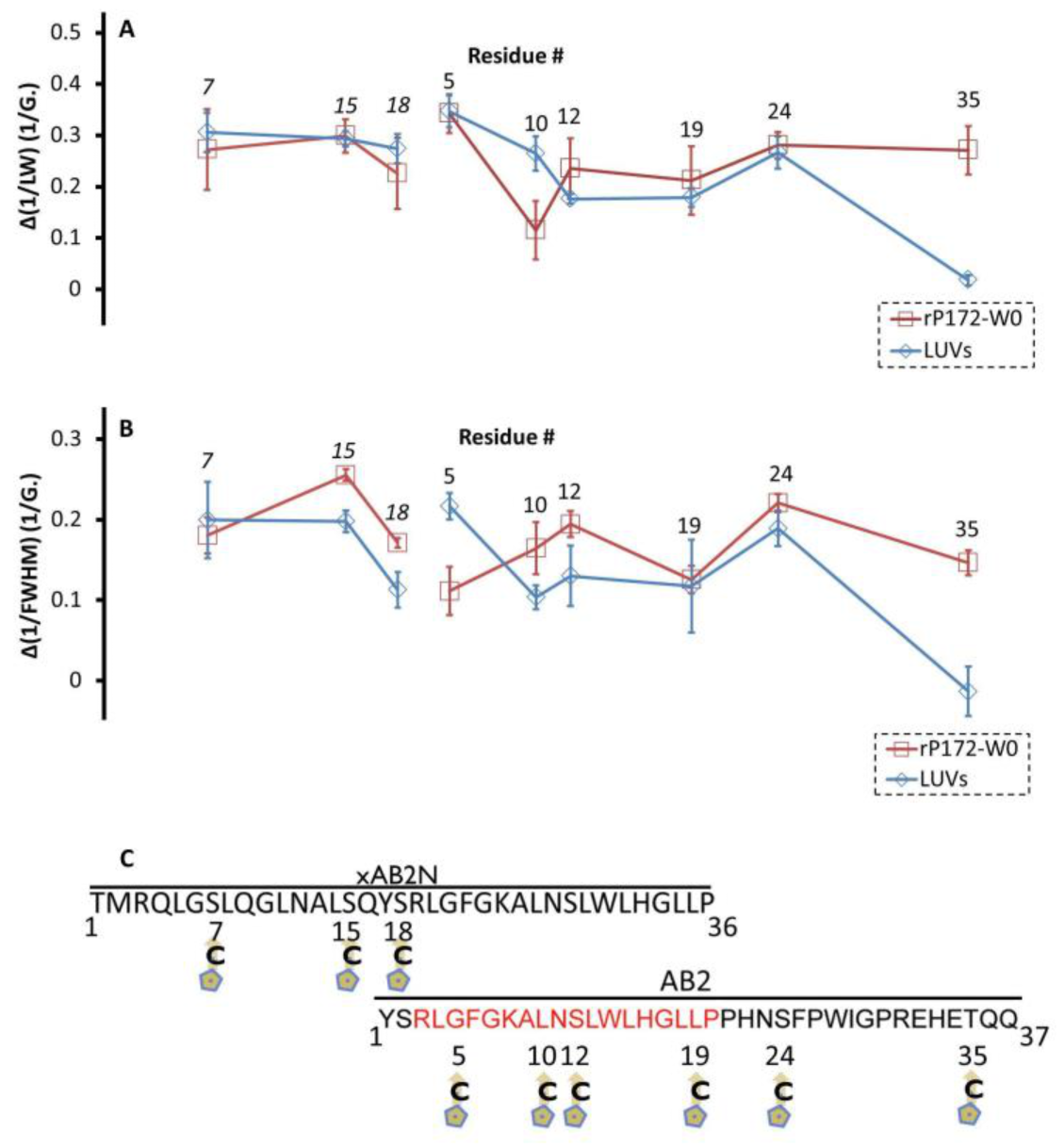
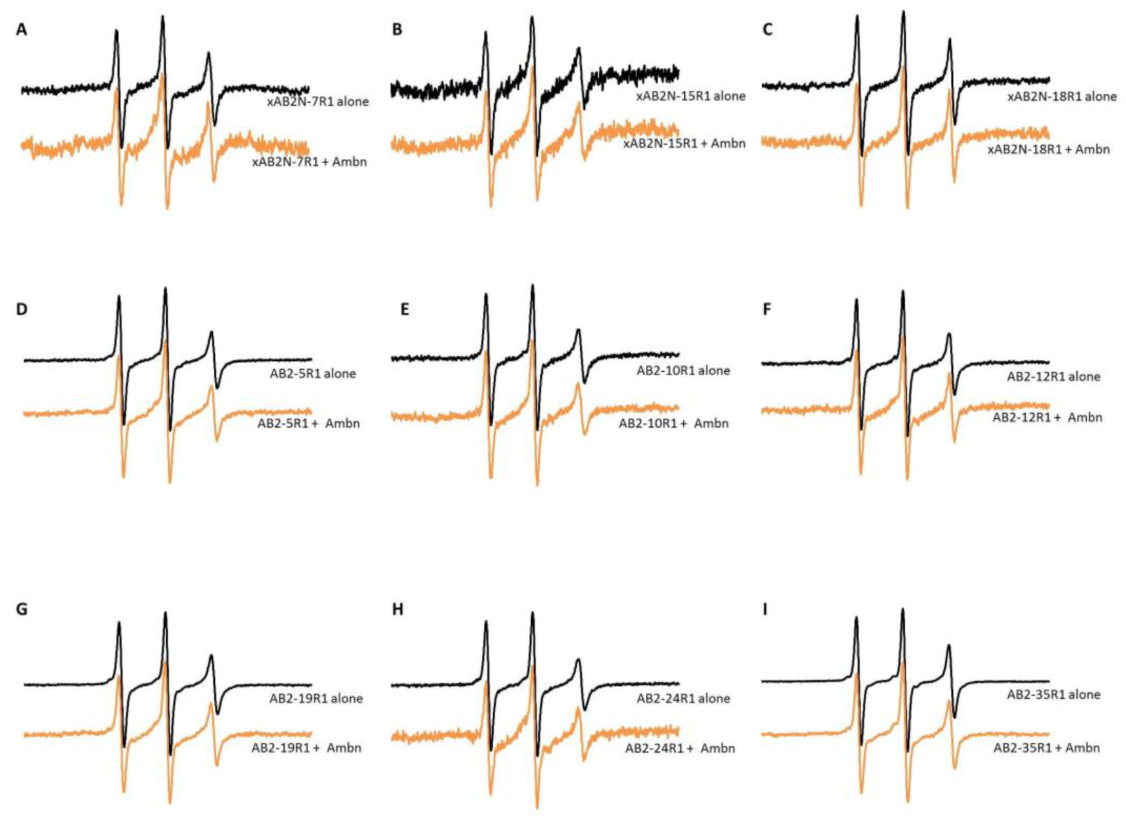
2.2. EPR Revealed That Ambn Peptides Gain Secondary Structure upon Interaction with Liposomes
2.3. EPR Revealed That Ambn Peptides Gain Tertiary/Quaternary Contacts upon Interaction with Full-Length Amel
2.4. EPR Revealed That Ambn Peptides Gain Structural Order upon Interaction with Full-Length Ambn
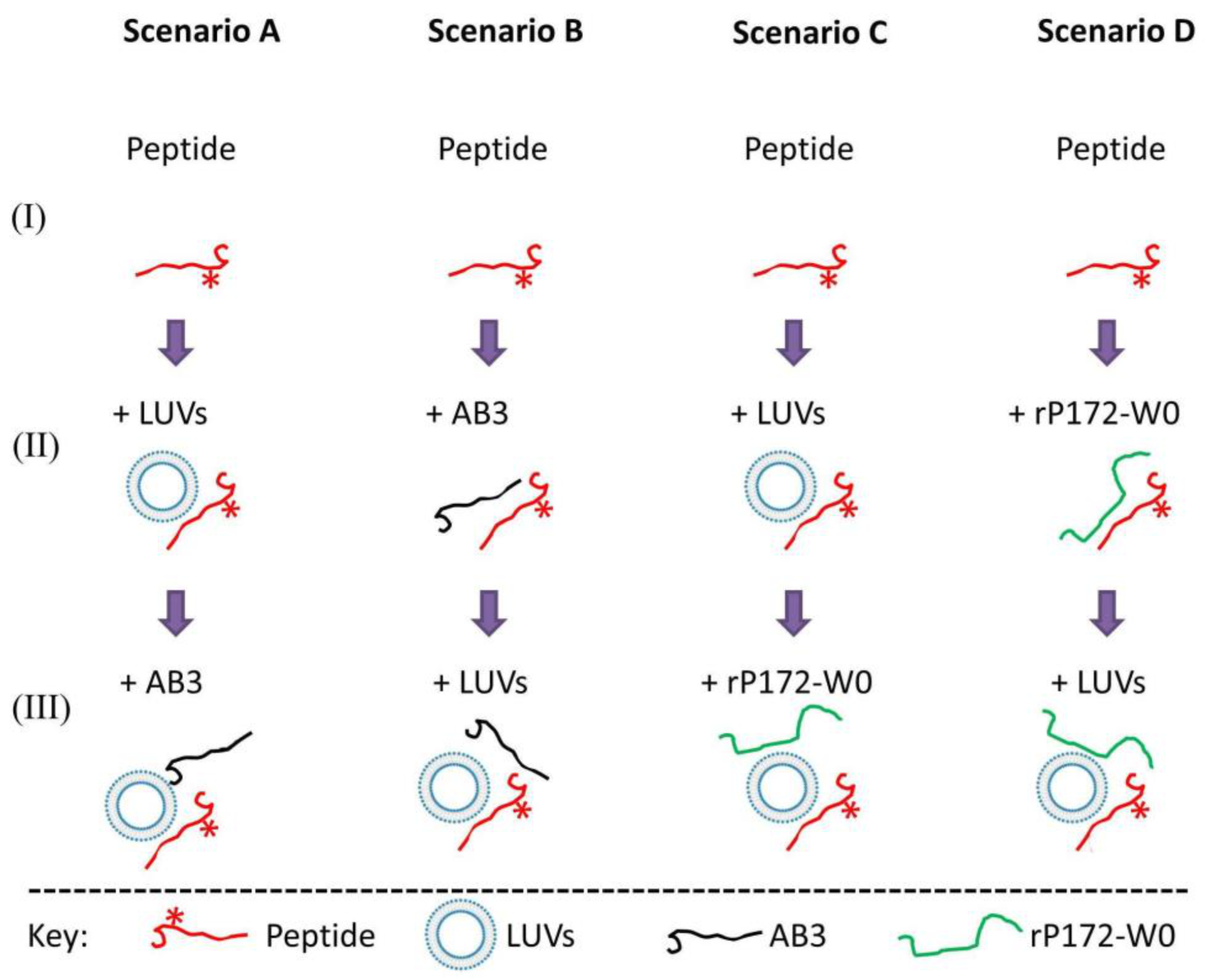
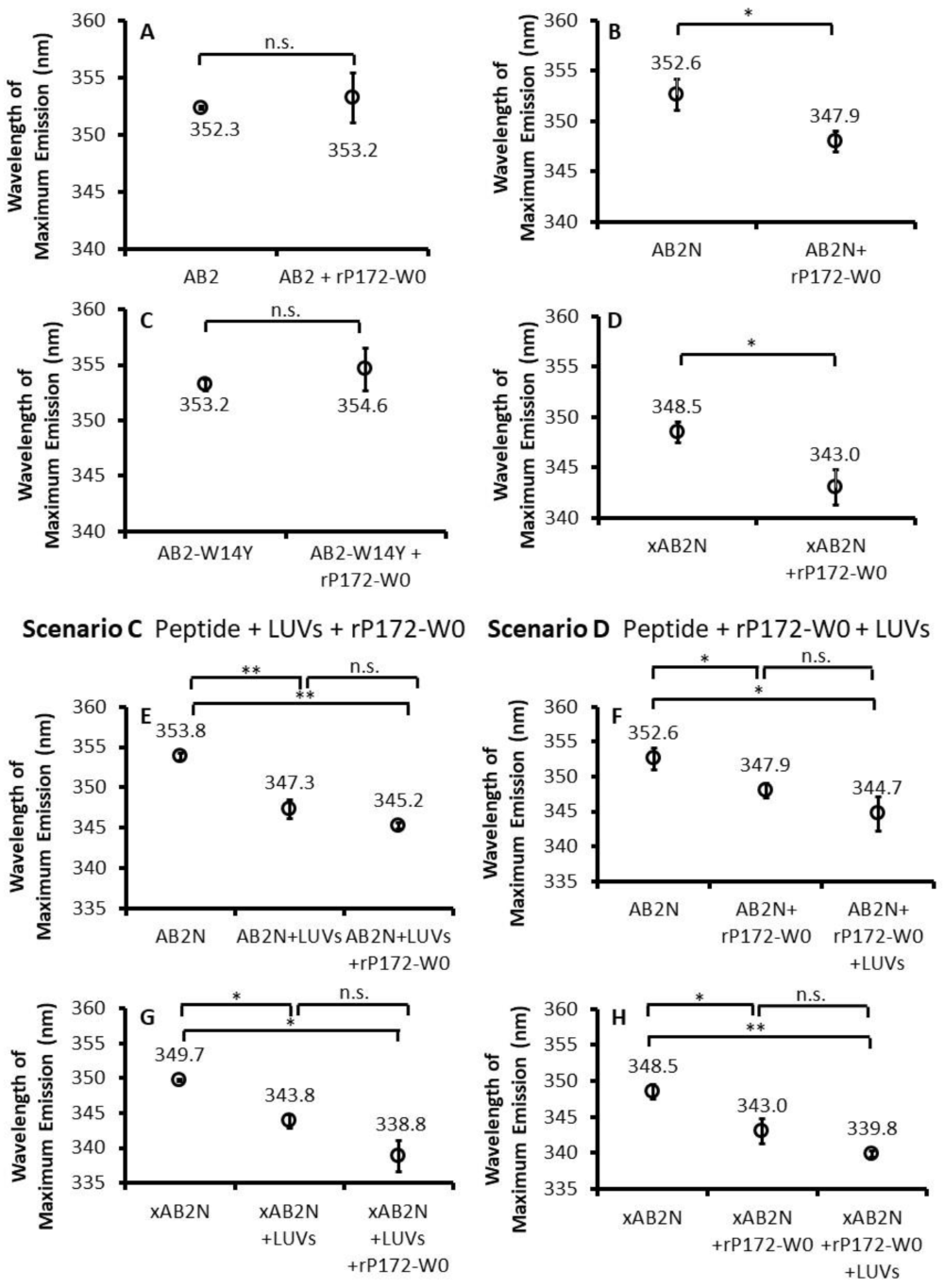
2.5. Experimental Scenarios to Examine Simultaneous Ambn Peptide Interactions with Its Targets
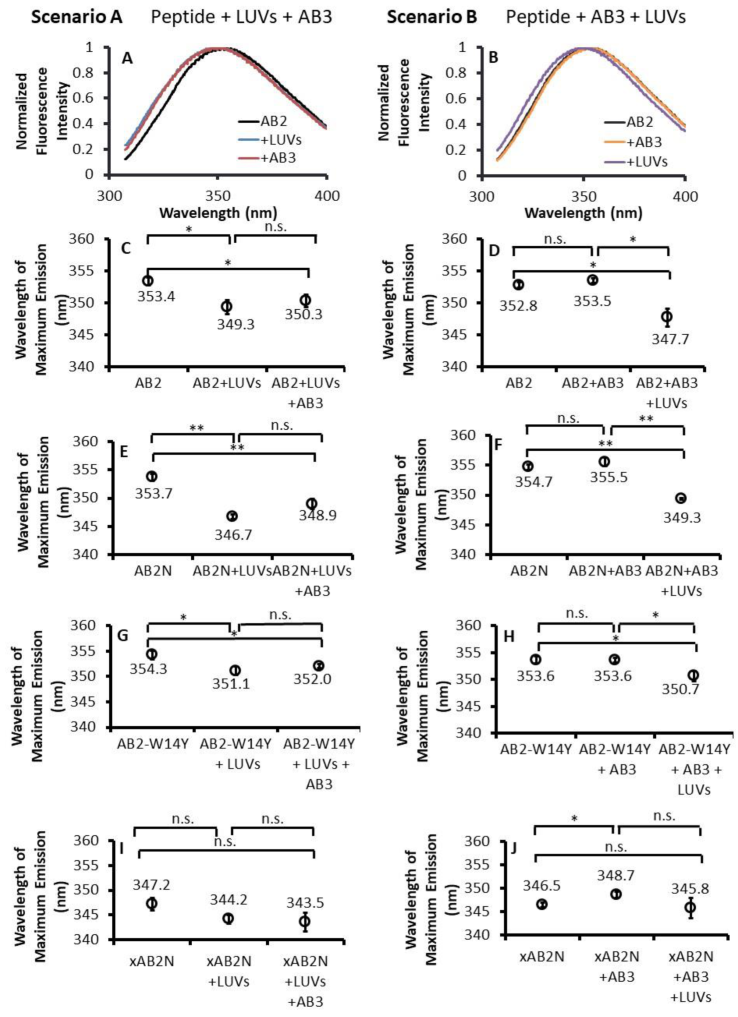
2.6. Fluorescence Spectroscopy Showed Concomitant Ambn Self-Association and Ambn–Membrane Interaction
2.7. AB2–Membrane Interactions Are Independent of AB2 Self-Association
2.8. Ambn has Dynamic Interactions with Amel and Membranes near Its Self-Assembly Region
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification and Peptide Synthesis
4.2. LUV Preparation
4.3. CD
4.4. EPR
4.5. Intrinsic Tryptophan Fluorescence
4.6. Ambn–Membrane Interactions by Leakage Assay
4.7. Ambn–Membrane Interactions by Clearance Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AB1 | Peptide composed of the 40 amino acids encoded by mouse Ambn exons 3 and 4 |
| AB2 | Peptide composed of the 37 amino acids encoded by mouse Ambn exon 5 |
| AB2N | AB2 N-terminus |
| AB3 | Tryptophan-free AB2 |
| AH | Amphipathic helix |
| Ambn | Ameloblastin |
| Amel | Amelogenin |
| ANTS | 8-Aminonaphthalene-1,3,6-trisulfonic acid |
| Amtn | Amelotin |
| BSA | Bovine serum albumin |
| CD | Circular dichroism |
| DPX | P-xylene-bis(pyridinium bromide) |
| EMP | Enamel matrix protein |
| Enam | Enamelin |
| EPR | Electron paramagnetic resonance |
| FWHM | Full width at half maximum |
| G. | Gauss |
| HPLC | High-performance liquid chromatography |
| LUV | Large unilamellar vesicle |
| LW | Linewidth |
| MLV | Multilamellar vesicle |
| MRE | Mean residue ellipticity |
| MTSL | (1-Oxyl-2,2,5,5-tetramethyl-∆3-pyrroline-3-methyl) methanethiosulfonate |
| PI | Phosphatidylinositol |
| POPC | 1-Palmitoyl-2-oleoyl-glycero-3-phosphocholine |
| POPE | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine |
| POPS | 1-Palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine |
| rP172-W0 | Tryptophan-free recombinant porcine amelogenin |
| SDSL | Site-directed spin-labeling |
| SM | Sphingomyelin |
| xAB2N | Extended AB2 N-terminus |
References
- Ravindran, S.; George, A. Multifunctional ECM proteins in bone and teeth. Exp. Cell Res 2014, 325, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Moradian-Oldak, J.; George, A. Biomineralization of Enamel and Dentin Mediated by Matrix Proteins. J. Dent. Res. 2021, 100, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, S.; Kiba, T.; Hall, B.; Iehara, N.; Nakamura, T.; Longenecker, G.; Krebsbach, P.H.; Nanci, A.; Kulkarni, A.B.; Yamada, Y. Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J. Cell Biol. 2004, 167, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Hoang, A.M.; Klebe, R.J.; Steffensen, B.; Ryu, O.H.; Simmer, J.P.; Cochran, D.L. Amelogenin is a cell adhesion protein. J. Dent. Res. 2002, 81, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Fijneman, A.; Wan, J.; Prajapati, S.; Mukherjee, K.; Fernandez-Martinez, A.; Moradian-Oldak, J.; De Yoreo, J.J. Control of Calcium Phosphate Nucleation and Transformation through Interactions of Enamelin and Amelogenin Exhibits the “Goldilocks Effect”. Cryst. Growth Des. 2018, 18, 7391–7400. [Google Scholar] [CrossRef]
- Mazumder, P.; Prajapati, S.; Bapat, R.; Moradian-Oldak, J. Amelogenin-Ameloblastin Spatial Interaction around Maturing Enamel Rods. J. Dent. Res. 2016, 95, 1042–1048. [Google Scholar] [CrossRef]
- Su, J.; Bapat, R.A.; Visakan, G.; Moradian-Oldak, J. An Evolutionarily Conserved Helix Mediates Ameloblastin-Cell Interaction. J. Dent. Res. 2020, 99, 1072–1081. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kang, J.; Seymen, F.; Koruyucu, M.; Zhang, H.; Kasimoglu, Y.; Bayram, M.; Tuna-Ince, E.B.; Bayrak, S.; Tuloglu, N.; et al. Alteration of Exon Definition Causes Amelogenesis Imperfecta. J. Dent. Res. 2020, 99, 410–418. [Google Scholar] [CrossRef]
- Lu, T.; Li, M.; Xu, X.; Xiong, J.; Huang, C.; Zhang, X.; Hu, A.; Peng, L.; Cai, D.; Zhang, L.; et al. Whole exome sequencing identifies an AMBN missense mutation causing severe autosomal-dominant amelogenesis imperfecta and dentin disorders. Int. J. Oral Sci. 2018, 10, 26. [Google Scholar] [CrossRef]
- Brookes, S.J.; Barron, M.J.; Smith, C.E.L.; Poulter, J.A.; Mighell, A.J.; Inglehearn, C.F.; Brown, C.J.; Rodd, H.; Kirkham, J.; Dixon, M.J. Amelogenesis imperfecta caused by N-terminal enamelin point mutations in mice and men is driven by endoplasmic reticulum stress. Hum. Mol. Genet. 2017, 26, 1863–1876. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.E.; Murillo, G.; Brookes, S.J.; Poulter, J.A.; Silva, S.; Kirkham, J.; Inglehearn, C.F.; Mighell, A.J. Deletion of amelotin exons 3-6 is associated with amelogenesis imperfecta. Hum. Mol. Genet. 2016, 25, 3578–3587. [Google Scholar] [CrossRef]
- Crawford, P.J.; Aldred, M.; Bloch-Zupan, A. Amelogenesis imperfecta. Orphanet. J. Rare Dis 2007, 2, 17. [Google Scholar] [CrossRef]
- Boskey, A.L.; Villarreal-Ramirez, E. Intrinsically disordered proteins and biomineralization. Matrix Biol. 2016, 52–54, 43–59. [Google Scholar] [CrossRef]
- Kawasaki, K.; Weiss, K.M. Mineralized tissue and vertebrate evolution: The secretory calcium-binding phosphoprotein gene cluster. Proc. Natl. Acad. Sci. USA 2003, 100, 4060–4065. [Google Scholar] [CrossRef]
- Krebsbach, P.H.; Lee, S.K.; Matsuki, Y.; Kozak, C.A.; Yamada, K.M.; Yamada, Y. Full-length sequence, localization, and chromosomal mapping of ameloblastin. A novel tooth-specific gene. J. Biol. Chem. 1996, 271, 4431–4435. [Google Scholar] [CrossRef]
- Lacruz, R.S.; Habelitz, S.; Wright, J.T.; Paine, M.L. Dental Enamel Formation and Implications for Oral Health and Disease. Physiol. Rev. 2017, 97, 939–993. [Google Scholar] [CrossRef]
- Bartlett, J.D.; Simmer, J.P. Proteinases in developing dental enamel. Crit. Rev. Oral Biol. Med. 1999, 10, 425–441. [Google Scholar] [CrossRef]
- Smith, C.E. Cellular and chemical events during enamel maturation. Crit. Rev. Oral Biol. Med. 1998, 9, 128–161. [Google Scholar] [CrossRef]
- Fincham, A.G.; Moradian-Oldak, J.; Diekwisch, T.G.; Lyaruu, D.M.; Wright, J.T.; Bringas, P., Jr.; Slavkin, H.C. Evidence for amelogenin “nanospheres” as functional components of secretory-stage enamel matrix. J. Struct. Biol. 1995, 115, 50–59. [Google Scholar] [CrossRef]
- Fang, P.A.; Conway, J.F.; Margolis, H.C.; Simmer, J.P.; Beniash, E. Hierarchical self-assembly of amelogenin and the regulation of biomineralization at the nanoscale. Proc. Natl. Acad. Sci. USA 2011, 108, 14097–14102. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, L.; Qin, Y.; Sun, Z.; Henneman, Z.J.; Moradian-Oldak, J.; Nancollas, G.H. How amelogenin orchestrates the organization of hierarchical elongated microstructures of apatite. J. Phys. Chem. B 2010, 114, 2293–2300. [Google Scholar] [CrossRef] [PubMed]
- Jacques, J.; Hotton, D.; De la Dure-Molla, M.; Petit, S.; Asselin, A.; Kulkarni, A.B.; Gibson, C.W.; Brookes, S.J.; Berdal, A.; Isaac, J. Tracking endogenous amelogenin and ameloblastin in vivo. PLoS ONE 2014, 9, e99626. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Bapat, R.A.; Su, J.; Moradian-Oldak, J. Regulation of Hydroxyapatite Nucleation In Vitro through Ameloblastin-Amelogenin Interactions. ACS Biomater. Sci. Eng. 2022. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, P.; Prajapati, S.; Lokappa, S.B.; Gallon, V.; Moradian-Oldak, J. Analysis of co-assembly and co-localization of ameloblastin and amelogenin. Front. Physiol. 2014, 5, 274. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Kegulian, N.C.; Arun Bapat, R.; Moradian-Oldak, J. Ameloblastin Binds to Phospholipid Bilayers via a Helix-Forming Motif within the Sequence Encoded by Exon 5. ACS Omega 2019, 4, 4405–4416. [Google Scholar] [CrossRef]
- Su, J.; Arun Bapat, R.; Visakan, G.; Moradian-Oldak, J. Co-emergence of the amphipathic helix on ameloblastin with mammalian prismatic enamel. Mol. Biol. Evol. 2022, 39, msac205. [Google Scholar] [CrossRef]
- Wald, T.; Spoutil, F.; Osickova, A.; Prochazkova, M.; Benada, O.; Kasparek, P.; Bumba, L.; Klein, O.D.; Sedlacek, R.; Sebo, P.; et al. Intrinsically disordered proteins drive enamel formation via an evolutionarily conserved self-assembly motif. Proc. Natl. Acad. Sci. USA 2017, 114, E1641–E1650. [Google Scholar] [CrossRef]
- Bapat, R.A.; Su, J.; Moradian-Oldak, J. Co-Immunoprecipitation Reveals Interactions Between Amelogenin and Ameloblastin via Their Self-Assembly Domains. Front. Physiol. 2020, 11, 622086. [Google Scholar] [CrossRef]
- Ravindranath, H.H.; Chen, L.S.; Zeichner-David, M.; Ishima, R.; Ravindranath, R.M. Interaction between the enamel matrix proteins amelogenin and ameloblastin. Biochem. Biophys. Res. Commun. 2004, 323, 1075–1083. [Google Scholar] [CrossRef]
- Su, J.; Chandrababu, K.B.; Moradian-Oldak, J. Ameloblastin peptide encoded by exon 5 interacts with amelogenin N-terminus. Biochem. Biophys. Rep. 2016, 7, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Marquez, M.G.; Nieto, F.L.; Fernandez-Tome, M.C.; Favale, N.O.; Sterin-Speziale, N. Membrane lipid composition plays a central role in the maintenance of epithelial cell adhesion to the extracellular matrix. Lipids 2008, 43, 343–352. [Google Scholar] [CrossRef]
- Sahu, I.D.; Lorigan, G.A. Electron Paramagnetic Resonance as a Tool for Studying Membrane Proteins. Biomolecules 2020, 10, 763. [Google Scholar] [CrossRef]
- Jao, C.C.; Hegde, B.G.; Chen, J.; Haworth, I.S.; Langen, R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA 2008, 105, 19666–19671. [Google Scholar] [CrossRef]
- Chen, D.; Wang, J.M.; Lanyi, J.K. Electron paramagnetic resonance study of structural changes in the O photointermediate of bacteriorhodopsin. J. Mol. Biol. 2007, 366, 790–805. [Google Scholar] [CrossRef]
- Feix, J.B.; Klug, C.S. Site-Directed Spin Labeling of Membrane Proteins and Peptide-Membrane Interactions. In Biological Magnetic Resonance; Spin Labeling: The Next Millennium; Berliner, L.J., Ed.; Plenum Press: New York, NY, USA, 1998; Volume 14, pp. 251–281. [Google Scholar]
- Margittai, M.; Langen, R. Spin labeling analysis of amyloids and other protein aggregates. Methods Enzym. 2006, 413, 122–139. [Google Scholar]
- Mobius, K.; Lubitz, W.; Savitsky, A. High-field EPR on membrane proteins—Crossing the gap to NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 75, 1–49. [Google Scholar] [CrossRef]
- Schuck, P. Use of surface plasmon resonance to probe the equilibrium and dynamic aspects of interactions between biological macromolecules. Annu. Rev. Biophys. Biomol. Struct. 1997, 26, 541–566. [Google Scholar] [CrossRef]
- Siemer, A.B. Advances in studying protein disorder with solid-state NMR. Solid State Nucl. Magn. Reson. 2020, 106, 101643. [Google Scholar] [CrossRef]
- Tao, M.; Pandey, N.K.; Barnes, R.; Han, S.; Langen, R. Structure of Membrane-Bound Huntingtin Exon 1 Reveals Membrane Interaction and Aggregation Mechanisms. Structure 2019, 27, 1570–1580.e1574. [Google Scholar] [CrossRef]
- Xia, Y.; Fischer, A.W.; Teixeira, P.; Weiner, B.; Meiler, J. Integrated Structural Biology for alpha-Helical Membrane Protein Structure Determination. Structure 2018, 26, 657–666.e652. [Google Scholar] [CrossRef]
- Rustad, M.D.; Roopnarine, O.; Cornea, R.L.; Thomas, D.D. Interaction of DWORF with SERCA and PLB as determined by EPR spectroscopy. Biochem. Biophys. Res. Commun. 2023, 645, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, F.F.; Sanchania, V.A.; Tolner, B.; Wright, Z.V.; Ryan, C.P.; Smith, M.E.; Ward, J.M.; Caddick, S.; Kay, C.W.; Aeppli, G.; et al. Homogeneous antibody fragment conjugation by disulfide bridging introduces ‘spinostics’. Sci. Rep. 2013, 3, 1525. [Google Scholar] [CrossRef] [PubMed]
- Haeri, H.H.; Blaffert, J.; Schoffmann, F.A.; Blech, M.; Hartl, J.; Garidel, P.; Hinderberger, D. Concentration Effects in the Interaction of Monoclonal Antibodies (mAbs) with their Immediate Environment Characterized by EPR Spectroscopy. Molecules 2019, 24, 2528. [Google Scholar] [CrossRef] [PubMed]
- Stakkestad, O.; Lyngstadaas, S.P.; Thiede, B.; Vondrasek, J.; Skalhegg, B.S.; Reseland, J.E. Phosphorylation Modulates Ameloblastin Self-assembly and Ca (2+) Binding. Front. Physiol. 2017, 8, 531. [Google Scholar] [CrossRef]
- Wald, T.; Osickova, A.; Sulc, M.; Benada, O.; Semeradtova, A.; Rezabkova, L.; Veverka, V.; Bednarova, L.; Maly, J.; Macek, P.; et al. Intrinsically disordered enamel matrix protein ameloblastin forms ribbon-like supramolecular structures via an N-terminal segment encoded by exon 5. J. Biol. Chem. 2013, 288, 22333–22345. [Google Scholar] [CrossRef]
- Raghuraman, H.; Chatterjee, S.; Das, A. Site-Directed Fluorescence Approaches for Dynamic Structural Biology of Membrane Peptides and Proteins. Front. Mol. Biosci. 2019, 6, 96. [Google Scholar] [CrossRef]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic protein-membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 2010, 49, 5628–5654. [Google Scholar] [CrossRef]
- Kegulian, N.C.; Sankhagowit, S.; Apostolidou, M.; Jayasinghe, S.A.; Malmstadt, N.; Butler, P.C.; Langen, R. Membrane Curvature-sensing and Curvature-inducing Activity of Islet Amyloid Polypeptide and Its Implications for Membrane Disruption. J. Biol. Chem. 2015, 290, 25782–25793. [Google Scholar] [CrossRef]
- Moradian-Oldak, J. Protein-mediated enamel mineralization. Front. Biosci. (Landmark Ed.) 2012, 17, 1996–2023. [Google Scholar] [CrossRef]
- Wilman, H.R.; Shi, J.; Deane, C.M. Helix kinks are equally prevalent in soluble and membrane proteins. Proteins 2014, 82, 1960–1970. [Google Scholar] [CrossRef]
- Li, S.C.; Goto, N.K.; Williams, K.A.; Deber, C.M. Alpha-helical, but not beta-sheet, propensity of proline is determined by peptide environment. Proc. Natl. Acad. Sci. USA 1996, 93, 6676–6681. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Barkley, M.D. Toward understanding tryptophan fluorescence in proteins. Biochemistry 1998, 37, 9976–9982. [Google Scholar] [CrossRef]
- Ghisaidoobe, A.B.; Chung, S.J. Intrinsic tryptophan fluorescence in the detection and analysis of proteins: A focus on Forster resonance energy transfer techniques. Int. J. Mol. Sci. 2014, 15, 22518–22538. [Google Scholar] [CrossRef]
- Vetyskova, V.; Zouharova, M.; Bednarova, L.; Vanek, O.; Sazelova, P.; Kasicka, V.; Vymetal, J.; Srp, J.; Rumlova, M.; Charnavets, T.; et al. Characterization of AMBN I and II Isoforms and Study of Their Ca(2+)-Binding Properties. Int. J. Mol. Sci. 2020, 21, 9293. [Google Scholar] [CrossRef]
- Ambroso, M.R.; Hegde, B.G.; Langen, R. Endophilin A1 induces different membrane shapes using a conformational switch that is regulated by phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, 6982–6987. [Google Scholar] [CrossRef]
- Isas, J.M.; Ambroso, M.R.; Hegde, P.B.; Langen, J.; Langen, R. Tubulation by amphiphysin requires concentration-dependent switching from wedging to scaffolding. Structure 2015, 23, 873–881. [Google Scholar] [CrossRef]
- Michalek, M.; Salnikov, E.S.; Bechinger, B. Structure and topology of the huntingtin 1–17 membrane anchor by a combined solution and solid-state NMR approach. Biophys. J. 2013, 105, 699–710. [Google Scholar] [CrossRef]
- Varkey, J.; Isas, J.M.; Mizuno, N.; Jensen, M.B.; Bhatia, V.K.; Jao, C.C.; Petrlova, J.; Voss, J.C.; Stamou, D.G.; Steven, A.C.; et al. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem. 2010, 285, 32486–32493. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef]
- Zhang, X.; Diekwisch, T.G.; Luan, X. Structure and function of ameloblastin as an extracellular matrix protein: Adhesion, calcium binding, and CD63 interaction in human and mouse. Eur. J. Oral Sci. 2011, 119 (Suppl. S1), 270–279. [Google Scholar] [CrossRef]
- Cerny, R.; Slaby, I.; Hammarstrom, L.; Wurtz, T. A novel gene expressed in rat ameloblasts codes for proteins with cell binding domains. J. Bone Miner. Res. 1996, 11, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Beyeler, M.; Schild, C.; Lutz, R.; Chiquet, M.; Trueb, B. Identification of a fibronectin interaction site in the extracellular matrix protein ameloblastin. Exp. Cell Res. 2010, 316, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Lokappa, S.B.; Chandrababu, K.B.; Dutta, K.; Perovic, I.; Evans, J.S.; Moradian-Oldak, J. Interactions of amelogenin with phospholipids. Biopolymers 2015, 103, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Lokappa, S.B.; Chandrababu, K.B.; Moradian-Oldak, J. Tooth enamel protein amelogenin binds to ameloblast cell membrane-mimicking vesicles via its N-terminus. Biochem. Biophys. Res. Commun. 2015, 464, 956–961. [Google Scholar] [CrossRef]
- Wazen, R.M.; Moffatt, P.; Zalzal, S.F.; Yamada, Y.; Nanci, A. A mouse model expressing a truncated form of ameloblastin exhibits dental and junctional epithelium defects. Matrix Biol. 2009, 28, 292–303. [Google Scholar] [CrossRef]
- Poulter, J.A.; Murillo, G.; Brookes, S.J.; Smith, C.E.; Parry, D.A.; Silva, S.; Kirkham, J.; Inglehearn, C.F.; Mighell, A.J. Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Hum. Mol. Genet. 2014, 23, 5317–5324. [Google Scholar] [CrossRef]
- Visakan, G.; Su, J.; Moradian-Oldak, J. Ameloblastin promotes polarization of ameloblast cell lines in a 3-D cell culture system. Matrix Biol. 2022, 105, 72–86. [Google Scholar] [CrossRef]
- Hatakeyama, J.; Fukumoto, S.; Nakamura, T.; Haruyama, N.; Suzuki, S.; Hatakeyama, Y.; Shum, L.; Gibson, C.W.; Yamada, Y.; Kulkarni, A.B. Synergistic roles of amelogenin and ameloblastin. J. Dent. Res. 2009, 88, 318–322. [Google Scholar] [CrossRef]
- Zalzal, S.F.; Smith, C.E.; Nanci, A. Ameloblastin and amelogenin share a common secretory pathway and are co-secreted during enamel formation. Matrix Biol. 2008, 27, 352–359. [Google Scholar] [CrossRef]
- Ravindranath, R.M.; Devarajan, A.; Uchida, T. Spatiotemporal expression of ameloblastin isoforms during murine tooth development. J. Biol. Chem. 2007, 282, 36370–36376. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yamakoshi, Y.; Hu, J.C.; Gomi, K.; Arai, T.; Fukae, M.; Krebsbach, P.H.; Simmer, J.P. Splicing determines the glycosylation state of ameloblastin. J. Dent. Res. 2007, 86, 962–967. [Google Scholar] [CrossRef]
- MacDougall, M.; Simmons, D.; Gu, T.T.; Forsman-Semb, K.; Mardh, C.K.; Mesbah, M.; Forest, N.; Krebsbach, P.H.; Yamada, Y.; Berdal, A. Cloning, characterization and immunolocalization of human ameloblastin. Eur. J. Oral Sci. 2000, 108, 303–310. [Google Scholar] [CrossRef]
- Chun, Y.H.; Yamakoshi, Y.; Yamakoshi, F.; Fukae, M.; Hu, J.C.; Bartlett, J.D.; Simmer, J.P. Cleavage site specificity of MMP-20 for secretory-stage ameloblastin. J. Dent. Res. 2010, 89, 785–790. [Google Scholar] [CrossRef]
- Moss-Salentijn, L.; Moss, M.L.; Yuan, M.S.-T. The ontogeny of mammalian enamel. In Tooth Enamel Microstructure; von Koenigswald, W., Sander, P.M., Eds.; CRC Press: London, UK, 1997; pp. 5–30. [Google Scholar]
- Nikoloudaki, G. Functions of Matricellular Proteins in Dental Tissues and Their Emerging Roles in Orofacial Tissue Development, Maintenance, and Disease. Int. J. Mol. Sci. 2021, 22, 6626. [Google Scholar] [CrossRef]
- Bromley, K.M.; Kiss, A.S.; Lokappa, S.B.; Lakshminarayanan, R.; Fan, D.; Ndao, M.; Evans, J.S.; Moradian-Oldak, J. Dissecting amelogenin protein nanospheres: Characterization of metastable oligomers. J. Biol. Chem. 2011, 286, 34643–34653. [Google Scholar] [CrossRef]
- Lakshminarayanan, R.; Yoon, I.; Hegde, B.G.; Fan, D.; Du, C.; Moradian-Oldak, J. Analysis of secondary structure and self-assembly of amelogenin by variable temperature circular dichroism and isothermal titration calorimetry. Proteins 2009, 76, 560–569. [Google Scholar] [CrossRef]
- Su, J.; Bapat, R.A.; Moradian-Oldak, J. The Expression and Purification of Recombinant Mouse Ameloblastin in E. coli. Methods Mol. Biol. 2019, 1922, 229–236. [Google Scholar]
- Ladokhin, A.S.; Wimley, W.C.; White, S.H. Leakage of membrane vesicle contents: Determination of mechanism using fluorescence requenching. Biophys. J. 1995, 69, 1964–1971. [Google Scholar] [CrossRef]
- Greenfield, N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kegulian, N.C.; Langen, R.; Moradian-Oldak, J. The Dynamic Interactions of a Multitargeting Domain in Ameloblastin Protein with Amelogenin and Membrane. Int. J. Mol. Sci. 2023, 24, 3484. https://doi.org/10.3390/ijms24043484
Kegulian NC, Langen R, Moradian-Oldak J. The Dynamic Interactions of a Multitargeting Domain in Ameloblastin Protein with Amelogenin and Membrane. International Journal of Molecular Sciences. 2023; 24(4):3484. https://doi.org/10.3390/ijms24043484
Chicago/Turabian StyleKegulian, Natalie C., Ralf Langen, and Janet Moradian-Oldak. 2023. "The Dynamic Interactions of a Multitargeting Domain in Ameloblastin Protein with Amelogenin and Membrane" International Journal of Molecular Sciences 24, no. 4: 3484. https://doi.org/10.3390/ijms24043484