
How Can Insulin Resistance Cause Alzheimer’s Disease?


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Definition and Epidemiology of AD
3. Pathophysiology of AD
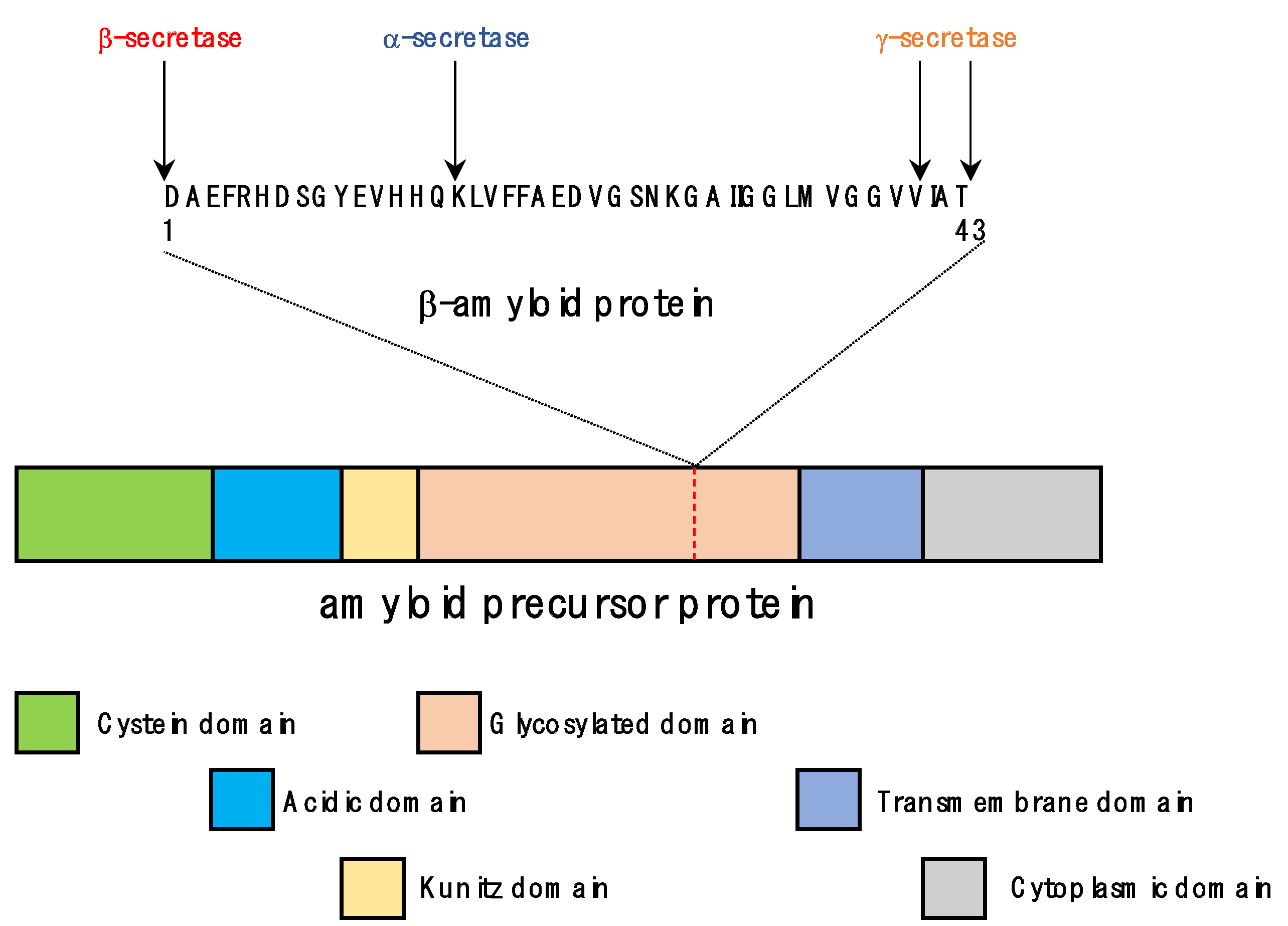
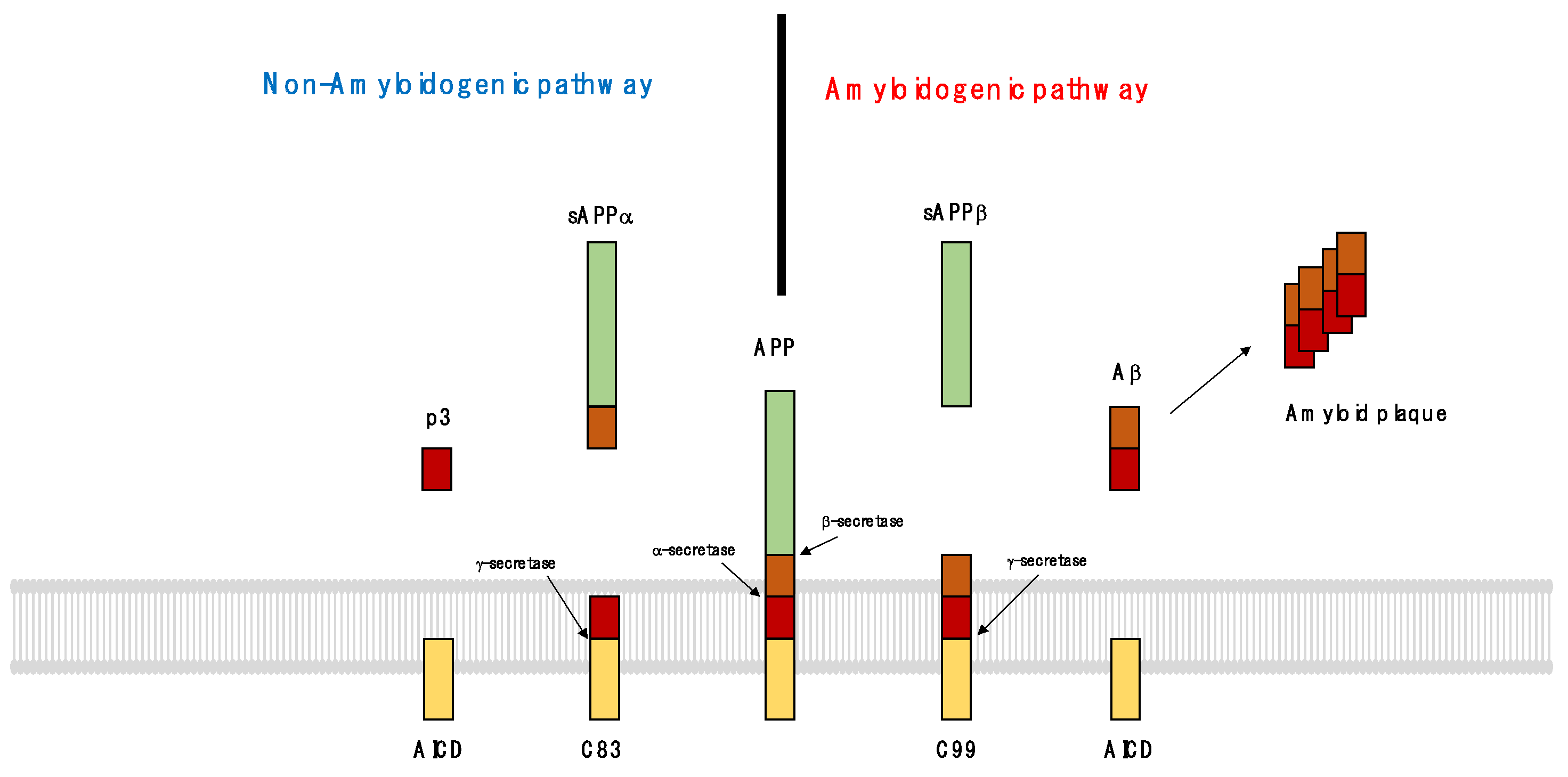
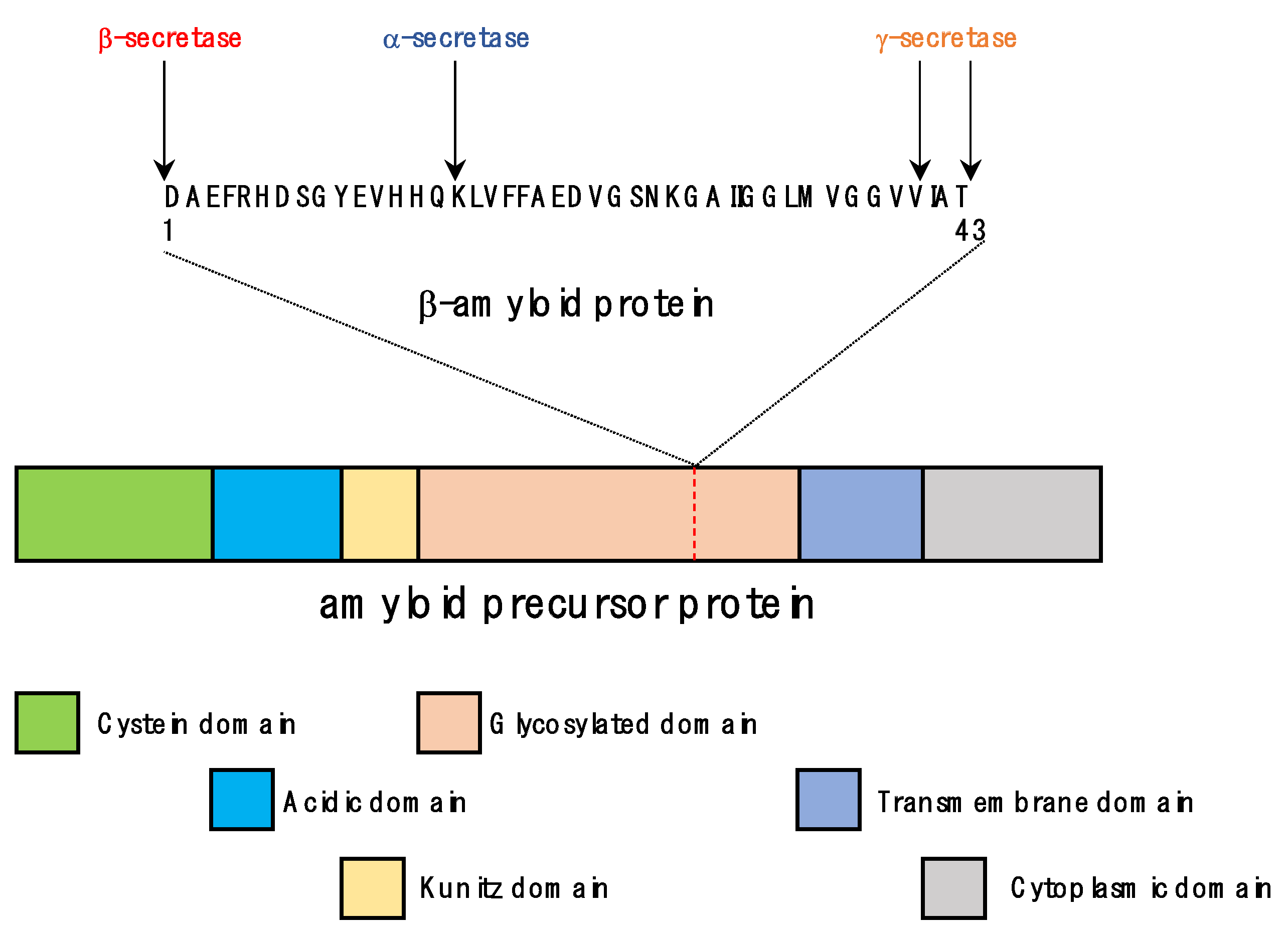
3.1. Functions of APP and Tau
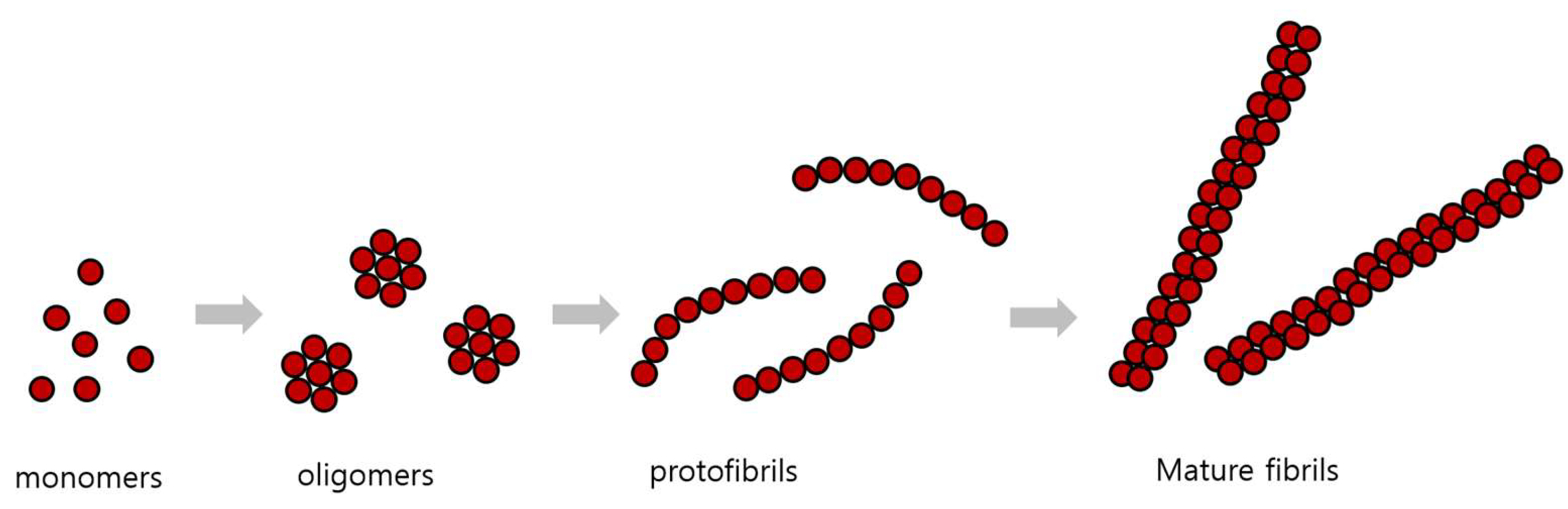
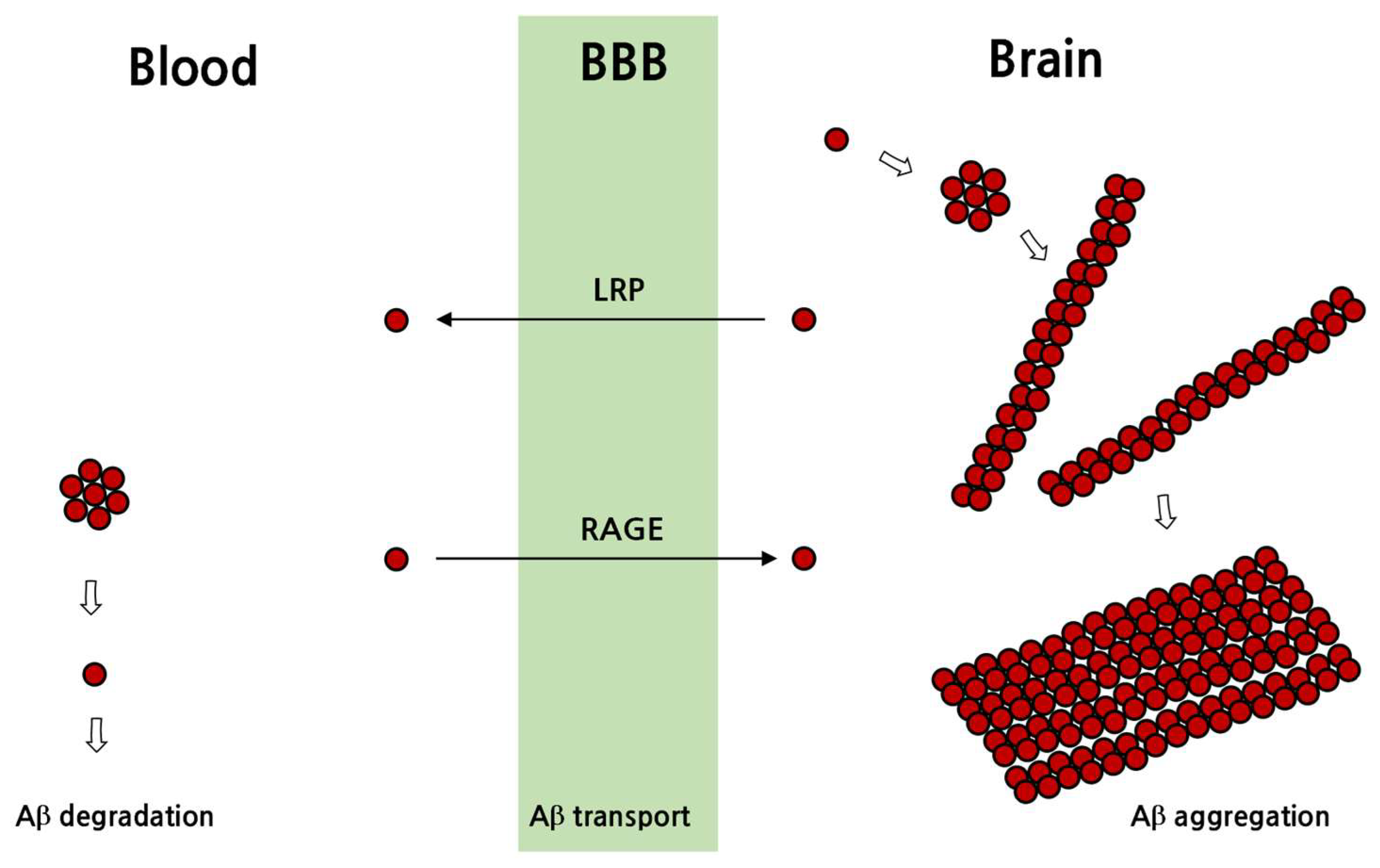
3.2. Pathological Processes in the Formation of Senile Plaques and Neurofibrillary Tangles
3.3. AD Treatment Development Status
4. Why Insulin Resistance Is Linked to AD
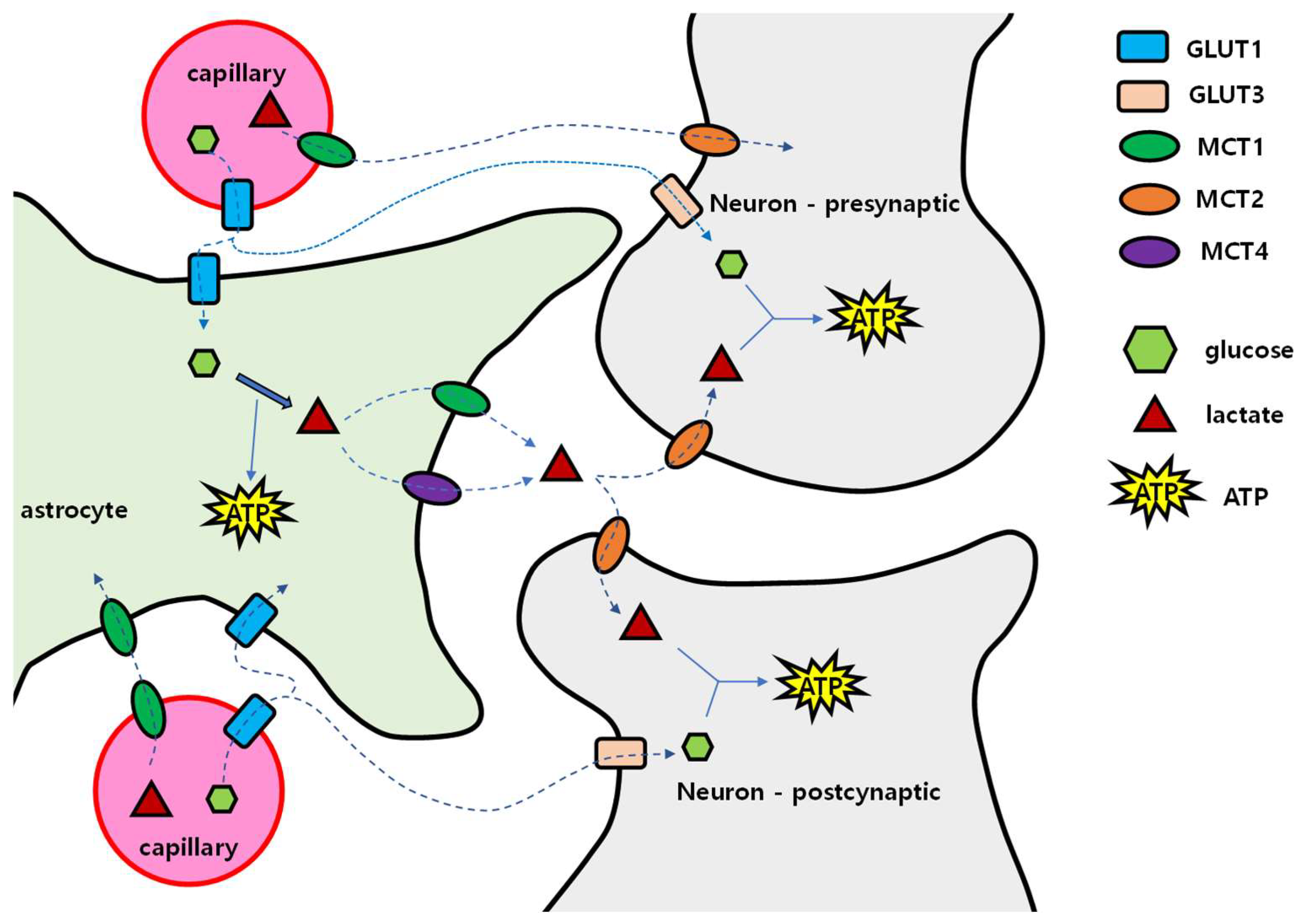
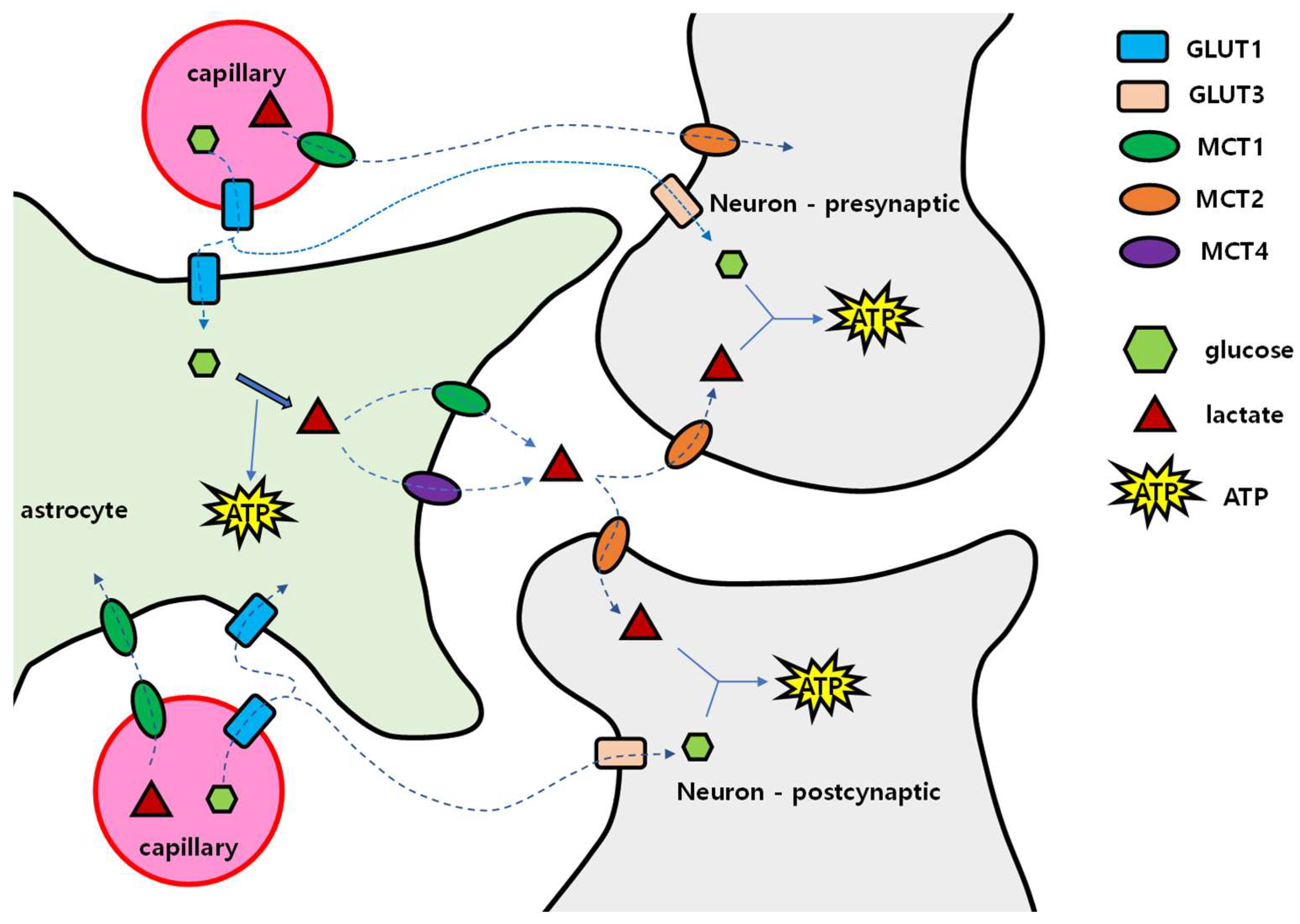
4.1. Brain Energy Mobilization
4.2. When and How the Brain Deals with the Lack of Glucose
5. Insulin and Insulin Resistance
5.1. Synthesis, Actions, and Degradation of Insulin
5.2. How Does Insulin Resistance Emerge?
5.3. Insulin Action on the Brain
5.4. Brain Insulin Resistance
6. How Insulin Resistance Causes Dementia
6.1. Brain Glucose Insufficiency
6.2. Brain Insulin Insufficiency
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin resistance and pathological brain ageing. Diabet. Med. 2011, 28, 1463–1475. [Google Scholar]
- van der Flier, W.M.; Scheltens, P. Epidemiology and risk factors of dementia. J. Neurol. Neurosurg. Psychiatry 2005, 76 (Suppl. S5), v2–v7. [Google Scholar] [CrossRef]
- Grundman, M.; Thal, L.J. Treatment of Alzheimer’s disease: Rationale and strategies. Neurol. Clin. 2000, 18, 807–828. [Google Scholar]
- O’Brien, J.T.; Burns, A.; BAP Dementia Consensus Group. Clinical practice with anti-dementia drugs: A revised (second) consensus statement from the British Association for Psychopharmacology. J. Psychopharmacol. 2011, 25, 997–1019. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Neff, R.A.; Wang, M.; Vatansever, S.; Guo, L.; Ming, C.; Wang, Q.; Wang, E.; Horgusluoglu-Moloch, E.; Song, W.M.; Li, A.; et al. Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci. Adv. 2021, 7, eabb5398. [Google Scholar] [CrossRef] [PubMed]
- Iacono, D.; Resnick, S.M.; O’Brien, R.; Zonderman, A.B.; An, Y.; Pletnikova, O.; Rudow, G.; Crain, B.; Troncoso, J.C. Mild cognitive impairment and asymptomatic Alzheimer disease subjects: Equivalent beta-amyloid and tau loads with divergent cognitive outcomes. J. Neuropathol. Exp. Neurol. 2014, 73, 295–304. [Google Scholar] [PubMed]
- Bredesen, D.E. Metabolic profiling distinguishes three subtypes of Alzheimer’s disease. Aging 2015, 7, 595–600. [Google Scholar] [CrossRef]
- Phillips, J.S.; Da Re, F.; Dratch, L.; Xie, S.X.; Irwin, D.J.; McMillan, C.T.; Vaishnavi, S.N.; Ferrarese, C.; Lee, E.B.; Shaw, L.M.; et al. Neocortical origin and progression of gray matter atrophy in nonamnestic Alzheimer’s disease. Neurobiol. Aging 2018, 63, 75–87. [Google Scholar] [CrossRef]
- Qian, W.; Fischer, C.E.; Schweizer, T.A.; Munoz, D.G. Association Between Psychosis Phenotype and APOE Genotype on the Clinical Profiles of Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 187–194. [Google Scholar] [CrossRef]
- Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [PubMed]
- De la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- WHO Report Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 20 September 2022).
- Huang, L.K.; Chao, S.P.; Hu, C.J. Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [PubMed]
- Garn, H.; Coronel, C.; Waser, M.; Caravias, G.; Ransmayr, G. Differential diagnosis between patients with probable Alzheimer’s disease, Parkinson’s disease dementia, or dementia with Lewy bodies and frontotemporal dementia, behavioral variant, using quantitative electroencephalographic features. J. Neural. Transm. 2017, 124, 569–581. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef]
- Association, A. Medications for Memory, Cognition and Dementia-Related Behaviors. Available online: https://www.alz.org/alzheimers-dementia/treatments/medications-for-memory (accessed on 20 September 2022).
- Huang, H.C.; Jiang, Z.F. Accumulated amyloid-beta peptide and hyperphosphorylated tau protein: Relationship and links in Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 15–27. [Google Scholar] [CrossRef]
- Goedert, M. Tau protein and neurodegeneration. Semin. Cell. Dev. Biol. 2004, 15, 45–49. [Google Scholar] [CrossRef]
- Coulson, E.J.; Paliga, K.; Beyreuther, K.; Masters, C.L. What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem. Int. 2000, 36, 175–184. [Google Scholar]
- Tanzi, R.E.; Kovacs, D.M.; Kim, T.W.; Moir, R.D.; Guenette, S.Y.; Wasco, W. The gene defects responsible for familial Alzheimer’s disease. Neurobiol. Dis. 1996, 3, 159–168. [Google Scholar] [CrossRef]
- Jakob-Roetne, R.; Jacobsen, H. Alzheimer’s disease: From pathology to therapeutic approaches. Angew. Chem. Int. Ed. Engl. 2009, 48, 3030–3059. [Google Scholar] [PubMed]
- Mattson, M.P. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol. Rev. 1997, 77, 1081–1132. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Sundsmo, M.; Roch, J.M.; Kimura, N.; Cole, G.; Schubert, D.; Oltersdorf, T.; Schenk, D.B. Secreted form of amyloid beta protein precursor is involved in the growth regulation of fibroblasts. Cell 1989, 58, 615–622. [Google Scholar] [CrossRef]
- Roch, J.M.; Masliah, E.; Roch-Levecq, A.C.; Sundsmo, M.P.; Otero, D.A.; Veinbergs, I.; Saitoh, T. Increase of synaptic density and memory retention by a peptide representing the trophic domain of the amyloid beta/A4 protein precursor. Proc. Natl. Acad. Sci. USA 1994, 91, 7450–7454. [Google Scholar] [CrossRef] [PubMed]
- Meziane, H.; Dodart, J.C.; Mathis, C.; Little, S.; Clemens, J.; Paul, S.M.; Ungerer, A. Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc. Natl. Acad. Sci. USA 1998, 95, 12683–12688. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, J.; Rostagno, A.; Gardella, J.E.; Liem, L.; Gorevic, P.D.; Frangione, B. A 109-amino-acid C-terminal fragment of Alzheimer’s-disease amyloid precursor protein contains a sequence, -RHDS-, that promotes cell adhesion. Biochem. J. 1992, 288 Pt 3, 1053–1059. [Google Scholar] [CrossRef]
- Ho, A.; Sudhof, T.C. Binding of F-spondin to amyloid-beta precursor protein: A candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc. Natl. Acad. Sci. USA 2004, 101, 2548–2553. [Google Scholar] [CrossRef]
- Zheng, H.; Jiang, M.; Trumbauer, M.E.; Sirinathsinghji, D.J.; Hopkins, R.; Smith, D.W.; Heavens, R.P.; Dawson, G.R.; Boyce, S.; Conner, M.W.; et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 1995, 81, 525–531. [Google Scholar]
- Anliker, B.; Muller, U. The functions of mammalian amyloid precursor protein and related amyloid precursor-like proteins. Neurodegener. Dis. 2006, 3, 239–246. [Google Scholar] [CrossRef]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Small, S.A.; Gandy, S. Sorting through the cell biology of Alzheimer’s disease: Intracellular pathways to pathogenesis. Neuron 2006, 52, 15–31. [Google Scholar] [PubMed]
- Nunan, J.; Small, D.H. Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [PubMed]
- Sisodia, S.S. Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl. Acad. Sci. USA 1992, 89, 6075–6079. [Google Scholar] [CrossRef]
- Allinson, T.M.; Parkin, E.T.; Turner, A.J.; Hooper, N.M. ADAMs family members as amyloid precursor protein alpha-secretases. J. Neurosci. Res. 2003, 74, 342–352. [Google Scholar] [CrossRef]
- Koo, E.H.; Squazzo, S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994, 269, 17386–17389. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R. BACE1: The beta-secretase enzyme in Alzheimer’s disease. J. Mol. Neurosci. 2004, 23, 105–114. [Google Scholar] [CrossRef]
- Iwatsubo, T. The gamma-secretase complex: Machinery for intramembrane proteolysis. Curr. Opin. Neurobiol. 2004, 14, 379–383. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Wolfe, M.S. Presenilin: Running with scissors in the membrane. Cell 2007, 131, 215–221. [Google Scholar]
- Buchhave, P.; Minthon, L.; Zetterberg, H.; Wallin, A.K.; Blennow, K.; Hansson, O. Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 2012, 69, 98–106. [Google Scholar] [CrossRef]
- Robakis, N.K. Mechanisms of AD neurodegeneration may be independent of Abeta and its derivatives. Neurobiol. Aging 2011, 32, 372–379. [Google Scholar]
- Zhu, J.; Li, W.; Mao, Z. Cdk5: Mediator of neuronal development, death and the response to DNA damage. Mech. Ageing Dev. 2011, 132, 389–394. [Google Scholar] [PubMed]
- Liraz, O.; Boehm-Cagan, A.; Michaelson, D.M. ApoE4 induces Abeta42, tau, and neuronal pathology in the hippocampus of young targeted replacement apoE4 mice. Mol. Neurodegener. 2013, 8, 16. [Google Scholar] [PubMed]
- Matus, A. Microtubule-associated proteins: Their potential role in determining neuronal morphology. Annu. Rev. Neurosci. 1988, 11, 29–44. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Novak, M.; Edwards, P.C.; Klug, A.; Tichelaar, W.; Crowther, R.A. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef] [PubMed]
- Drubin, D.G.; Kirschner, M.W. Tau protein function in living cells. J. Cell Biol. 1986, 103 Pt 2, 2739–2746. [Google Scholar] [CrossRef] [Green Version]
- Lacovich, V.; Espindola, S.L.; Alloatti, M.; Pozo Devoto, V.; Cromberg, L.E.; Carna, M.E.; Forte, G.; Gallo, J.M.; Bruno, L.; Stokin, G.B.; et al. Tau Isoforms Imbalance Impairs the Axonal Transport of the Amyloid Precursor Protein in Human Neurons. J. Neurosci. 2017, 37, 58–69. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295. [Google Scholar]
- Sayas, C.L.; Tortosa, E.; Bollati, F.; Ramirez-Rios, S.; Arnal, I.; Avila, J. Tau regulates the localization and function of End-binding proteins 1 and 3 in developing neuronal cells. J. Neurochem. 2015, 133, 653–667. [Google Scholar]
- Medina, M.; Avila, J. Further understanding of tau phosphorylation: Implications for therapy. Expert. Rev. Neurother. 2015, 15, 115–122. [Google Scholar] [CrossRef]
- Regan, P.; Piers, T.; Yi, J.H.; Kim, D.H.; Huh, S.; Park, S.J.; Ryu, J.H.; Whitcomb, D.J.; Cho, K. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J. Neurosci. 2015, 35, 4804–4812. [Google Scholar] [CrossRef] [PubMed]
- Menkes-Caspi, N.; Yamin, H.G.; Kellner, V.; Spires-Jones, T.L.; Cohen, D.; Stern, E.A. Pathological tau disrupts ongoing network activity. Neuron 2015, 85, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369, 20130144. [Google Scholar]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Sultan, A.; Nesslany, F.; Violet, M.; Begard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef]
- Marciniak, E.; Leboucher, A.; Caron, E.; Ahmed, T.; Tailleux, A.; Dumont, J.; Issad, T.; Gerhardt, E.; Pagesy, P.; Vileno, M.; et al. Tau deletion promotes brain insulin resistance. J. Exp. Med. 2017, 214, 2257–2269. [Google Scholar]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [PubMed]
- Conrad, C.; Vianna, C.; Freeman, M.; Davies, P. A polymorphic gene nested within an intron of the tau gene: Implications for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 7751–7756. [Google Scholar] [CrossRef]
- Patterson, D.; Costa, A.C. Down syndrome and genetics—A case of linked histories. Nat. Rev. Genet. 2005, 6, 137–147. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B.; et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef]
- Hensley, K.; Carney, J.M.; Mattson, M.P.; Aksenova, M.; Harris, M.; Wu, J.F.; Floyd, R.A.; Butterfield, D.A. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 3270–3274. [Google Scholar] [CrossRef]
- Atwood, C.S.; Scarpa, R.C.; Huang, X.; Moir, R.D.; Jones, W.D.; Fairlie, D.P.; Tanzi, R.E.; Bush, A.I. Characterization of copper interactions with alzheimer amyloid beta peptides: Identification of an attomolar-affinity copper binding site on amyloid beta1-42. J. Neurochem. 2000, 75, 1219–1233. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gonzalez, I.; Edwards Iii, G.; Salvadores, N.; Shahnawaz, M.; Diaz-Espinoza, R.; Soto, C. Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol. Psychiatry 2017, 22, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Hoppener, J.W.; Ahren, B.; Lips, C.J. Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med. 2000, 343, 411–419. [Google Scholar] [CrossRef]
- DeToma, A.S.; Salamekh, S.; Ramamoorthy, A.; Lim, M.H. Misfolded proteins in Alzheimer’s disease and type II diabetes. Chem. Soc. Rev. 2012, 41, 608–621. [Google Scholar] [CrossRef]
- Ono, K.; Takahashi, R.; Ikeda, T.; Mizuguchi, M.; Hamaguchi, T.; Yamada, M. Exogenous amyloidogenic proteins function as seeds in amyloid beta-protein aggregation. Biochim. Biophys. Acta 2014, 1842, 646–653. [Google Scholar] [CrossRef]
- Oskarsson, M.E.; Paulsson, J.F.; Schultz, S.W.; Ingelsson, M.; Westermark, P.; Westermark, G.T. In vivo seeding and cross-seeding of localized amyloidosis: A molecular link between type 2 diabetes and Alzheimer disease. Am. J. Pathol. 2015, 185, 834–846. [Google Scholar] [CrossRef]
- Blurton-Jones, M.; Laferla, F.M. Pathways by which Abeta facilitates tau pathology. Curr. Alzheimer Res. 2006, 3, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Sun, K.; Suo, W.H.; Yao, L.Y.; Fu, Q.; Cui, Y.Y.; Fu, G.H.; Chen, H.Z.; Lu, Y. N-stearoyltyrosine protects primary neurons from Abeta-induced apoptosis through modulating mitogen-activated protein kinase activity. Neuroscience 2010, 169, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.C.; Tranel, D.; Damasio, A.R.; Van Hoesen, G.W. The autonomic-related cortex: Pathology in Alzheimer’s disease. Cereb. Cortex 1997, 7, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Yatin, S.M.; Yatin, M.; Aulick, T.; Ain, K.B.; Butterfield, D.A. Alzheimer’s amyloid beta-peptide associated free radicals increase rat embryonic neuronal polyamine uptake and ornithine decarboxylase activity: Protective effect of vitamin E. Neurosci. Lett. 1999, 263, 17–20. [Google Scholar] [CrossRef]
- Neniskyte, U.; Neher, J.J.; Brown, G.C. Neuronal death induced by nanomolar amyloid beta is mediated by primary phagocytosis of neurons by microglia. J. Biol. Chem. 2011, 286, 39904–39913. [Google Scholar] [CrossRef]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Graber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef]
- Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q. Comparative epitope analysis of neuronal cytoskeletal proteins in Alzheimer’s disease senile plaque neurites and neuropil threads. Lab. Investig. 1991, 64, 352–357. [Google Scholar] [PubMed]
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef]
- Xiong, K.; Cai, H.; Luo, X.G.; Struble, R.G.; Clough, R.W.; Yan, X.X. Mitochondrial respiratory inhibition and oxidative stress elevate beta-secretase (BACE1) proteins and activity in vivo in the rat retina. Exp. Brain Res. 2007, 181, 435–446. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Xu, H.; Shi, Q.; Chen, L.H.; Pedrini, S.; Pechman, D.; Baker, H.; Beal, M.F.; Gandy, S.E.; Gibson, G.E. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer’s mouse model. Neurobiol. Aging 2009, 30, 1587–1600. [Google Scholar] [CrossRef]
- Wang, Y.J.; Zhou, H.D.; Zhou, X.F. Clearance of amyloid-beta in Alzheimer’s disease: Progress, problems and perspectives. Drug Discov. Today 2006, 11, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.T., 3rd; Koelsch, G.; Hong, L.; Castanheira, P.; Ermolieff, J.; Ghosh, A.K.; Tang, J. Subsite specificity of memapsin 2 (beta-secretase): Implications for inhibitor design. Biochemistry 2001, 40, 10001–10006. [Google Scholar] [CrossRef] [PubMed]
- Eckman, E.A.; Reed, D.K.; Eckman, C.B. Degradation of the Alzheimer’s amyloid beta peptide by endothelin-converting enzyme. J. Biol. Chem. 2001, 276, 24540–24548. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef]
- Tucker, H.M.; Kihiko, M.; Caldwell, J.N.; Wright, S.; Kawarabayashi, T.; Price, D.; Walker, D.; Scheff, S.; McGillis, J.P.; Rydel, R.E.; et al. The plasmin system is induced by and degrades amyloid-beta aggregates. J. Neurosci. 2000, 20, 3937–3946. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharm. Sin 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Shibata, M.; Yamada, S.; Kumar, S.R.; Calero, M.; Bading, J.; Frangione, B.; Holtzman, D.M.; Miller, C.A.; Strickland, D.K.; Ghiso, J.; et al. Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Investig. 2000, 106, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Lowe, L.C.; Gaser, C.; Franke, K.; Alzheimer’s Disease Neuroimaging, I. The Effect of the APOE Genotype on Individual BrainAGE in Normal Aging, Mild Cognitive Impairment, and Alzheimer’s Disease. PLoS ONE 2016, 11, e0157514. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M. Role of apoe/Abeta interactions in the pathogenesis of Alzheimer’s disease and cerebral amyloid angiopathy. J. Mol. Neurosci. 2001, 17, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 Mediates Brain Abeta Clearance and Impacts Amyloid Deposition. J. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef] [PubMed]
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Appukuttan, A.T.; Volitakis, I.; Adlard, P.A.; Finkelstein, D.I.; Bush, A.I. Clioquinol rescues Parkinsonism and dementia phenotypes of the tau knockout mouse. Neurobiol. Dis. 2015, 81, 168–175. [Google Scholar] [CrossRef]
- Medina, M.; Avila, J. The role of extracellular Tau in the spreading of neurofibrillary pathology. Front. Cell. Neurosci. 2014, 8, 113. [Google Scholar] [CrossRef]
- Liu, K.Y.; Howard, R. Can we learn lessons from the FDA’s approval of aducanumab? Nat. Rev. Neurol. 2021, 17, 715–722. [Google Scholar] [CrossRef]
- Woodruff-Pak, D.S.; Tobia, M.J.; Jiao, X.; Beck, K.D.; Servatius, R.J. Preclinical investigation of the functional effects of memantine and memantine combined with galantamine or donepezil. Neuropsychopharmacology 2007, 32, 1284–1294. [Google Scholar] [CrossRef]
- Winslow, B.T.; Onysko, M.K.; Stob, C.M.; Hazlewood, K.A. Treatment of Alzheimer disease. Am. Fam. Physician 2011, 83, 1403–1412. [Google Scholar] [PubMed]
- Del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andres, M.V.; Gomez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; Leon, T. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: A pilot study. J. Alzheimers Dis. 2013, 33, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Seripa, D.; Solfrizzi, V.; Imbimbo, B.P.; Daniele, A.; Santamato, A.; Lozupone, M.; Zuliani, G.; Greco, A.; Logroscino, G.; Panza, F. Tau-directed approaches for the treatment of Alzheimer’s disease: Focus on leuco-methylthioninium. Expert. Rev. Neurother. 2016, 16, 259–277. [Google Scholar] [CrossRef]
- Sankaranarayanan, S.; Barten, D.M.; Vana, L.; Devidze, N.; Yang, L.; Cadelina, G.; Hoque, N.; DeCarr, L.; Keenan, S.; Lin, A.; et al. Passive immunization with phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse tauopathy models. PLoS ONE 2015, 10, e0125614. [Google Scholar] [CrossRef]
- Kontsekova, E.; Zilka, N.; Kovacech, B.; Novak, P.; Novak, M. First-in-man tau vaccine targeting structural determinants essential for pathological tau-tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers Res. 2014, 6, 44. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sodium Oligomannate: First Approval. Drugs 2020, 80, 441–444. [Google Scholar] [CrossRef]
- Tuligenga, R.H.; Dugravot, A.; Tabak, A.G.; Elbaz, A.; Brunner, E.J.; Kivimaki, M.; Singh-Manoux, A. Midlife type 2 diabetes and poor glycaemic control as risk factors for cognitive decline in early old age: A post-hoc analysis of the Whitehall II cohort study. Lancet Diabetes Endocrinol. 2014, 2, 228–235. [Google Scholar] [CrossRef]
- Sokoloff, L. Energetics of functional activation in neural tissues. Neurochem. Res. 1999, 24, 321–329. [Google Scholar] [CrossRef]
- Yu, L.; Yu, Y. Energy-efficient neural information processing in individual neurons and neuronal networks. J. Neurosci. Res. 2017, 95, 2253–2266. [Google Scholar] [CrossRef]
- Lying-Tunell, U.; Lindblad, B.S.; Malmlund, H.O.; Persson, B. Cerebral blood flow and metabolic rate of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids. Acta Neurol. Scand. 1980, 62, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Holliday, M.A. Metabolic rate and organ size during growth from infancy to maturity and during late gastation and early infancy. Pediatrics 1971, 47 (Suppl. S2), 169. [Google Scholar]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflug. Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, H.E. Insulin resistance: Definition and consequences. Exp. Clin. Endocrinol. Diabetes 2001, 109 (Suppl. S2), S135–S148. [Google Scholar] [CrossRef]
- Leybaert, L.; De Bock, M.; Van Moorhem, M.; Decrock, E.; De Vuyst, E. Neurobarrier coupling in the brain: Adjusting glucose entry with demand. J. Neurosci. Res. 2007, 85, 3213–3220. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, M.; Spriet, L.L. Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020, 2, 817–828. [Google Scholar] [CrossRef]
- Bergersen, L.H. Is lactate food for neurons? Comparison of monocarboxylate transporter subtypes in brain and muscle. Neuroscience 2007, 145, 11–19. [Google Scholar] [CrossRef]
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessieres, B.; Gao, V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia 2018, 66, 1244–1262. [Google Scholar] [CrossRef]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef]
- Morris, A.A. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005, 28, 109–121. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef]
- Wilkins, J.M.; Trushina, E. Application of Metabolomics in Alzheimer’s Disease. Front. Neurol. 2017, 8, 719. [Google Scholar] [CrossRef] [Green Version]
- Chassard, C.; Scott, K.P.; Marquet, P.; Martin, J.C.; Del’homme, C.; Dapoigny, M.; Flint, H.J.; Bernalier-Donadille, A. Assessment of metabolic diversity within the intestinal microbiota from healthy humans using combined molecular and cultural approaches. FEMS Microbiol. Ecol. 2008, 66, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, M. Occurrence, absorption and metabolism of short chain fatty acids in the digestive tract of mammals. Comp. Biochem. Physiol. B 1987, 86, 439–472. [Google Scholar] [CrossRef] [PubMed]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 461–478. [Google Scholar] [CrossRef]
- Rhea, E.M.; Banks, W.A. Role of the Blood-Brain Barrier in Central Nervous System Insulin Resistance. Front. Neurosci. 2019, 13, 521. [Google Scholar] [CrossRef] [PubMed]
- Newington, J.T.; Harris, R.A.; Cumming, R.C. Reevaluating Metabolism in Alzheimer’s Disease from the Perspective of the Astrocyte-Neuron Lactate Shuttle Model. J. Neurodegener. Dis. 2013, 2013, 234572. [Google Scholar] [CrossRef] [PubMed]
- Rhea, E.M.; Salameh, T.S.; Banks, W.A. Routes for the delivery of insulin to the central nervous system: A comparative review. Exp. Neurol. 2019, 313, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Seaquist, E.R.; Tkac, I.; Damberg, G.; Thomas, W.; Gruetter, R. Brain glucose concentrations in poorly controlled diabetes mellitus as measured by high-field magnetic resonance spectroscopy. Metabolism 2005, 54, 1008–1013. [Google Scholar] [CrossRef]
- Berthet, C.; Lei, H.; Thevenet, J.; Gruetter, R.; Magistretti, P.J.; Hirt, L. Neuroprotective role of lactate after cerebral ischemia. J. Cereb. Blood Flow. Metab. 2009, 29, 1780–1789. [Google Scholar] [CrossRef]
- Schousboe, A.; Westergaard, N.; Sonnewald, U.; Petersen, S.B.; Huang, R.; Peng, L.; Hertz, L. Glutamate and glutamine metabolism and compartmentation in astrocytes. Dev. Neurosci. 1993, 15, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C.G. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer’s disease: Preclinical evidence. Int. J. Geriatr. Psychiatry 2003, 18 (Suppl. S1), S23–S32. [Google Scholar] [CrossRef] [PubMed]
- Souza, D.G.; Almeida, R.F.; Souza, D.O.; Zimmer, E.R. The astrocyte biochemistry. Semin. Cell. Dev. Biol. 2019, 95, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ke, Z.; Luo, J. Thiamine Deficiency and Neurodegeneration: The Interplay Among Oxidative Stress, Endoplasmic Reticulum Stress, and Autophagy. Mol. Neurobiol. 2017, 54, 5440–5448. [Google Scholar] [CrossRef]
- Alexander-Kaufman, K.; Harper, C. Transketolase: Observations in alcohol-related brain damage research. Int. J. Biochem. Cell. Biol. 2009, 41, 717–720. [Google Scholar] [CrossRef]
- Green, P.H. Alcohol, nutrition and malabsorption. Clin. Gastroenterol. 1983, 12, 563–574. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, G.; Li, W.; Fan, Z.; Sun, A.; Luo, J.; Ke, Z.J. Thiamine deficiency increases beta-secretase activity and accumulation of beta-amyloid peptides. Neurobiol. Aging 2011, 32, 42–53. [Google Scholar] [CrossRef]
- Zubaran, C.; Fernandes, J.G.; Rodnight, R. Wernicke-Korsakoff syndrome. Postgrad. Med. J. 1997, 73, 27–31. [Google Scholar] [CrossRef]
- Gibson, G.E.; Hirsch, J.A.; Fonzetti, P.; Jordan, B.D.; Cirio, R.T.; Elder, J. Vitamin B1 (thiamine) and dementia. Ann. N. Y. Acad. Sci. 2016, 1367, 21–30. [Google Scholar] [CrossRef]
- Gold, M.; Hauser, R.A.; Chen, M.F. Plasma thiamine deficiency associated with Alzheimer’s disease but not Parkinson’s disease. Metab. Brain Dis. 1998, 13, 43–53. [Google Scholar] [CrossRef]
- Wilkinson, T.J.; Hanger, H.C.; George, P.M.; Sainsbury, R. Is thiamine deficiency in elderly people related to age or co-morbidity? Age Ageing 2000, 29, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Lazarov, J. Resorption of vitamin B1—XII. Changes in the resorption and the phosphorylation of thiamine in rats in relation to age. Exp. Gerontol. 1977, 12, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A. The regulation of the release of ketone bodies by the liver. Adv. Enzym. Regul. 1966, 4, 339–354. [Google Scholar] [CrossRef]
- Walsh, E.I.; Shaw, M.; Sachdev, P.; Anstey, K.J.; Cherbuin, N. Brain atrophy in ageing: Estimating effects of blood glucose levels vs. other type 2 diabetes effects. Diabetes Metab. 2018, 44, 80–83. [Google Scholar] [CrossRef]
- Nedelska, Z.; Schwarz, C.G.; Boeve, B.F.; Lowe, V.J.; Reid, R.I.; Przybelski, S.A.; Lesnick, T.G.; Gunter, J.L.; Senjem, M.L.; Ferman, T.J.; et al. White matter integrity in dementia with Lewy bodies: A voxel-based analysis of diffusion tensor imaging. Neurobiol. Aging 2015, 36, 2010–2017. [Google Scholar] [CrossRef]
- Ivanov, A.I.; Malkov, A.E.; Waseem, T.; Mukhtarov, M.; Buldakova, S.; Gubkina, O.; Zilberter, M.; Zilberter, Y. Glycolysis and oxidative phosphorylation in neurons and astrocytes during network activity in hippocampal slices. J. Cereb. Blood Flow. Metab. 2014, 34, 397–407. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Neuroscience. Let there be (NADH) light. Science 2004, 305, 50–52. [Google Scholar] [CrossRef]
- Demetrius, L.A.; Simon, D.K. An inverse-Warburg effect and the origin of Alzheimer’s disease. Biogerontology 2012, 13, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Demetrius, L.A.; Magistretti, P.J.; Pellerin, L. Alzheimer’s disease: The amyloid hypothesis and the Inverse Warburg effect. Front. Physiol. 2014, 5, 522. [Google Scholar] [CrossRef]
- Chandrasekaran, K.; Hatanpaa, K.; Brady, D.R.; Rapoport, S.I. Evidence for physiological down-regulation of brain oxidative phosphorylation in Alzheimer’s disease. Exp. Neurol. 1996, 142, 80–88. [Google Scholar] [CrossRef]
- Lehmann, M.; Ghosh, P.M.; Madison, C.; Karydas, A.; Coppola, G.; O’Neil, J.P.; Huang, Y.; Miller, B.L.; Jagust, W.J.; Rabinovici, G.D. Greater medial temporal hypometabolism and lower cortical amyloid burden in ApoE4-positive AD patients. J. Neurol. Neurosurg. Psychiatry 2014, 85, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhou, S.; Zhao, H.; Wang, Y.; Chen, X.; Sun, X. Effects of apolipoprotein E gene polymorphism on the intracellular Ca2+ concentration of astrocytes in the early stages post injury. Exp. Med. 2018, 15, 1417–1423. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.; Esiri, M.M.; Jobst, K.A.; Johnston, C.; Litchfield, S.; Sim, E.; Smith, A.D. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease. Neuroscience 1995, 69, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Lange, M.L. Multifunctional roles of enolase in Alzheimer’s disease brain: Beyond altered glucose metabolism. J. Neurochem. 2009, 111, 915–933. [Google Scholar] [CrossRef] [PubMed]
- Castegna, A.; Aksenov, M.; Aksenova, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: Creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic. Biol. Med. 2002, 33, 562–571. [Google Scholar] [CrossRef]
- Castegna, A.; Aksenov, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: Dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J. Neurochem. 2002, 82, 1524–1532. [Google Scholar] [CrossRef]
- Inoue, Y.; Tasaki, M.; Masuda, T.; Misumi, Y.; Nomura, T.; Ando, Y.; Ueda, M. alpha-Enolase reduces cerebrovascular Abeta deposits by protecting Abeta amyloid formation. Cell. Mol. Life Sci. 2022, 79, 462. [Google Scholar] [CrossRef]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar]
- De Meyts, P. Insulin and its receptor: Structure, function and evolution. Bioessays 2004, 26, 1351–1362. [Google Scholar] [CrossRef]
- Henquin, J.C. Regulation of insulin secretion: A matter of phase control and amplitude modulation. Diabetologia 2009, 52, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Wortham, M.; Sander, M. Mechanisms of beta-cell functional adaptation to changes in workload. Diabetes Obes. Metab. 2016, 18 (Suppl. S1), 78–86. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.; Mitrou, P.; Lambadiari, V.; Maratou, E.; Raptis, S.A. Insulin effects in muscle and adipose tissue. Diabetes Res. Clin. Pract. 2011, 93 (Suppl. S1), S52–S59. [Google Scholar] [CrossRef]
- Lee, J.; Pilch, P.F. The insulin receptor: Structure, function, and signaling. Am. J. Physiol. 1994, 266 Pt 1, C319–C334. [Google Scholar] [CrossRef]
- Burks, D.J.; White, M.F. IRS proteins and beta-cell function. Diabetes 2001, 50 (Suppl. S1), S140–S145. [Google Scholar] [CrossRef] [PubMed]
- White, M.F.; Yenush, L. The IRS-signaling system: A network of docking proteins that mediate insulin and cytokine action. Curr. Top. Microbiol. Immunol. 1998, 228, 179–208. [Google Scholar] [PubMed]
- Giorgetti, S.; Pelicci, P.G.; Pelicci, G.; Van Obberghen, E. Involvement of Src-homology/collagen (SHC) proteins in signaling through the insulin receptor and the insulin-like-growth-factor-I-receptor. Eur. J. Biochem. 1994, 223, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Folstein, M.F. Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: Review and hypothesis. Neurobiol. Aging 2006, 27, 190–198. [Google Scholar] [CrossRef]
- Pivovarova, O.; Hohn, A.; Grune, T.; Pfeiffer, A.F.; Rudovich, N. Insulin-degrading enzyme: New therapeutic target for diabetes and Alzheimer’s disease? Ann. Med. 2016, 48, 614–624. [Google Scholar] [CrossRef]
- Taylor, R. Insulin resistance and type 2 diabetes. Diabetes 2012, 61, 778–779. [Google Scholar] [CrossRef]
- Frolich, L.; Blum-Degen, D.; Bernstein, H.G.; Engelsberger, S.; Humrich, J.; Laufer, S.; Muschner, D.; Thalheimer, A.; Turk, A.; Hoyer, S.; et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural. Transm. 1998, 105, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Fantus, I.G. Inhibition of the protein tyrosine phosphatase PTP1B: Potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 621–640. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. FEBS Lett. 2008, 582, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sesti, G. Pathophysiology of insulin resistance. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Hou, Y.; Mattson, M.P. Mitochondria and neuroplasticity. ASN Neuro 2010, 2, e00045. [Google Scholar] [CrossRef]
- Salvado, L.; Palomer, X.; Barroso, E.; Vazquez-Carrera, M. Targeting endoplasmic reticulum stress in insulin resistance. Trends. Endocrinol. Metab. 2015, 26, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Adochio, R.L.; Leitner, J.W.; Gray, K.; Draznin, B.; Cornier, M.A. Early responses of insulin signaling to high-carbohydrate and high-fat overfeeding. Nutr. Metab. 2009, 6, 37. [Google Scholar] [CrossRef]
- Bray, G.A.; Lovejoy, J.C.; Smith, S.R.; DeLany, J.P.; Lefevre, M.; Hwang, D.; Ryan, D.H.; York, D.A. The influence of different fats and fatty acids on obesity, insulin resistance and inflammation. J. Nutr. 2002, 132, 2488–2491. [Google Scholar] [CrossRef]
- Linn, T.; Santosa, B.; Gronemeyer, D.; Aygen, S.; Scholz, N.; Busch, M.; Bretzel, R.G. Effect of long-term dietary protein intake on glucose metabolism in humans. Diabetologia 2000, 43, 1257–1265. [Google Scholar] [CrossRef]
- Toida, S.; Takahashi, M.; Shimizu, H.; Sato, N.; Shimomura, Y.; Kobayashi, I. Effect of high sucrose feeding on fat accumulation in the male Wistar rat. Obes. Res. 1996, 4, 561–568. [Google Scholar] [CrossRef]
- Xu, E.; Schwab, M.; Marette, A. Role of protein tyrosine phosphatases in the modulation of insulin signaling and their implication in the pathogenesis of obesity-linked insulin resistance. Rev. Endocr. Metab. Disord. 2014, 15, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Hall, A.M. Does Diacylglycerol Accumulation in Fatty Liver Disease Cause Hepatic Insulin Resistance? Biomed. Res. Int. 2015, 2015, 104132. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.I. Cellular mechanisms of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, Y.H.; Hannun, Y.A. The acid sphingomyelinase/ceramide pathway: Biomedical significance and mechanisms of regulation. Curr. Mol. Med. 2010, 10, 454–466. [Google Scholar] [CrossRef]
- Calderon-DuPont, D.; Torre-Villalvazo, I.; Diaz-Villasenor, A. Is insulin resistance tissue-dependent and substrate-specific? The role of white adipose tissue and skeletal muscle. Biochimie 2023, 204, 48–68. [Google Scholar] [CrossRef]
- Hirose, M.; Kaneki, M.; Sugita, H.; Yasuhara, S.; Martyn, J.A. Immobilization depresses insulin signaling in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1235–E1241. [Google Scholar] [CrossRef]
- Ropelle, E.R.; Flores, M.B.; Cintra, D.E.; Rocha, G.Z.; Pauli, J.R.; Morari, J.; de Souza, C.T.; Moraes, J.C.; Prada, P.O.; Guadagnini, D.; et al. IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS Biol. 2010, 8, e1000465. [Google Scholar] [CrossRef]
- Grigsby, R.J.; Dobrowsky, R.T. Inhibition of ceramide production reverses TNF-induced insulin resistance. Biochem. Biophys. Res. Commun. 2001, 287, 1121–1124. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R. Insulin receptor and cancer. Endocr. Relat. Cancer 2011, 18, R125–R147. [Google Scholar] [CrossRef]
- Havrankova, J.; Brownstein, M.; Roth, J. Insulin and insulin receptors in rodent brain. Diabetologia 1981, 20 (Suppl. S1), 268–273. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 2015, 64, 3927–3936. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Chen, H.; Xu, H.; Moore, E.; Meiri, N.; Quon, M.J.; Alkon, D.L. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J. Biol. Chem. 1999, 274, 34893–34902. [Google Scholar] [CrossRef] [Green Version]
- Park, C.R.; Seeley, R.J.; Craft, S.; Woods, S.C. Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol. Behav. 2000, 68, 509–514. [Google Scholar] [CrossRef]
- Salameh, T.S.; Bullock, K.M.; Hujoel, I.A.; Niehoff, M.L.; Wolden-Hanson, T.; Kim, J.; Morley, J.E.; Farr, S.A.; Banks, W.A. Central Nervous System Delivery of Intranasal Insulin: Mechanisms of Uptake and Effects on Cognition. J. Alzheimers Dis. 2015, 47, 715–728. [Google Scholar] [CrossRef]
- Swaminathan, S.K.; Ahlschwede, K.M.; Sarma, V.; Curran, G.L.; Omtri, R.S.; Decklever, T.; Lowe, V.J.; Poduslo, J.F.; Kandimalla, K.K. Insulin differentially affects the distribution kinetics of amyloid beta 40 and 42 in plasma and brain. J. Cereb. Blood Flow. Metab. 2018, 38, 904–918. [Google Scholar] [CrossRef] [PubMed]
- Uemura, E.; Greenlee, H.W. Insulin regulates neuronal glucose uptake by promoting translocation of glucose transporter GLUT3. Exp. Neurol. 2006, 198, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Griffith, C.M.; Macklin, L.N.; Cai, Y.; Sharp, A.A.; Yan, X.X.; Reagan, L.P.; Strader, A.D.; Rose, G.M.; Patrylo, P.R. Impaired Glucose Tolerance and Reduced Plasma Insulin Precede Decreased AKT Phosphorylation and GLUT3 Translocation in the Hippocampus of Old 3xTg-AD Mice. J. Alzheimers Dis. 2019, 68, 809–837. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Leary, J.; McNay, E.C. Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory. J. Neurosci. 2016, 36, 11851–11864. [Google Scholar] [CrossRef]
- Komori, T.; Morikawa, Y.; Tamura, S.; Doi, A.; Nanjo, K.; Senba, E. Subcellular localization of glucose transporter 4 in the hypothalamic arcuate nucleus of ob/ob mice under basal conditions. Brain Res. 2005, 1049, 34–42. [Google Scholar] [CrossRef]
- Air, E.L.; Benoit, S.C.; Blake Smith, K.A.; Clegg, D.J.; Woods, S.C. Acute third ventricular administration of insulin decreases food intake in two paradigms. Pharm. Biochem. Behav. 2002, 72, 423–429. [Google Scholar] [CrossRef]
- Scherer, T.; O’Hare, J.; Diggs-Andrews, K.; Schweiger, M.; Cheng, B.; Lindtner, C.; Zielinski, E.; Vempati, P.; Su, K.; Dighe, S.; et al. Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab. 2011, 13, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.C.; Fasshauer, M.; Filatova, N.; Grundell, L.A.; Zielinski, E.; Zhou, J.Y.; Scherer, T.; Lindtner, C.; White, P.J.; Lapworth, A.L.; et al. Brain insulin lowers circulating BCAA levels by inducing hepatic BCAA catabolism. Cell Metab. 2014, 20, 898–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheline, Y.I.; Barch, D.M.; Price, J.L.; Rundle, M.M.; Vaishnavi, S.N.; Snyder, A.Z.; Mintun, M.A.; Wang, S.; Coalson, R.S.; Raichle, M.E. The default mode network and self-referential processes in depression. Proc. Natl. Acad. Sci. USA 2009, 106, 1942–1947. [Google Scholar] [CrossRef] [PubMed]
- Muniyappa, R.; Quon, M.J. Insulin action and insulin resistance in vascular endothelium. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 523–530. [Google Scholar] [CrossRef]
- Richards, O.C.; Raines, S.M.; Attie, A.D. The role of blood vessels, endothelial cells, and vascular pericytes in insulin secretion and peripheral insulin action. Endocr. Rev. 2010, 31, 343–363. [Google Scholar] [CrossRef]
- Takechi, R.; Lam, V.; Brook, E.; Giles, C.; Fimognari, N.; Mooranian, A.; Al-Salami, H.; Coulson, S.H.; Nesbit, M.; Mamo, J.C.L. Blood-Brain Barrier Dysfunction Precedes Cognitive Decline and Neurodegeneration in Diabetic Insulin Resistant Mouse Model: An Implication for Causal Link. Front. Aging Neurosci. 2017, 9, 399. [Google Scholar] [CrossRef]
- Mielke, J.G.; Wang, Y.T. Insulin, synaptic function, and opportunities for neuroprotection. Prog. Mol. Biol. Transl. Sci. 2011, 98, 133–186. [Google Scholar]
- Willette, A.A.; Johnson, S.C.; Birdsill, A.C.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P.; et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement. 2015, 11, 504–510.e1. [Google Scholar] [CrossRef]
- Heni, M.; Hennige, A.M.; Peter, A.; Siegel-Axel, D.; Ordelheide, A.M.; Krebs, N.; Machicao, F.; Fritsche, A.; Haring, H.U.; Staiger, H. Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS ONE 2011, 6, e21594. [Google Scholar] [CrossRef]
- Garcia-Caceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef] [PubMed]
- King, G.L.; Buzney, S.M.; Kahn, C.R.; Hetu, N.; Buchwald, S.; Macdonald, S.G.; Rand, L.I. Differential responsiveness to insulin of endothelial and support cells from micro- and macrovessels. J. Clin. Investig. 1983, 71, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.L.; Fragoso, G.; Miron, V.E.; Darlington, P.J.; Mushynski, W.E.; Antel, J.; Almazan, G. Response of human oligodendrocyte progenitors to growth factors and axon signals. J. Neuropathol. Exp. Neurol. 2010, 69, 930–944. [Google Scholar] [CrossRef] [PubMed]
- Spielman, L.J.; Bahniwal, M.; Little, J.P.; Walker, D.G.; Klegeris, A. Insulin Modulates In Vitro Secretion of Cytokines and Cytotoxins by Human Glial Cells. Curr. Alzheimer Res. 2015, 12, 684–693. [Google Scholar] [CrossRef]
- Mielke, J.G.; Taghibiglou, C.; Liu, L.; Zhang, Y.; Jia, Z.; Adeli, K.; Wang, Y.T. A biochemical and functional characterization of diet-induced brain insulin resistance. J. Neurochem. 2005, 93, 1568–1578. [Google Scholar] [CrossRef]
- Banks, W.A. The source of cerebral insulin. Eur. J. Pharm. 2004, 490, 5–12. [Google Scholar] [CrossRef]
- Dorn, A.; Rinne, A.; Hahn, H.J.; Bernstein, H.G.; Ziegler, M. C-peptide immunoreactive neurons in human brain. Acta Histochem. 1982, 70, 326–330. [Google Scholar] [CrossRef]
- Mehran, A.E.; Templeman, N.M.; Brigidi, G.S.; Lim, G.E.; Chu, K.Y.; Hu, X.; Botezelli, J.D.; Asadi, A.; Hoffman, B.G.; Kieffer, T.J.; et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012, 16, 723–737. [Google Scholar] [CrossRef]
- Baskin, D.G.; Stein, L.J.; Ikeda, H.; Woods, S.C.; Figlewicz, D.P.; Porte, D., Jr.; Greenwood, M.R.; Dorsa, D.M. Genetically obese Zucker rats have abnormally low brain insulin content. Life Sci. 1985, 36, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Kaiyala, K.J.; Prigeon, R.L.; Kahn, S.E.; Woods, S.C.; Schwartz, M.W. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes 2000, 49, 1525–1533. [Google Scholar] [CrossRef]
- Heni, M.; Schopfer, P.; Peter, A.; Sartorius, T.; Fritsche, A.; Synofzik, M.; Haring, H.U.; Maetzler, W.; Hennige, A.M. Evidence for altered transport of insulin across the blood-brain barrier in insulin-resistant humans. Acta Diabetol. 2014, 51, 679–681. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, T.; Peter, A.; Heni, M.; Maetzler, W.; Fritsche, A.; Haring, H.U.; Hennige, A.M. The brain response to peripheral insulin declines with age: A contribution of the blood-brain barrier? PloS ONE 2015, 10, e0126804. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.; Macauley, S.L.; Holtzman, D.M. Changes in insulin and insulin signaling in Alzheimer’s disease: Cause or consequence? J. Exp. Med. 2016, 213, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Begg, D.P.; Mul, J.D.; Liu, M.; Reedy, B.M.; D’Alessio, D.A.; Seeley, R.J.; Woods, S.C. Reversal of diet-induced obesity increases insulin transport into cerebrospinal fluid and restores sensitivity to the anorexic action of central insulin in male rats. Endocrinology 2013, 154, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Asthana, S.; Schellenberg, G.; Baker, L.; Cherrier, M.; Boyt, A.A.; Martins, R.N.; Raskind, M.; Peskind, E.; Plymate, S. Insulin effects on glucose metabolism, memory, and plasma amyloid precursor protein in Alzheimer’s disease differ according to apolipoprotein-E genotype. Ann. N. Y. Acad. Sci. 2000, 903, 222–228. [Google Scholar] [CrossRef]
- Floras, J.S.; Meneilly, G. Insulin-mediated blood flow and glucose uptake. Can. J. Cardiol. 2001, 17 (Suppl. A), 7A–10A. [Google Scholar]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Ropelle, E.R.; Pauli, J.R.; Prada, P.O.; de Souza, C.T.; Picardi, P.K.; Faria, M.C.; Cintra, D.E.; Fernandes, M.F.; Flores, M.B.; Velloso, L.A.; et al. Reversal of diet-induced insulin resistance with a single bout of exercise in the rat: The role of PTP1B and IRS-1 serine phosphorylation. J. Physiol. 2006, 577 Pt 3, 997–1007. [Google Scholar] [CrossRef]
- Dandona, P.; Aljada, A.; Bandyopadhyay, A. Inflammation: The link between insulin resistance, obesity and diabetes. Trends. Immunol. 2004, 25, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Peraldi, P.; Spiegelman, B. TNF-alpha and insulin resistance: Summary and future prospects. Mol. Cell. Biochem. 1998, 182, 169–175. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends. Endocrinol. Metab. 2008, 19, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Maciejczyk, M.; Zebrowska, E.; Chabowski, A. Insulin Resistance and Oxidative Stress in the Brain: What’s New? Int. J. Mol. Sci. 2019, 20, 874. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.L.; Maddux, B.A.; Goldfine, I.D. The molecular basis for oxidative stress-induced insulin resistance. Antioxid. Redox. Signal. 2005, 7, 1040–1052. [Google Scholar] [CrossRef]
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar] [CrossRef]
- Vanitallie, T.B. Preclinical sporadic Alzheimer’s disease: Target for personalized diagnosis and preventive intervention. Metabolism 2013, 62 (Suppl. S1), S30–S33. [Google Scholar] [CrossRef]
- Mosconi, L.; Brys, M.; Switalski, R.; Mistur, R.; Glodzik, L.; Pirraglia, E.; Tsui, W.; De Santi, S.; de Leon, M.J. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc. Natl. Acad. Sci. USA 2007, 104, 19067–19072. [Google Scholar] [CrossRef]
- Mosconi, L.; Tsui, W.H.; De Santi, S.; Li, J.; Rusinek, H.; Convit, A.; Li, Y.; Boppana, M.; de Leon, M.J. Reduced hippocampal metabolism in MCI and AD: Automated FDG-PET image analysis. Neurology 2005, 64, 1860–1867. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Mullins, R.J.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 118. [Google Scholar] [CrossRef]
- Ogata, S.; Ito, S.; Masuda, T.; Ohtsuki, S. Changes of Blood-Brain Barrier and Brain Parenchymal Protein Expression Levels of Mice under Different Insulin-Resistance Conditions Induced by High-Fat Diet. Pharm. Res. 2019, 36, 141. [Google Scholar] [CrossRef] [PubMed]
- Soto, M.; Cai, W.; Konishi, M.; Kahn, C.R. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc. Natl. Acad. Sci. USA 2019, 116, 6379–6384. [Google Scholar] [CrossRef] [PubMed]
- Sathya, M.; Premkumar, P.; Karthick, C.; Moorthi, P.; Jayachandran, K.S.; Anusuyadevi, M. BACE1 in Alzheimer’s disease. Clin. Chim. Acta 2012, 414, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Cong, W.N.; Ji, S.; Rothman, S.; Maudsley, S.; Martin, B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr. Alzheimer Res. 2012, 9, 5–17. [Google Scholar] [CrossRef]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef]
- Frankiewicz, T.; Parsons, C.G. Memantine restores long term potentiation impaired by tonic N-methyl-D-aspartate (NMDA) receptor activation following reduction of Mg2+ in hippocampal slices. Neuropharmacology 1999, 38, 1253–1259. [Google Scholar] [CrossRef]
- Tominaga-Yoshino, K.; Uetsuki, T.; Yoshikawa, K.; Ogura, A. Neurotoxic and neuroprotective effects of glutamate are enhanced by introduction of amyloid precursor protein cDNA. Brain Res. 2001, 918, 121–130. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E.; Ohm, T.; Bohl, J. Alzheimer’s disease: Mismatch between amyloid plaques and neuritic plaques. Neurosci. Lett. 1989, 103, 24–28. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J. Neurochem. 2009, 111, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef]
- Medina, D.; DeToledo-Morrell, L.; Urresta, F.; Gabrieli, J.D.; Moseley, M.; Fleischman, D.; Bennett, D.A.; Leurgans, S.; Turner, D.A.; Stebbins, G.T. White matter changes in mild cognitive impairment and AD: A diffusion tensor imaging study. Neurobiol. Aging 2006, 27, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, B.; Targan, E.; Madison, C.; Weiner, M.; Zhang, Y.; Reed, B.; Chui, H.C.; Jagust, W. White matter integrity and cortical metabolic associations in aging and dementia. Alzheimers Dement. 2010, 6, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dwyer, L.; Lamberton, F.; Bokde, A.L.; Ewers, M.; Faluyi, Y.O.; Tanner, C.; Mazoyer, B.; O’Neill, D.; Bartley, M.; Collins, D.R.; et al. Multiple indices of diffusion identifies white matter damage in mild cognitive impairment and Alzheimer’s disease. PLoS ONE 2011, 6, e21745. [Google Scholar] [CrossRef]
- Goldman-Rakic, P.S.; Selemon, L.D.; Schwartz, M.L. Dual pathways connecting the dorsolateral prefrontal cortex with the hippocampal formation and parahippocampal cortex in the rhesus monkey. Neuroscience 1984, 12, 719–743. [Google Scholar] [CrossRef] [PubMed]
- Risacher, S.L.; Saykin, A.J.; West, J.D.; Shen, L.; Firpi, H.A.; McDonald, B.C.; Alzheimer’s Disease Neuroimaging, I. Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Curr. Alzheimer Res. 2009, 6, 347–361. [Google Scholar] [CrossRef]
- Villain, N.; Fouquet, M.; Baron, J.C.; Mezenge, F.; Landeau, B.; de La Sayette, V.; Viader, F.; Eustache, F.; Desgranges, B.; Chetelat, G. Sequential relationships between grey matter and white matter atrophy and brain metabolic abnormalities in early Alzheimer’s disease. Brain 2010, 133, 3301–3314. [Google Scholar] [CrossRef]
- Sokoloff, L. Metabolism of ketone bodies by the brain. Annu. Rev. Med. 1973, 24, 271–280. [Google Scholar] [CrossRef]
- Young, K.J.; Bennett, J.P. The mitochondrial secret(ase) of Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S381–S400. [Google Scholar] [CrossRef]
- Hatting, M.; Tavares, C.D.J.; Sharabi, K.; Rines, A.K.; Puigserver, P. Insulin regulation of gluconeogenesis. Ann. N. Y. Acad. Sci. 2018, 1411, 21–35. [Google Scholar] [CrossRef]
- Fields, R.D. White matter in learning, cognition and psychiatric disorders. Trends. Neurosci. 2008, 31, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Calingasan, N.Y.; Gibson, G.E. Vascular endothelium is a site of free radical production and inflammation in areas of neuronal loss in thiamine-deficient brain. Ann. N. Y. Acad. Sci. 2000, 903, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Hazell, A.S.; Butterworth, R.F. Update of cell damage mechanisms in thiamine deficiency: Focus on oxidative stress, excitotoxicity and inflammation. Alcohol. Alcohol. 2009, 44, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef]
- Moley, K.H.; Mueckler, M.M. Glucose transport and apoptosis. Apoptosis 2000, 5, 99–105. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Soininen, H.; Hiltunen, M. AMP-activated protein kinase: A potential player in Alzheimer’s disease. J. Neurochem. 2011, 118, 460–474. [Google Scholar] [CrossRef]
- Dice, J.F. Chaperone-mediated autophagy. Autophagy 2007, 3, 295–299. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar]
- Lafay-Chebassier, C.; Paccalin, M.; Page, G.; Barc-Pain, S.; Perault-Pochat, M.C.; Gil, R.; Pradier, L.; Hugon, J. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem. 2005, 94, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Bove, J.; Martinez-Vicente, M.; Vila, M. Fighting neurodegeneration with rapamycin: Mechanistic insights. Nat. Rev. Neurosci. 2011, 12, 437–452. [Google Scholar] [CrossRef] [PubMed]
- Hamano, T.; Gendron, T.F.; Causevic, E.; Yen, S.H.; Lin, W.L.; Isidoro, C.; Deture, M.; Ko, L.W. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur. J. Neurosci. 2008, 27, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Freude, S.; Schilbach, K.; Schubert, M. The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer’s disease: From model organisms to human disease. Curr. Alzheimer Res. 2009, 6, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Figlewicz, D.F.; Kahn, S.E.; Baskin, D.G.; Greenwood, M.R.; Porte, D., Jr. Insulin binding to brain capillaries is reduced in genetically obese, hyperinsulinemic Zucker rats. Peptides 1990, 11, 467–472. [Google Scholar] [CrossRef]
- Hoyer, S. The aging brain. Changes in the neuronal insulin/insulin receptor signal transduction cascade trigger late-onset sporadic Alzheimer disease (SAD). A mini-review. J. Neural. Transm. 2002, 109, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Subramanian, S.; John, M. Intranasal administration of insulin lowers amyloid-beta levels in rat model of diabetes. Indian J. Exp. Biol. 2012, 50, 41–44. [Google Scholar]
- Van den Berg, E.; Kloppenborg, R.P.; Kessels, R.P.; Kappelle, L.J.; Biessels, G.J. Type 2 diabetes mellitus, hypertension, dyslipidemia and obesity: A systematic comparison of their impact on cognition. Biochim. Biophys. Acta 2009, 1792, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Bruning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Muller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef]
- Fisher, S.J.; Bruning, J.C.; Lannon, S.; Kahn, C.R. Insulin signaling in the central nervous system is critical for the normal sympathoadrenal response to hypoglycemia. Diabetes 2005, 54, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Kappeler, L.; De Magalhaes Filho, C.; Dupont, J.; Leneuve, P.; Cervera, P.; Perin, L.; Loudes, C.; Blaise, A.; Klein, R.; Epelbaum, J.; et al. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 2008, 6, e254. [Google Scholar] [CrossRef]
- Heidenreich, K.A.; Toledo, S.P.; Kenner, K.A. Regulation of protein phosphorylation by insulin and insulin-like growth factors in cultured fetal neurons. Adv. Exp. Med. Biol. 1991, 293, 379–384. [Google Scholar]
- Lee, C.C.; Huang, C.C.; Wu, M.Y.; Hsu, K.S. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J. Biol. Chem. 2005, 280, 18543–18550. [Google Scholar] [CrossRef]
- Bramham, C.R.; Messaoudi, E. BDNF function in adult synaptic plasticity: The synaptic consolidation hypothesis. Prog. Neurobiol. 2005, 76, 99–125. [Google Scholar] [CrossRef]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef]
- Prakash, A.; Kumar, A. Role of nuclear receptor on regulation of BDNF and neuroinflammation in hippocampus of beta-amyloid animal model of Alzheimer’s disease. Neurotox. Res. 2014, 25, 335–347. [Google Scholar] [CrossRef]
- Schur, E.A.; Melhorn, S.J.; Oh, S.K.; Lacy, J.M.; Berkseth, K.E.; Guyenet, S.J.; Sonnen, J.A.; Tyagi, V.; Rosalynn, M.; De Leon, B.; et al. Radiologic evidence that hypothalamic gliosis is associated with obesity and insulin resistance in humans. Obesity 2015, 23, 2142–2148. [Google Scholar] [CrossRef] [PubMed]
- Draznin, B. Molecular mechanisms of insulin resistance: Serine phosphorylation of insulin receptor substrate-1 and increased expression of p85alpha: The two sides of a coin. Diabetes 2006, 55, 2392–2397. [Google Scholar] [CrossRef]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 2010, 93, 546–553. [Google Scholar] [CrossRef]
- Sivanesan, S.; Mundugaru, R.; Rajadas, J. Possible Clues for Brain Energy Translation via Endolysosomal Trafficking of APP-CTFs in Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2018, 2018, 2764831. [Google Scholar] [CrossRef] [PubMed]
- Velliquette, R.A.; O’Connor, T.; Vassar, R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: Possible early events in Alzheimer’s disease pathogenesis. J. Neurosci. 2005, 25, 10874–10883. [Google Scholar] [CrossRef]
- Moreira, P.I.; Cardoso, S.M.; Pereira, C.M.; Santos, M.S.; Oliveira, C.R. Mitochondria as a therapeutic target in Alzheimer’s disease and diabetes. CNS Neurol. Disord. Drug Targets 2009, 8, 492–511. [Google Scholar] [CrossRef]
- Giese, K.P. GSK-3: A key player in neurodegeneration and memory. IUBMB Life 2009, 61, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.; Klein, P.S. GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, G.; Munoz-Montano, J.R.; Satrustegui, J.; Avila, J.; Bogonez, E.; Diaz-Nido, J. Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett. 1999, 453, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Markevich, V.; Plattner, F.; Killick, R.; Schofield, E.; Engel, T.; Hernandez, F.; Anderton, B.; Rosenblum, K.; Bliss, T.; et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur. J. Neurosci. 2007, 25, 81–86. [Google Scholar] [CrossRef]
- Hoshi, M.; Takashima, A.; Noguchi, K.; Murayama, M.; Sato, M.; Kondo, S.; Saitoh, Y.; Ishiguro, K.; Hoshino, T.; Imahori, K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc. Natl. Acad. Sci. USA 1996, 93, 2719–2723. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci 2001, 21, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.; Mehta, T.; Selkoe, D.J. Soluble Abeta inhibits specific signal transduction cascades common to the insulin receptor pathway. J. Biol. Chem. 2007, 282, 33305–33312. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef]
- Lee, C.C.; Kuo, Y.M.; Huang, C.C.; Hsu, K.S. Insulin rescues amyloid beta-induced impairment of hippocampal long-term potentiation. Neurobiol. Aging 2009, 30, 377–387. [Google Scholar] [CrossRef]
- Zhao, L.; Teter, B.; Morihara, T.; Lim, G.P.; Ambegaokar, S.S.; Ubeda, O.J.; Frautschy, S.A.; Cole, G.M. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: Implications for Alzheimer’s disease intervention. J. Neurosci. 2004, 24, 11120–11126. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J. Pathol. 2011, 225, 54–62. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Braak, H.; Thal, D.R.; Ghebremedhin, E.; Del Tredici, K. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 2011, 70, 960–969. [Google Scholar] [CrossRef]
- Kerr, F.; Rickle, A.; Nayeem, N.; Brandner, S.; Cowburn, R.F.; Lovestone, S. PTEN, a negative regulator of PI3 kinase signalling, alters tau phosphorylation in cells by mechanisms independent of GSK-3. FEBS Lett. 2006, 580, 3121–3128. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, J.H.; Hwang, J.; Son, S.U.; Choi, J.; You, S.-W.; Park, H.; Cha, S.-Y.; Maeng, S. How Can Insulin Resistance Cause Alzheimer’s Disease? Int. J. Mol. Sci. 2023, 24, 3506. https://doi.org/10.3390/ijms24043506
Yoon JH, Hwang J, Son SU, Choi J, You S-W, Park H, Cha S-Y, Maeng S. How Can Insulin Resistance Cause Alzheimer’s Disease? International Journal of Molecular Sciences. 2023; 24(4):3506. https://doi.org/10.3390/ijms24043506
Chicago/Turabian StyleYoon, Ji Hye, JooHyun Hwang, Sung Un Son, Junhyuk Choi, Seung-Won You, Hyunwoo Park, Seung-Yun Cha, and Sungho Maeng. 2023. "How Can Insulin Resistance Cause Alzheimer’s Disease?" International Journal of Molecular Sciences 24, no. 4: 3506. https://doi.org/10.3390/ijms24043506
APA StyleYoon, J. H., Hwang, J., Son, S. U., Choi, J., You, S.-W., Park, H., Cha, S.-Y., & Maeng, S. (2023). How Can Insulin Resistance Cause Alzheimer’s Disease? International Journal of Molecular Sciences, 24(4), 3506. https://doi.org/10.3390/ijms24043506