
Structural and Functional Annotation of Hypothetical Proteins from the Microsporidia Species Vittaforma corneae ATCC 50505 Using in silico Approaches



Abstract
:1. Introduction
2. Results and Discussion
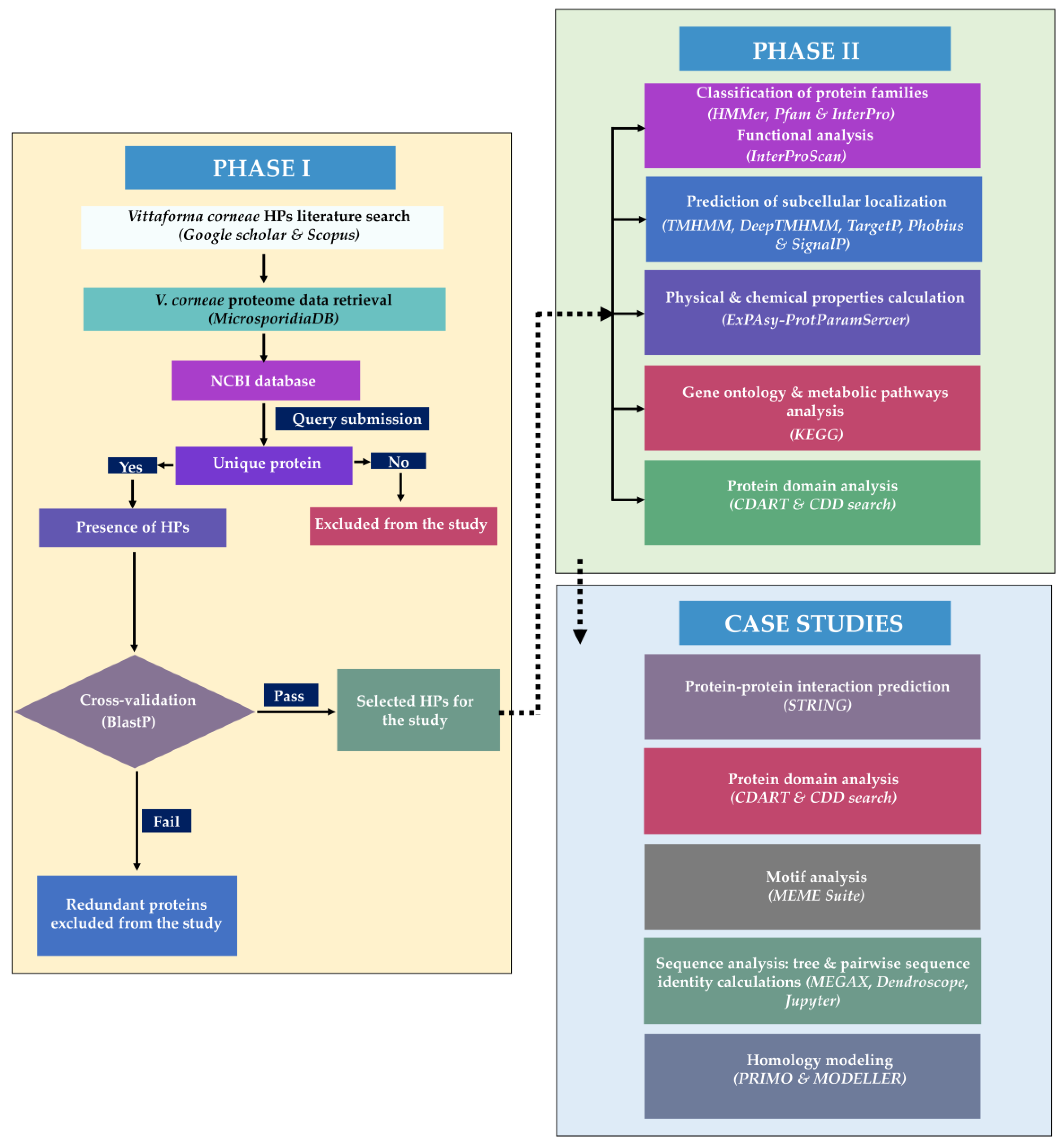
2.1. PHASE I
2.1.1. Sequence Retrieval
2.1.2. Sequence Similarity
2.2. PHASE II
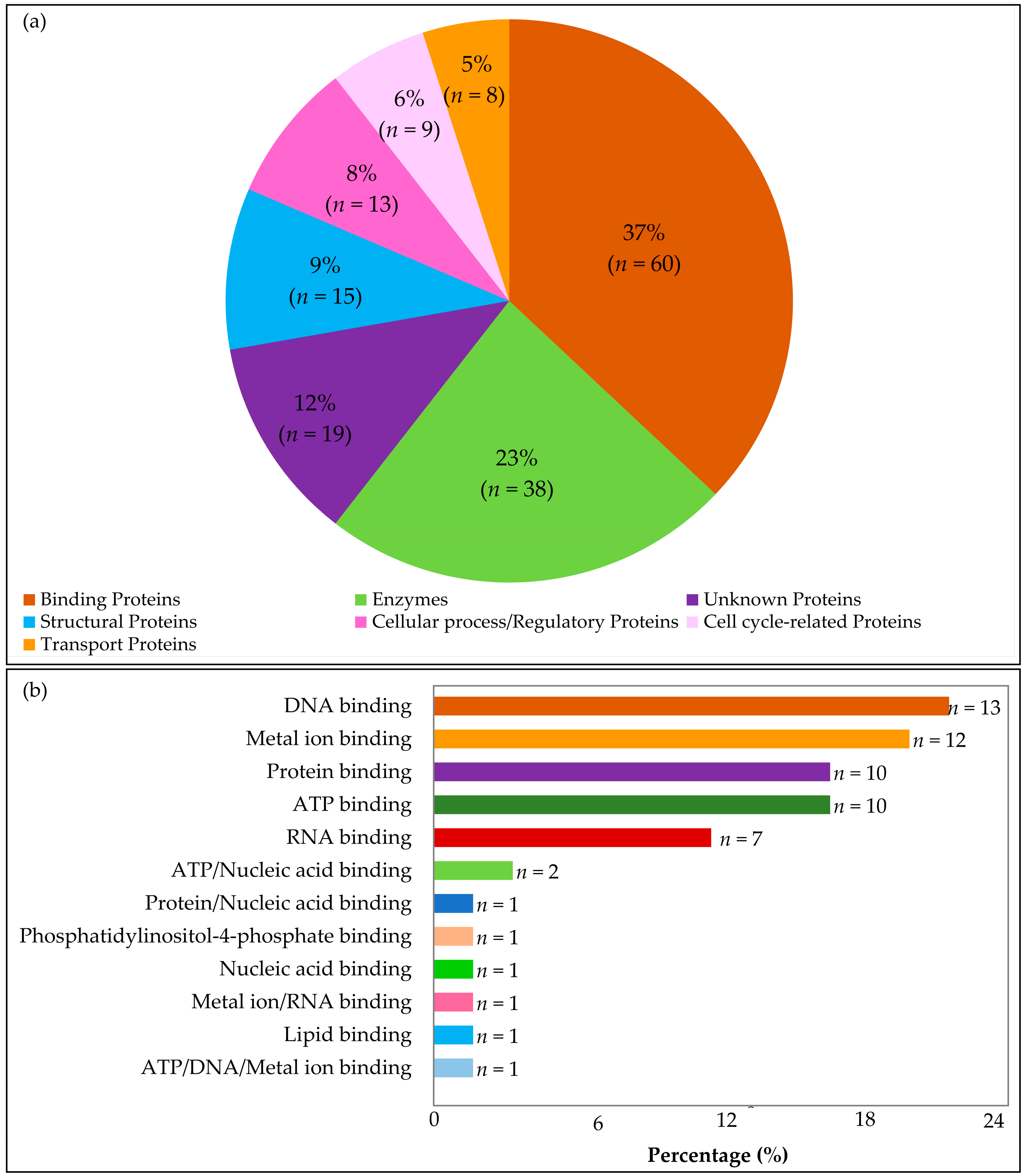
2.2.1. Classification of Protein Families
Enzymes
- a.
- Transferases
- b.
- Kinases
- c.
- Hydrolases
- d.
- Reductase
- e.
- Phosphatase
Binding Proteins
- a.
- Nucleic-acid-binding Proteins
- b.
- Protein-binding Proteins
- c.
- Metal-binding Proteins
Cellular/Regulatory Proteins
Transport Proteins
Structural Proteins
2.2.2. Subcellular Localization
2.2.3. Protein Characterization by Physicochemical Properties
2.2.4. Gene Ontology and Metabolic Pathway Analysis
2.3. Case Studies
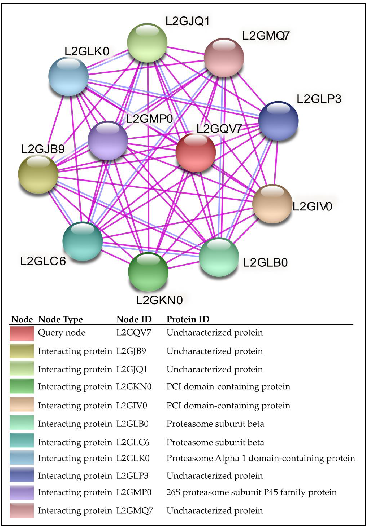
2.3.1. Case Study I: Functional and Structural Analysis of VICG00012
- a.
- Protein–Protein Interaction Analysis
- b.
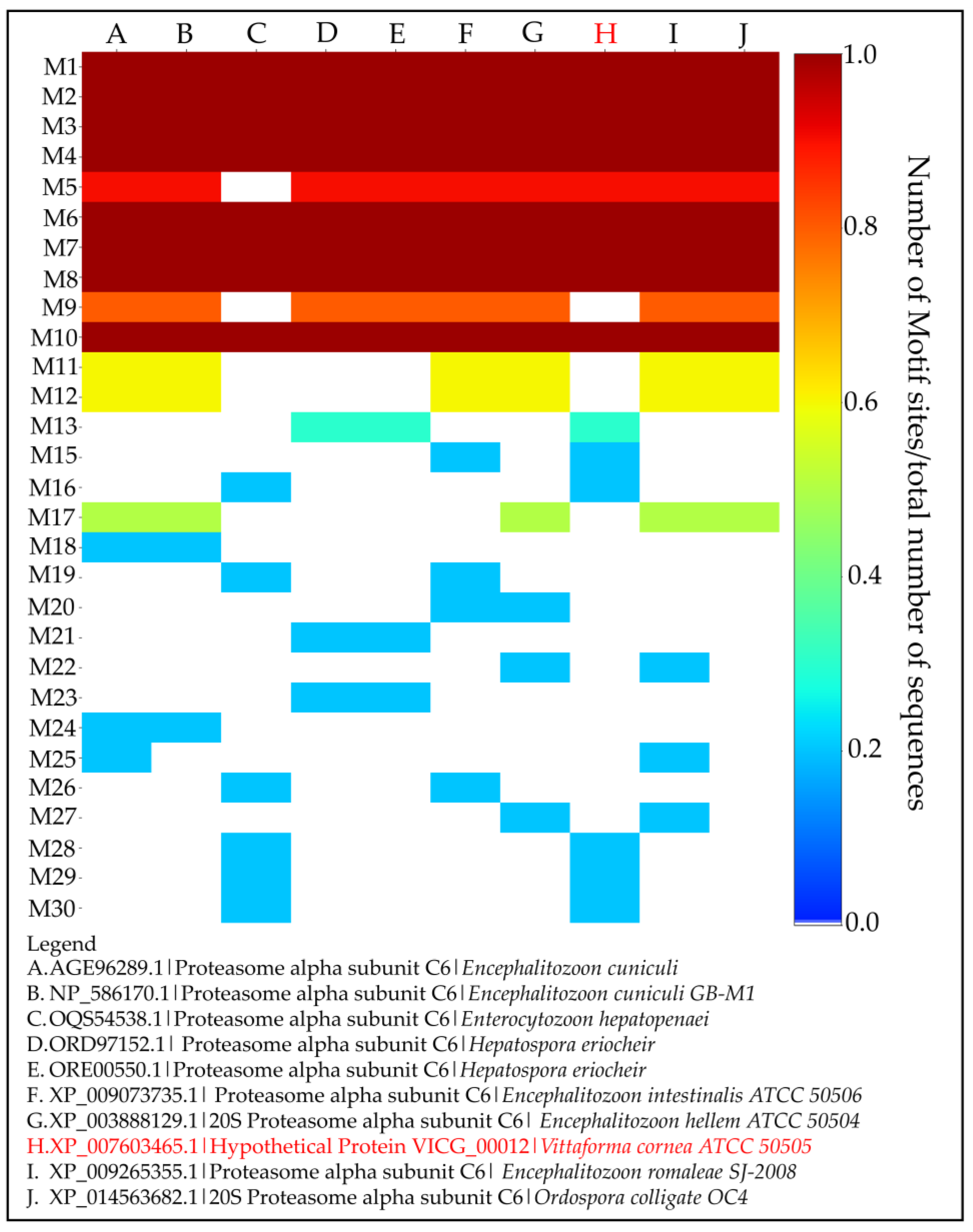
- Identification and Analysis of Conserved Motifs
- c.
- Multiple Sequence Alignment and Motif Mapping
- d.
- Motif Mapping to Homology Models
- e.
- Identification and Analysis of Key Domains
- f.
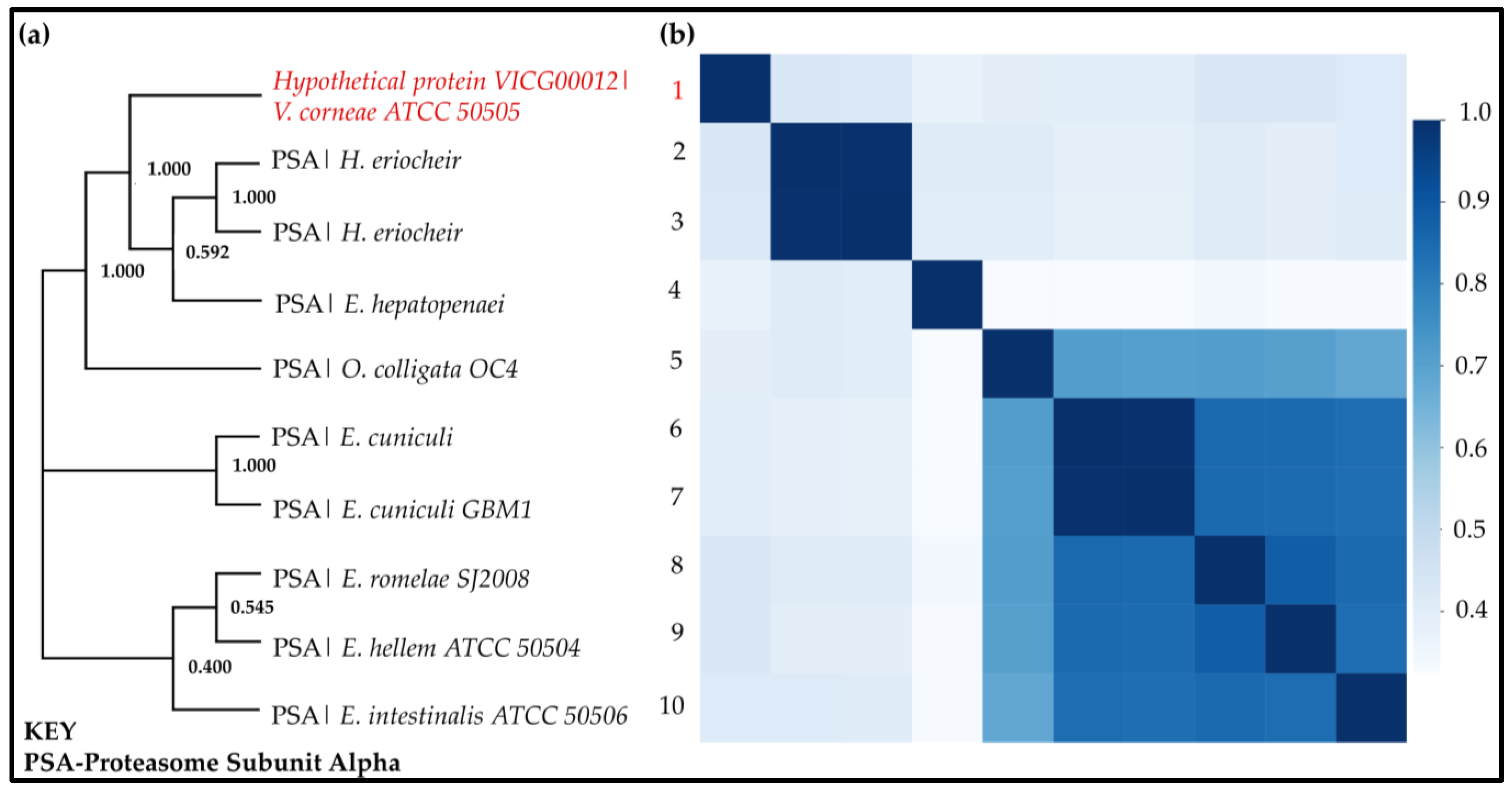
- Phylogenetic Tree Calculations and Pairwise Sequence Identity Calculations
2.3.2. Case Study II: Functional and Structural Analysis of VICG01314
2.3.3. Case Study III: Functional and Structural Analysis of VICG01349
2.3.4. Case Study IV: Functional and Structural Analysis of VICG01687
2.3.5. Case Study V: Functional and Structural Analysis of VICG01723
3. Materials and Methods
3.1. Sequence Retrieval
3.2. Sequence Similarity Search
3.3. Classification of Protein Families
3.4. Subcellular Localization
3.5. Protein Characterization by Physicochemical Properties
3.6. Protein Interaction Network Analysis
3.7. Phylogenetic Tree Calculations and Pairwise Sequence Identity
3.8. Functional Annotation-Functional Domains Prediction and Structural Analysis
3.9. Gene Ontology and Metabolic Pathway Analysis
3.10. Homology Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Han, B.; Weiss, L.M. Microsporidia: Obligate Intracellular Pathogens Within the Fungal Kingdom. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.M. Microsporidia 2003: IWOP-8. J. Eukaryot. Microbiol. 2003, 50, 566–568. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Marcet-Houben, M.; Gabaldón, T. Phylogenomics supports microsporidia as the earliest diverging clade of sequenced fungi. BMC Biol. 2012, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.E. The ecology and evolution of microsporidian parasites. Parasitology 2009, 136, 1901–1914. [Google Scholar] [CrossRef]
- Cuomo, C.A.; Desjardins, C.A.; Bakowski, M.A.; Goldberg, J.; Ma, A.T.; Becnel, J.J.; Didier, E.S.; Fan, L.; Heiman, D.I.; Levin, J.Z.; et al. Microsporidian genome analysis reveals evolutionary strategies for obligate intracellular growth. Genome Res. 2012, 22, 2478–2488. [Google Scholar] [CrossRef]
- Szumowski, S.C.; Troemel, E.R. Microsporidia-Host Interactions. Curr. Opin. Microbiol. 2015, 26, 10–16. [Google Scholar] [CrossRef]
- Han, B.; Takvorian, P.M.; Weiss, L.M. Invasion of Host Cells by Microsporidia. Front. Microbiol. 2020, 11, 172. [Google Scholar] [CrossRef]
- Texier, C.; Vidau, C.; Viguès, B.; El Alaoui, H.; Delbac, F. Microsporidia: A model for minimal parasite-host interactions. Curr. Opin. Microbiol. 2010, 13, 443–449. [Google Scholar] [CrossRef]
- Nakjang, S.; Williams, T.A.; Heinz, E.; Watson, A.K.; Foster, P.G.; Sendra, K.M.; Heaps, S.E.; Hirt, R.P.; Embley, T.M. Reduction and expansion inmicrosporidian genome evolution: New insights from comparative genomics. Genome Biol. Evol. 2013, 5, 2285–2303. [Google Scholar] [CrossRef]
- Katinka, M.D.; Duprat, S.; Cornillott, E.; Méténler, G.; Thomarat, F.; Prensier, G.; Barbe, V.; Peyretaillade, E.; Brottier, P.; Wincker, P.; et al. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 2001, 414, 450–453. [Google Scholar] [CrossRef] [Green Version]
- Polonais, V.; Belkorchia, A.; Roussel, M.; Peyretaillade, E.; Peyret, P.; Diogon, M.; Delbac, F. Identification of two new polar tube proteins related to polar tube protein 2 in the microsporidian Antonospora locustae. FEMS Microbiol. Lett. 2013, 346, 36–44. [Google Scholar] [CrossRef]
- Corradi, N.; Pombert, J.F.; Farinelli, L.; Didier, E.S.; Keeling, P.J. The complete sequence of the smallest known nuclear genome from the microsporidian Encephalitozoon intestinalis. Nat. Commun. 2010, 1, 77. [Google Scholar] [CrossRef]
- Williams, B.A.P.; Lee, R.C.H.; Becnel, J.J.; Weiss, L.M.; Fast, N.M.; Keeling, P.J. Genome sequence surveys of Brachiola algerae and Edhazardia aedis reveal microsporidia with low gene densities. BMC Genomics 2008, 9, 200. [Google Scholar] [CrossRef]
- Pombert, J.F.; Selman, M.; Burki, F.; Bardell, F.T.; Farinelli, L.; Solter, L.F.; Whitman, D.W.; Weiss, L.M.; Corradi, N.; Keeling, P.J. Gain and loss of multiple functionally related, horizontally transferred genes in the reduced genomes of two microsporidian parasites. Proc. Natl. Acad. Sci. USA 2012, 109, 12638–12643. [Google Scholar] [CrossRef]
- Vávra, J.; Lukeš, J. Microsporidia and ‘The Art of Living Together’; Elsevier: Amsterdam, The Netherlands, 2013; Volume 82, ISBN 9780124077065. [Google Scholar]
- Ascunce, M.S.; Valles, S.M.; Oi, D.H.; Shoemaker, D.W.; Plowes, R.; Gilbert, L.; LeBrun, E.G.; Sánchez-Arroyo, H.; Sanchez-Peña, S. Molecular diversity of the microsporidium Kneallhazia solenopsae reveals an expanded host range among fire ants in North America. J. Invertebr. Pathol. 2010, 105, 279–288. [Google Scholar] [CrossRef]
- Coyle, C.M.; Weiss, L.M.; Rhodes, L.V.; Cali, A.; Takvorian, P.M.; Brown, D.F.; Visvesvara, G.S.; Xiao, L.; Naktin, J.; Young, E.; et al. Fatal myositis due to the microsporidian Brachiola algerae, a mosquito pathogen. N. Engl. J. Med. 2004, 351, 42–47. [Google Scholar] [CrossRef]
- Herren, J.K.; Mbaisi, L.; Mararo, E.; Makhulu, E.E.; Mobegi, V.A.; Butungi, H.; Mancini, M.V.; Oundo, J.W.; Teal, E.T.; Pinaud, S.; et al. A microsporidian impairs Plasmodium falciparum transmission in Anopheles arabiensis mosquitoes. Nat. Commun. 2020, 11, 2187. [Google Scholar] [CrossRef]
- Pan, G.; Xu, J.; Li, T.; Xia, Q.; Liu, S.L.; Zhang, G.; Li, S.; Li, C.; Liu, H.; Yang, L.; et al. Comparative genomics of parasitic silkworm microsporidia reveal an association between genome expansion and host adaptation. BMC Genom. 2013, 14, 186. [Google Scholar] [CrossRef]
- Wadi, L.; Reinke, A.W. Evolution of microsporidia: An extremely successful group of eukaryotic intracellular parasites. PLoS Pathog. 2020, 16, e1008276. [Google Scholar] [CrossRef]
- Shahbaaz, M.; Hassan, M.I.; Ahmad, F. Functional annotation of conserved hypothetical proteins from Haemophilus influenzae Rd KW20. PLoS ONE 2013, 8, e84263. [Google Scholar] [CrossRef] [Green Version]
- Desler, C.; Suravajhala, P.; Sanderhoff, M.; Rasmussen, M.; Rasmussen, L.J. In Silico screening for functional candidates amongst hypothetical proteins. BMC Bioinform. 2009, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Eisenstein, E.; Gilliland, G.L.; Herzberg, O.; Moult, J.; Orban, J.; Poljak, R.J.; Banerjei, L.; Richardson, D.; Howard, A.J. Biological function made crystal clear—Annotation of hypothetical proteins via structural genomics. Curr. Opin. Biotechnol. 2000, 11, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Sivashankari, S.; Shanmughavel, P. Functional annotation of hypothetical proteins—A review. Bioinformation 2006, 1, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Galperin, M.Y. Conserved “hypothetical” proteins: New hints and new puzzles. Comp. Funct. Genom. 2001, 2, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Mazandu, G.K.; Mulder, N.J. Function prediction and analysis of mycobacterium tuberculosis hypothetical proteins. Int. J. Mol. Sci. 2012, 13, 7283–7302. [Google Scholar] [CrossRef]
- Kumar, K.; Prakash, A.; Tasleem, M.; Islam, A.; Ahmad, F.; Hassan, M.I. Functional annotation of putative hypothetical proteins from Candida dubliniensis. Gene 2014, 543, 93–100. [Google Scholar] [CrossRef]
- Gupta, S.; Singh, Y.; Kumar, H.; Raj, U.; Rao, A.R.; Varadwaj, P.K. Identification of Novel Abiotic Stress Proteins in Triticum aestivum Through Functional Annotation of Hypothetical Proteins. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 205–220. [Google Scholar] [CrossRef]
- Omeershffudin, U.N.M.; Kumar, S. In silico approach for mining of potential drug targets from hypothetical proteins of bacterial proteome. Int. J. Mol. Biol. 2019, 4, 145–152. [Google Scholar] [CrossRef]
- Texier, C.; Brosson, D.; El Alaoui, H.; Méténier, G.; Vivarès, C.P. Post-genomics of microsporidia, with emphasis on a model of minimal eukaryotic proteome: A review. Folia Parasitol. 2005, 52, 15–22. [Google Scholar] [CrossRef]
- Mittleider, D.; Green, L.C.; Mann, V.H.; Michael, S.F.; Didier, E.S.; Brindley, P.J. Sequence survey of the genome of the opportunistic microsporidian pathogen, Vittaforma corneae. J. Eukaryot. Microbiol. 2002, 49, 393–401. [Google Scholar] [CrossRef]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, 5–9. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2021, 49, D394–D403. [Google Scholar] [CrossRef]
- Attwood, T.K. The PRINTS database: A resource for identification of protein families. Brief. Bioinform. 2002, 3, 252–263. [Google Scholar] [CrossRef]
- Wu, C.H.; Nikolskaya, A.; Huang, H.; Yeh, L.S.; Natale, D.A.; Vinayaka, C.R.; Hu, Z.Z.; Mazumder, R.; Kumar, S.; Kourtesis, P.; et al. PIRSF: Family classification system at the Protein Information Resource. Nucleic Acids Res. 2004, 32, 112–114. [Google Scholar] [CrossRef]
- Lees, J.G.; Lee, D.; Studer, R.A.; Dawson, N.L.; Sillitoe, I.; Das, S.; Yeats, C.; Dessailly, B.H.; Rentzsch, R.; Orengo, C.A. Gene3D: Multi-domain annotations for protein sequence and comparative genome analysis. Nucleic Acids Res. 2014, 42, 240–245. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef]
- Sigrist, C.J.A.; Cerutti, L.; Hulo, N.; Gattiker, A.; Falquet, L.; Pagni, M.; Bairoch, A.; Bucher, P. PROSITE: A documented database using patterns and profiles as motif descriptors. Brief. Bioinform. 2002, 3, 265–274. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. The Proteomics Protocols Handbook. Proteomics Protoc. Handb. 2005, 571–608. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Salvatore, M.; Emanuelsson, O.; Winther, O.; Von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2019, 2, 429. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.L.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequence. Sixth Int. Conf. Intell. Syst. Mol. Biol. 2008, 8, 175–182. [Google Scholar]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Hatherley, R.; Brown, D.K.; Glenister, M.; Tastan Bishop, Ö. PRIMO: An interactive homology modeling pipeline. PLoS ONE 2016, 11, e166698. [Google Scholar] [CrossRef]
- Sen, T.; Verma, N.K. Functional annotation and curation of hypothetical proteins present in a newly emerged serotype 1c of Shigella flexneri: Emphasis on selecting targets for virulence and vaccine design studies. Genes 2020, 11, 340. [Google Scholar] [CrossRef] [Green Version]
- Thakur, C.J.; Saini, S.; Notra, A.; Chauhan, B. Deciphering the functional role of hypothetical proteins from Chloroflexus aurantiacs J-10-f1 using bioinformatics approach. Mol. Biol. Res. Commun. 2020, 9, 129–139. [Google Scholar] [CrossRef]
- Adams, M.A.; Suits, M.D.L.; Zheng, J.; Jia, Z. Piecing together the structure-function puzzle: Experiences in structure-based functional annotation of hypothetical proteins. Proteomics 2007, 7, 2920–2932. [Google Scholar] [CrossRef]
- Broad Institute Vittaforma. Corneae ATCC 50505 Genome Sequencing and Assembly. Available online: http://www.ncbi.nlm.nih.gov/bioproject/PRJNA63501 (accessed on 8 December 2022).
- Campbell, S.E.; Williams, T.A.; Yousuf, A.; Soanes, D.M.; Paszkiewicz, K.H.; Williams, B.A.P. The Genome of Spraguea lophii and the Basis of Host-Microsporidian Interactions. PLoS Genet. 2013, 9, e1003676. [Google Scholar] [CrossRef]
- Peyretaillade, E.; Parisot, N.; Polonais, V.; Terrat, S.; Denonfoux, J.; Dugat-Bony, E.; Wawrzyniak, I.; Biderre-Petit, C.; Mahul, A.; Rimour, S.; et al. Annotation of microsporidian genomes using transcriptional signals. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef]
- Eddy, S.R. HMMER User’ s Guide: Biological Sequence Analysis Using Profile Hidden Markov Models. Howard Hughes Medical Institute. 2020. Available online: http://eddylab.org/software/hmmer/Userguide.pdf (accessed on 8 December 2022).
- Senderskiy, I.V.; Timofeev, S.A.; Seliverstova, E.V.; Pavlova, O.A.; Dolgikh, V.V. Secretion of Antonospora (Paranosema) locustae proteins into infected cells suggests an active role of microsporidia in the control of host programs and metabolic processes. PLoS ONE 2014, 9, e93585. [Google Scholar] [CrossRef]
- Miranda-Saavedra, D.; Stark, M.J.R.; Packer, J.C.; Vivares, C.P.; Doerig, C.; Barton, G.J. The complement of protein kinases of the microsporidium Encephalitozoon cuniculi in relation to those of Saccharomyces cerevisiae and Schizosaccharomyces pombe. BMC Genom. 2007, 8, 1–21. [Google Scholar] [CrossRef]
- Li, Z.; Hao, Y.; Wang, L.; Xiang, H.; Zhou, Z. Genome-wide identification and comprehensive analyses of the kinomes in four pathogenic microsporidia species. PLoS ONE 2014, 9, e115890. [Google Scholar] [CrossRef]
- Koegl, M.; Hoppe, T.; Schlenker, S.; Ulrich, H.D.; Mayer, T.U.; Jentsch, S. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 1999, 96, 635–644. [Google Scholar] [CrossRef]
- Iuchi, S. Three classes of C2H2 zinc finger proteins. Cell. Mol. Life Sci. 2001, 58, 625–635. [Google Scholar] [CrossRef]
- Gonzalez, J.; Tinoco, I.J. Identification and characterization of metal ion binding sites in RNA. Methods Enzymol. 2002, 338, 421–443. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. The HORMA domain: A common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem. Sci. 1998, 23, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Tamim El Jarkass, H.; Reinke, A.W. The ins and outs of host-microsporidia interactions during invasion, proliferation and exit. Cell. Microbiol. 2020, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Vossbrinck, C.R.; Yang, Q.; Meng, X.Z.; Luo, J.; Pan, G.Q.; Zhou, Z.Y.; Li, T. Evolutionary and functional studies on microsporidian ATP-binding cassettes: Insights into the adaptation of microsporidia to obligated intracellular parasitism. Infect. Genet. Evol. 2019, 68, 136–144. [Google Scholar] [CrossRef]
- Schneider, E.; Hunke, S. ATP-binding-cassette (ABC) transport systems: Functional and structural aspects of the ATP-hydrolyzing subunits/domains. FEMS Microbiol. Rev. 1998, 22, 1–20. [Google Scholar] [CrossRef]
- Higgins, C.F. ABC transporters: Physiology, structure and mechanism—An overview. Res. Microbiol. 2001, 152, 205–210. [Google Scholar] [CrossRef]
- James, T.Y.; Pelin, A.; Bonen, L.; Ahrendt, S.; Sain, D.; Corradi, N.; Stajich, J.E. Shared signatures of parasitism and phylogenomics unite cryptomycota and microsporidia. Curr. Biol. 2013, 23, 1548–1553. [Google Scholar] [CrossRef]
- Jain, B.P.; Pandey, S. WD40 Repeat Proteins: Signalling Scaffold with Diverse Functions. Protein J. 2018, 37, 391–406. [Google Scholar] [CrossRef]
- Xu, C.; Min, J. Structure and function of WD40 domain proteins. Protein Cell 2011, 2, 202–214. [Google Scholar] [CrossRef]
- Kwan, A.H.Y.; Winefield, R.D.; Sunde, M.; Matthews, J.M.; Haverkamp, R.G.; Templeton, M.D.; Mackay, J.P. Structural basis for rodlet assembly in fungal hydrophobins. Proc. Natl. Acad. Sci. USA 2006, 103, 3621–3626. [Google Scholar] [CrossRef]
- Yang, D.; Pan, G.; Dang, X.; Shi, Y.; Li, C.; Peng, P.; Luo, B.; Bian, M.; Song, Y.; Ma, C.; et al. Interaction and assembly of two novel proteins in the spore wall of the microsporidian species Nosema bombycis and their roles in adherence to and infection of host cells. Infect. Immun. 2015, 83, 1715–1731. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Vandekerckhove, J. Structure and function of actin. Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 49–76. [Google Scholar] [CrossRef]
- Mahadevan, L.; Riera, C.S.; Shin, J.H. Structural dynamics of an actin spring. Biophys. J. 2011, 100, 839–844. [Google Scholar] [CrossRef]
- Möller, S.; Croning, M.D.R.; Apweiler, R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics 2001, 17, 646–653. [Google Scholar] [CrossRef]
- Korkuc, P.; Walther, D. Physicochemical characteristics of structurally determined metabolite-protein and drug-protein binding events with respect to binding specificity. Front. Mol. Biosci. 2015, 2, 1–20. [Google Scholar] [CrossRef]
- Moses, V.; Hatherley, R.; Tastan Bishop, Ö. Bioinformatic characterization of type-specific sequence and structural features in auxiliary activity family 9 proteins. Biotechnol. Biofuels 2016, 9, 1–17. [Google Scholar] [CrossRef]
- Nguyen, K.; Guo, X.; Pan, Y. Phylogeny in Multiple Sequence Alignments. In Multiple Biological Sequence Alignment: Scoring Functions, Algorithms and Applications; John Wiley & Sons, Inc.: London, UK, 2016; pp. 103–112. ISBN 9781119273769. [Google Scholar]
- Xiong, J. Essential Bioinformatics; Cambridge University Press: New York, NY, USA, 2006; Volume 3, ISBN 9780521840989. [Google Scholar]
- De La Cruz, J.; Kressler, D.; Linder, P. Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem. Sci. 1999, 24, 192–198. [Google Scholar] [CrossRef]
- Jaroenlak, P.; Boakye, D.W.; Vanichviriyakit, R.; Williams, B.A.P.; Sritunyalucksana, K.; Itsathitphaisarn, O. Identification, characterization and heparin binding capacity of a spore-wall, virulence protein from the shrimp microsporidian, Enterocytozoon hepatopenaei (EHP). Parasites Vectors 2018, 11, 1–15. [Google Scholar] [CrossRef]
- Kobe, B.; Kajava, A. V The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 2001, 11, 725–732. [Google Scholar] [CrossRef]
- Heinz, E.; Williams, T.A.; Nakjang, S.; Noël, C.J.; Swan, D.C.; Goldberg, A.V.; Harris, S.R.; Weinmaier, T.; Markert, S.; Becher, D.; et al. The Genome of the Obligate Intracellular Parasite Trachipleistophora hominis: New Insights into Microsporidian Genome Dynamics and Reductive Evolution. PLoS Pathog. 2012, 8, 979. [Google Scholar] [CrossRef]
- Delbac, F.; Peuvel, I.; Metenier, G.; Peyretaillade, E.; Vivares, C.P. Microsporidian invasion apparatus: Identification of a novel polar tube protein and evidence for clustering of ptp1 and ptp2 genes in three Encephalitozoon species. Infect. Immun. 2001, 69, 1016–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Poulin, R. Revisiting the phylogeny of microsporidia. Int. J. Parasitol. 2021, 51, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Drive, R.M.; Valley, M. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Luthy, R.; Bowei, J.; Einsenberg, D. Verify3D: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from Standard Atomic Volumes as a Quality Measure for Protein Crystal Structures. J. Mol. Biol. 1996, 264, 121–136. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, 407–410. [Google Scholar] [CrossRef]
- Löwe, J.; Stock, D.; Jap, B.; Zwickl, P.; Baumeister, W.; Hubert, R. Crystal Structure of the 20S Proteasome from the Archaeon acidophilum at 3.4 Å resolution. Science 1995, 268, 533–539. [Google Scholar] [CrossRef]
- Zwickl, P.; Grziwa, A.; Puehler, G.; Dahlmann, B.; Lottspeich, F.; Baumeister, W. Primary Structure of the Thermoplasma Proteasome and Its Implications for the Structure, Function, and Evolution of the Multicatalytic Proteinase. Biochemistry 1992, 31, 964–972. [Google Scholar] [CrossRef]
- Groll, M.; Bajorek, M.; Köhler, A.; Moroder, L.; Rubin, D.M.; Huber, R.; Glickman, M.H.; Finley, D. A gated channel into the proteasome core particle. Nat. Struct. Biol. 2000, 7, 1062–1067. [Google Scholar] [CrossRef]
- Vossbrinck, C.R.; Debrunner-Vossbrinck, B.A. Molecular phylogeny of the Microsporidia: Ecological, ultrastructural and taxonomic considerations. Folia Parasitol. 2005, 52, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Fraenkel, E.; Pabo, C.O.; Pavletich, N.P. Structural basis of DNA recognition by the heterodimeric cell cycle transcription factor E2F-DP. Genes Dev. 1999, 13, 666–674. [Google Scholar] [CrossRef]
- Fuller-Pace, F.V. DExD/H box RNA helicases: Multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 2006, 34, 4206–4215. [Google Scholar] [CrossRef]
- Keeling, P.J.; Palmer, J.D. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 2008, 9, 605–618. [Google Scholar] [CrossRef]
- Jacsó, P. Google scholar: The pros and the cons. Online Inf. Rev. 2005, 29, 208–214. [Google Scholar] [CrossRef]
- Burnham, J.F. Scopus database: A review. Biomed. Digit. Libr. 2006, 3, 1–8. [Google Scholar] [CrossRef]
- Aurrecoechea, C.; Barreto, A.; Brestelli, J.; Brunk, B.P.; Caler, E.V.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; et al. AmoebaDB and MicrosporidiaDB: Functional genomic resources for Amoebozoa and Microsporidia species. Nucleic Acids Res. 2011, 39, 612–619. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef]
- Kumar, A.; Prameela, T.P.; Bhai, R.S.; Siljo, A.; Biju, C.N.; Anandaraj, M.; Vinatzer, B.A. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Eur. J. plant Pathol. 2012, 132, 3389–3402. [Google Scholar] [CrossRef]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.; Grout, J.; Corlay, S.; et al. Jupyter Notebooks—A publishing format for reproducible computational workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas—Proceedings of the 20th International Conference on Electronic Publishing; IOS Press, Inc.: Amsterdam, The Netherlands, 2016; Volume 2016, pp. 87–90. ISBN 9781614996484. [Google Scholar]
- Silaparasetty, N. Python Programming in Jupyter Notebook. In Machine Learning Concepts with Python and the Jupyter Notebook Environment; Apress: Berkeley, CA, USA, 2020; pp. 119–145. ISBN 978-1-4842-5967-2. [Google Scholar]
- Van Rossum, G.; Drake, F.L. The Python Reference Manual; iUniverse: Indianapolis, IN, USA, 2000. [Google Scholar]
- Van Rossum, G.; Drake, F.L. Python Tutorial. Python Softw. Found. 2017, 42, 1–122. [Google Scholar]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 Update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallgren, J.; Tsirigos, K.D.; Damgaard Pedersen, M.; Juan, J.; Armenteros, A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Nielsen, H.; Engelbrecht, J.; Brunak, S.; von Heijne, G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Peterson, D.; Tamura, K. MEGA-CC: Computing core of molecular evolutionary genetics analysis program for automated and iterative data analysis. Bioinformatics 2012, 28, 2685–2686. [Google Scholar] [CrossRef]
- Huson, D.H.; Richter, D.C.; Rausch, C.; Dezulian, T.; Franz, M.; Rupp, R. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinform. 2007, 8, 1–6. [Google Scholar] [CrossRef]
- Faya, N.; Penkler, D.L.; Tastan Bishop, Ö. Human, vector and parasite Hsp90 proteins: A comparative bioinformatics analysis. FEBS Open Bio 2015, 5, 916–927. [Google Scholar] [CrossRef]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, 369–373. [Google Scholar] [CrossRef]
- Bailey, T.L.; Gribskov, M. Combining evidence using p-values: Application to sequence homology searches. Bioinformatics 1998, 14, 48–54. [Google Scholar] [CrossRef]
- Ross, C.; Knox, C.; Tastan Bishop, Ö. Interacting motif networks located in hotspots associated with RNA release are conserved in Enterovirus capsids. FEBS Lett. 2017, 591, 1687–1701. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- de Castro, E.; Sigrist, C.J.A.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, 362–365. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, 244–248. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, 1–10. [Google Scholar] [CrossRef]


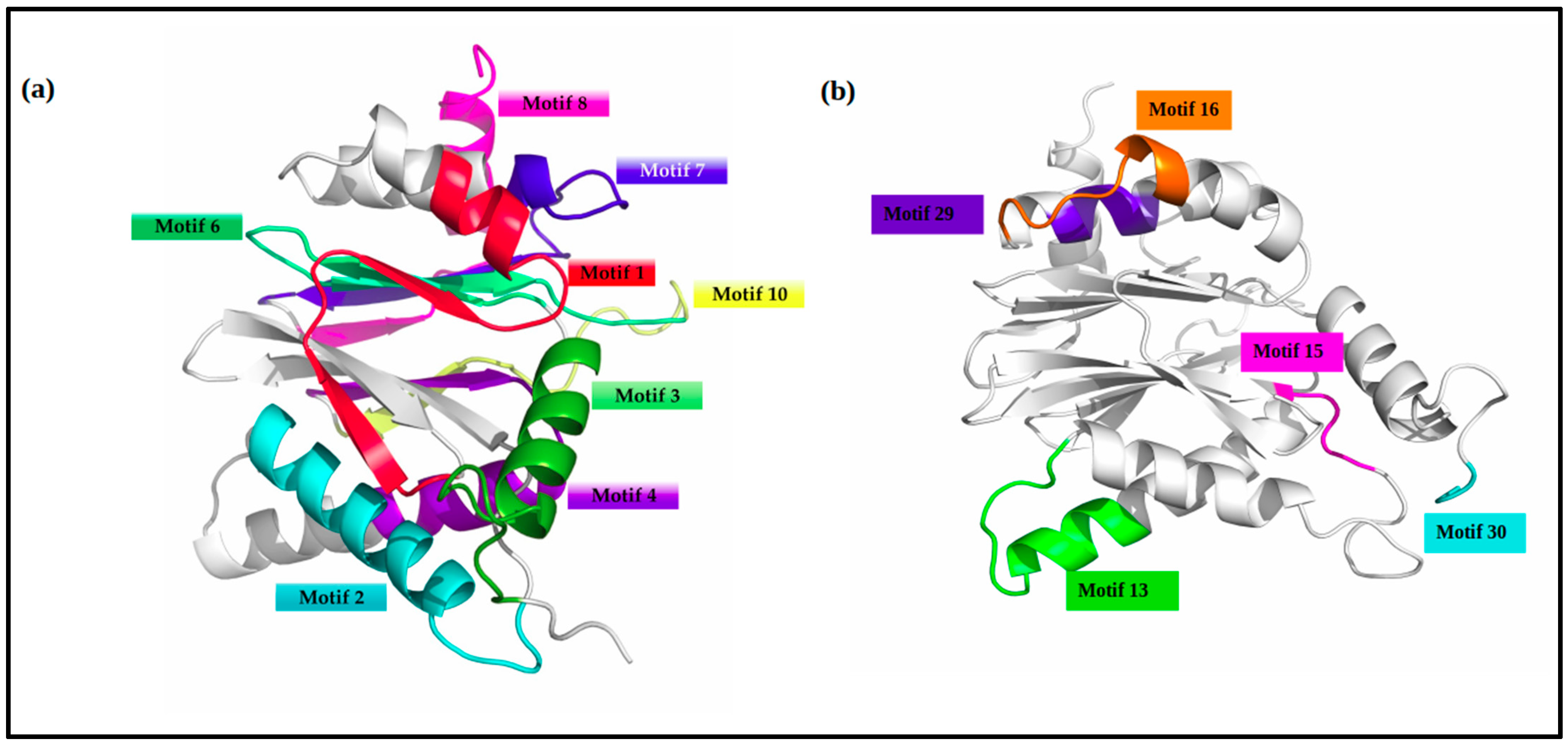

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Motif # | Regular Expression | Occurrence |
|---|---|---|
| Motif 1 | [ML]S[GA][IV][SE]Y[PQ]C[YF]A[TN]A[AS]G[EK][KD][HY][GLS]K | Present in all species |
| Motif 2 | TPDI[LF][CA]R[SV][FV][AG]DK[MIV]Q[KQ][LY]IQ[SRT] | Present in all species |
| Motif 3 | [NH]IF[ND][SA][DNE]G[KE][LI][LK]QIEY[GA]L[ES]A[VL] | Present in all species |
| Motif 4 | [KN][CL]Y[VL][AGQ][IM]TG[LR][PL][GD]D[IVMT][DN][YF][IV][VFI][KNR]R[IA | Present in all species |
| Motif 5 | AF[NA][AS][ST]IFG[FM][DQ][KDG][EG][KR][PAT][MV][IV]Y[QF]T | Absent only in E. hepatopenaei |
| Motif 6 | [PQ][IV][VI][TS][VA][KS]S[KDR][DSE][MEH][IV]V[CF][VA]S[KR]K[IVY][PN][QKR | Present in all species |
| Motif 7 | ALL[EM]SIG[ART][DET][SA][GE][CF][ST]E[IV][ED][VA][AFG][YV][LF | Present in all species |
| Motif 8 | [QE][DG]L[CEI][YHK]L[SE][AD][KQE]E[IV][DN][KR][IHV]LQ[DI][IV]A | Present in all species |
| Motif 9 | [KN][FY][IL]E[KN][NS]Y[RS][ES][DN][IKM][DES][DK][KE]EL[FI][EN][LV] | Absent in V. corneae and E. hepatopenaei |
| Motif 10 | [DE][KP]L[EQ]E[SED][ESV][AIS][ST][ST][FIV][YHK][KR][VI]S[ED | Present in all species |
| Motif 11 | KMVANRK[TA]FS[FL] | Absent in V. corneae |
| Motif 12 | MTTQEE[IV | Absent in V. corneae |
| Motif 13 | LA[SA][DS][KT]EY[ET]LGC | Present only in V. corneae and H. eriocheir |
| Motif 15 | RS | Present in V. corneae and E. intestinalis |
| Motif 16 | YLEK[HT][FY][DK] | Present only in V. corneae and E. hepatopenaei |
| Motif 17 | [AE]ER[SP] | Absent in V. corneae |
| Motif 18 | NG | Absent in V. corneae |
| Motif 19 | M[EH] | Absent in V. corneae |
| Motif 20 | RA | Absent in V. corneae |
| Motif 21 | NEEI | Absent in V. corneae |
| Motif 22 | NA | Absent in V. corneae |
| Motif 23 | NS | Absent in V. corneae |
| Motif 24 | FE | Absent in V. corneae |
| Motif 25 | KA | Absent in V. corneae |
| Motif 26 | N[NS] | Absent in V. corneae |
| Motif 27 | TE | Absent in V. corneae |
| Motif 28 | [FM]G | Present only in V. corneae and E. hepatopenaei |
| Motif 29 | [FN]A[IT] | Present only in V. corneae and E. hepatopenaei |
| Motif 30 | [NY]E[KQ][IY] | Present only in V. corneae and E. hepatopenaei |
| Sample ID/Motif # | M1 | M2 | M3 | M4 | M6 | M7 | M8 | M10 | M13 | M15 | M16 | M28 | M29 | M30 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VICG0012 | 148–168 | 103–123 | 9–29 | 68–88 | 32–52 | 190–210 | 212–231 | 52–68 | 90–102 | 125–128 | 170–178 | 1–4 | 182–186 | 4–9 |
| Encephalitozoon cuniculi | 148–168 | 103–123 | 9–29 | 68–88 | 32–52 | 191–211 | 214–234 | 52–68 | - | - | - | - | - | - |
| Encephalitozoon cuniculi GB-M1 | 148–168 | 103–123 | 9–29 | 68–88 | 32–52 | 191–211 | 214–234 | 52–68 | - | - | - | - | - | - |
| Enterocytozoon hepatopenaei | 151–171 | 101–121 | 7–27 | 66–86 | 30–50 | 194–214 | 216–236 | 50–66 | - | - | 173–181 | 132–135 | 190–194 | 2–7 |
| Hepatospora eriocheir | 146–166 | 101–121 | 7–27 | 66–86 | 30–50 | 189–209 | 211–231 | 50–66 | 88–100 | - | - | - | - | - |
| Hepatospora eriocheir | 146–166 | 101–121 | 7–27 | 66–86 | 30–50 | 189–209 | 211–231 | 50–66 | 88–100 | - | - | - | - | - |
| Encephalitozoon intestinalis ATCC 50506 | 148–168 | 103–123 | 9–29 | 68–88 | 32–52 | 191–211 | 214–234 | 52–68 | - | 125–128 | - | - | - | - |
| Encephalitozoon hellem ATCC 50504 | 148–168 | 103–123 | 9–29 | 68–88 | 32–52 | 191–211 | 214–234 | 52–68 | - | - | - | - | - | - |
| Encephalitozoon romaleae SJ-2008 | 148–168 | 103–123 | 9–29 | 68–88 | 32–52 | 191–211 | 214–234 | 52–68 | - | - | - | - | - | - |
| Ordospora colligata OC4 | 148–168 | 103–123 | 9–29 | 68–88 | 32–52 | 191–211 | 214–234 | 52–68 | - | - | - | - | - | - |
| HP ID | Template PDB ID | Sequence Identity (%) | Coverage (%) | Resolution (Å) |
|---|---|---|---|---|
| VICG00012 | 6QM7 | 25 | 98 | 2.80 |
| VICG01314 | 6AZ3 | 60 | 98 | 2.50 |
| VICG01349 | 1CF7 | 19 | 35 | 2.60 |
| 5TUU | 24 | 41 | 2.25 | |
| VICG01687 | 2WJY_A | 43 | 98 | 2.50 |
| VICG01723 | 4BUJ | 36 | 93 | 3.70 |
| 5MC6 | 38 | 93 | 3.80 | |
| 4A4Z | 38 | 93 | 2.40 | |
| 6IEH | 44 | 98 | 2.89 | |
| 6BB8 | 45 | 94 | 3.49 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ang’ang’o, L.M.; Herren, J.K.; Tastan Bishop, Ö. Structural and Functional Annotation of Hypothetical Proteins from the Microsporidia Species Vittaforma corneae ATCC 50505 Using in silico Approaches. Int. J. Mol. Sci. 2023, 24, 3507. https://doi.org/10.3390/ijms24043507
Ang’ang’o LM, Herren JK, Tastan Bishop Ö. Structural and Functional Annotation of Hypothetical Proteins from the Microsporidia Species Vittaforma corneae ATCC 50505 Using in silico Approaches. International Journal of Molecular Sciences. 2023; 24(4):3507. https://doi.org/10.3390/ijms24043507
Chicago/Turabian StyleAng’ang’o, Lilian Mbaisi, Jeremy Keith Herren, and Özlem Tastan Bishop. 2023. "Structural and Functional Annotation of Hypothetical Proteins from the Microsporidia Species Vittaforma corneae ATCC 50505 Using in silico Approaches" International Journal of Molecular Sciences 24, no. 4: 3507. https://doi.org/10.3390/ijms24043507
APA StyleAng’ang’o, L. M., Herren, J. K., & Tastan Bishop, Ö. (2023). Structural and Functional Annotation of Hypothetical Proteins from the Microsporidia Species Vittaforma corneae ATCC 50505 Using in silico Approaches. International Journal of Molecular Sciences, 24(4), 3507. https://doi.org/10.3390/ijms24043507