
Extracellular Vesicles Released from Cancer Cells Promote Tumorigenesis by Inducing Epithelial to Mesenchymal Transition via β-Catenin Signaling

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
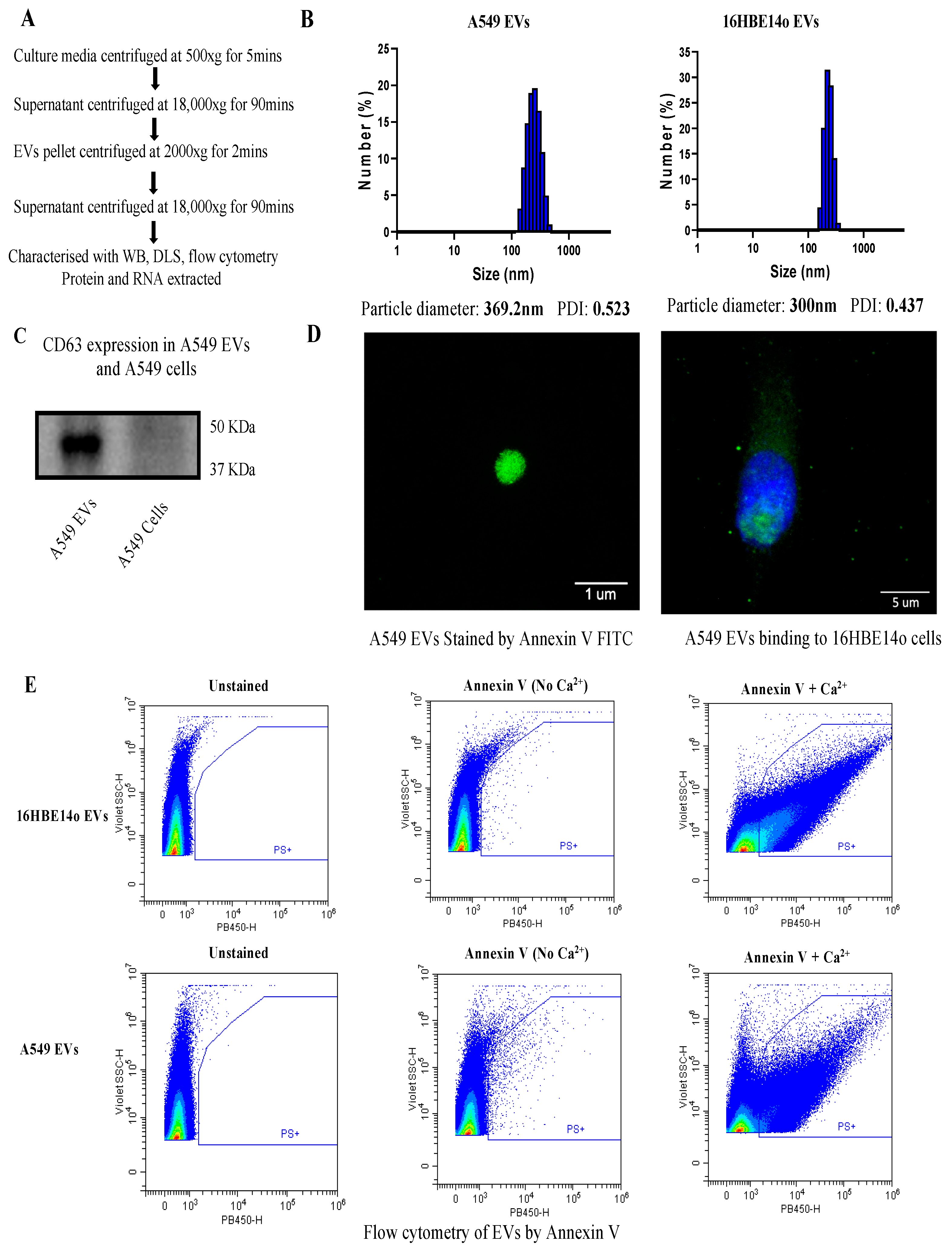
2.1. Isolation and Characterization of EVs
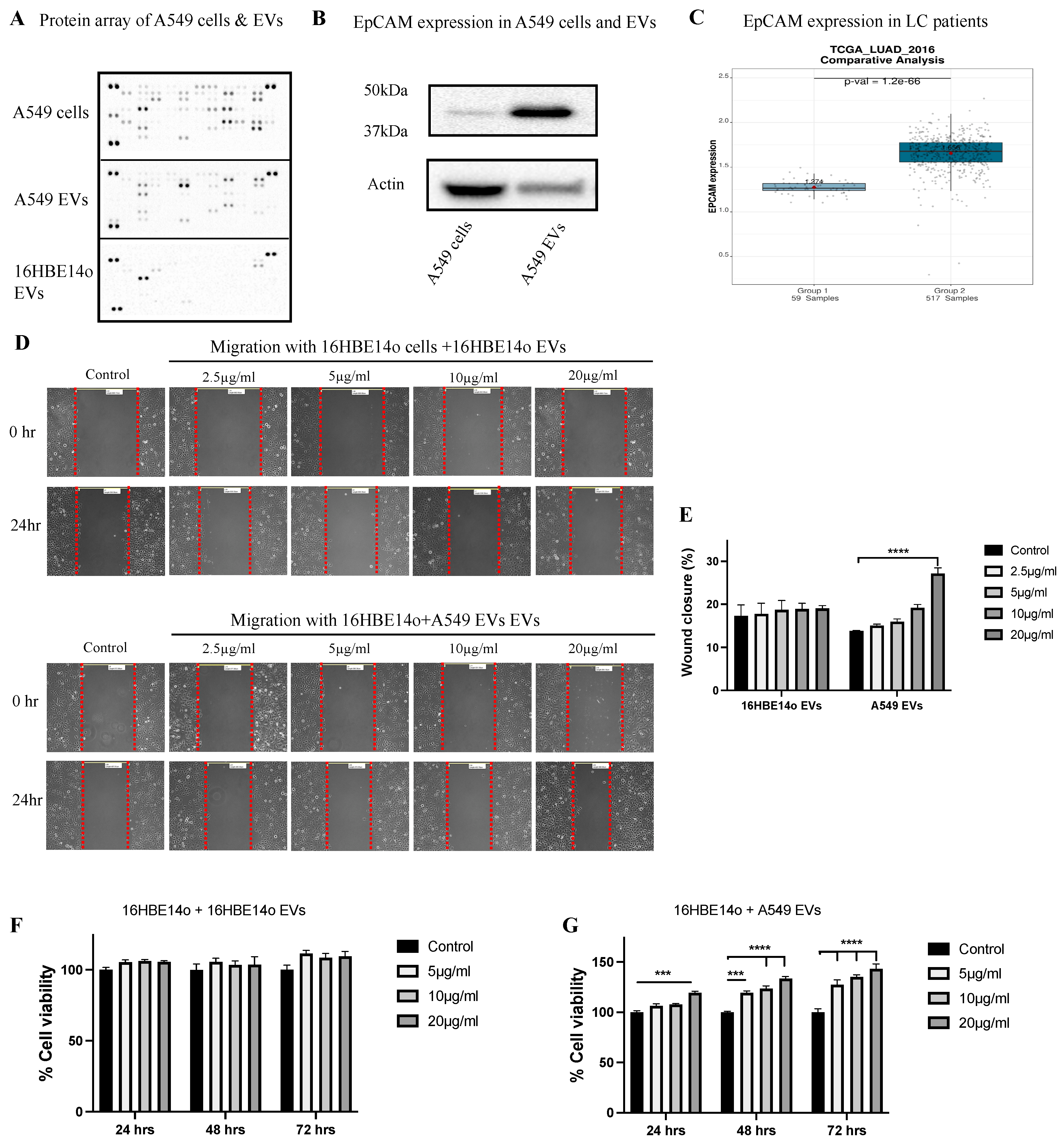
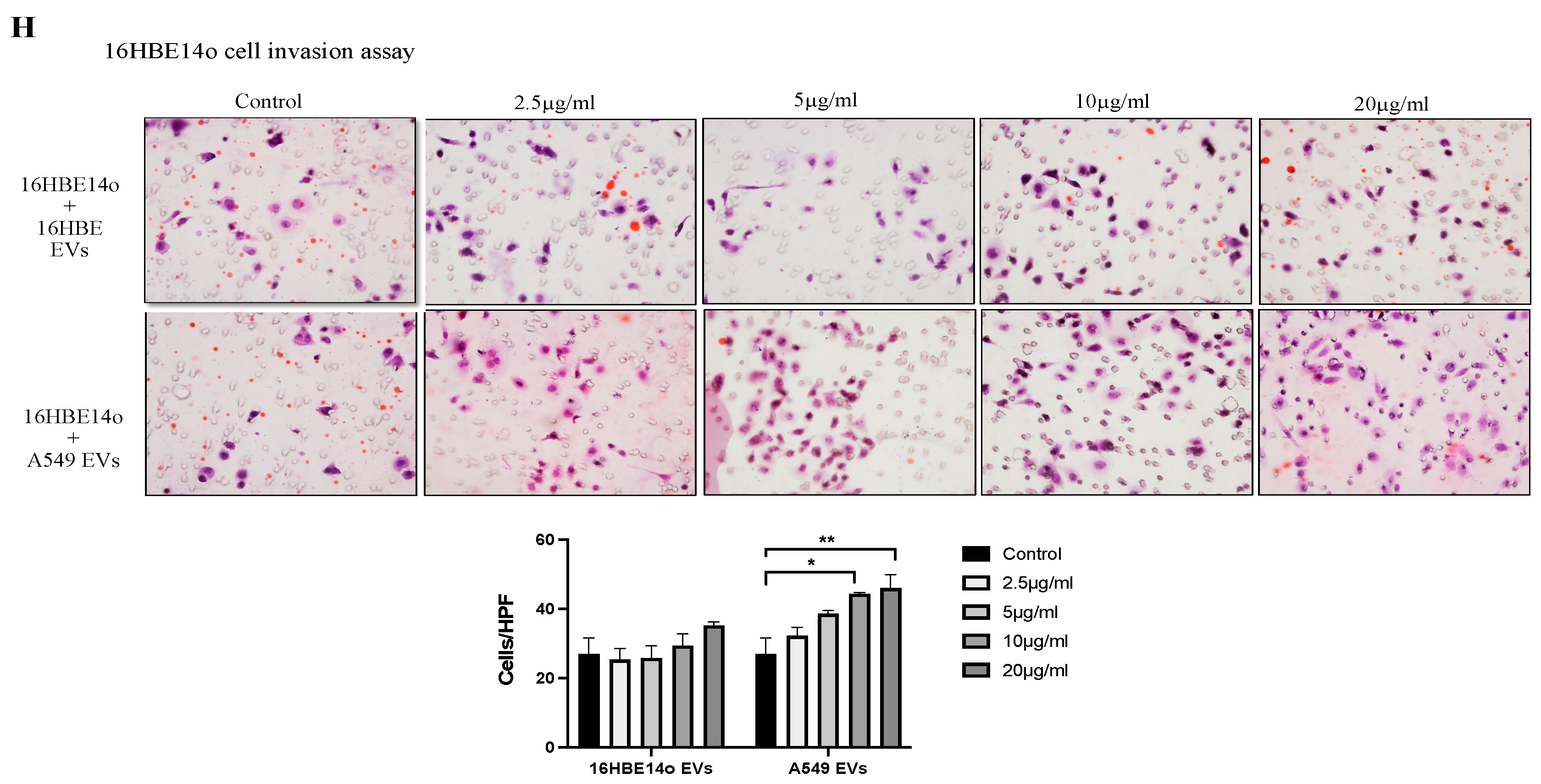
2.2. Profiling A549 EVs for Oncogenic Protein Expression and Effects on Cell Migration, Invasion, and Proliferation
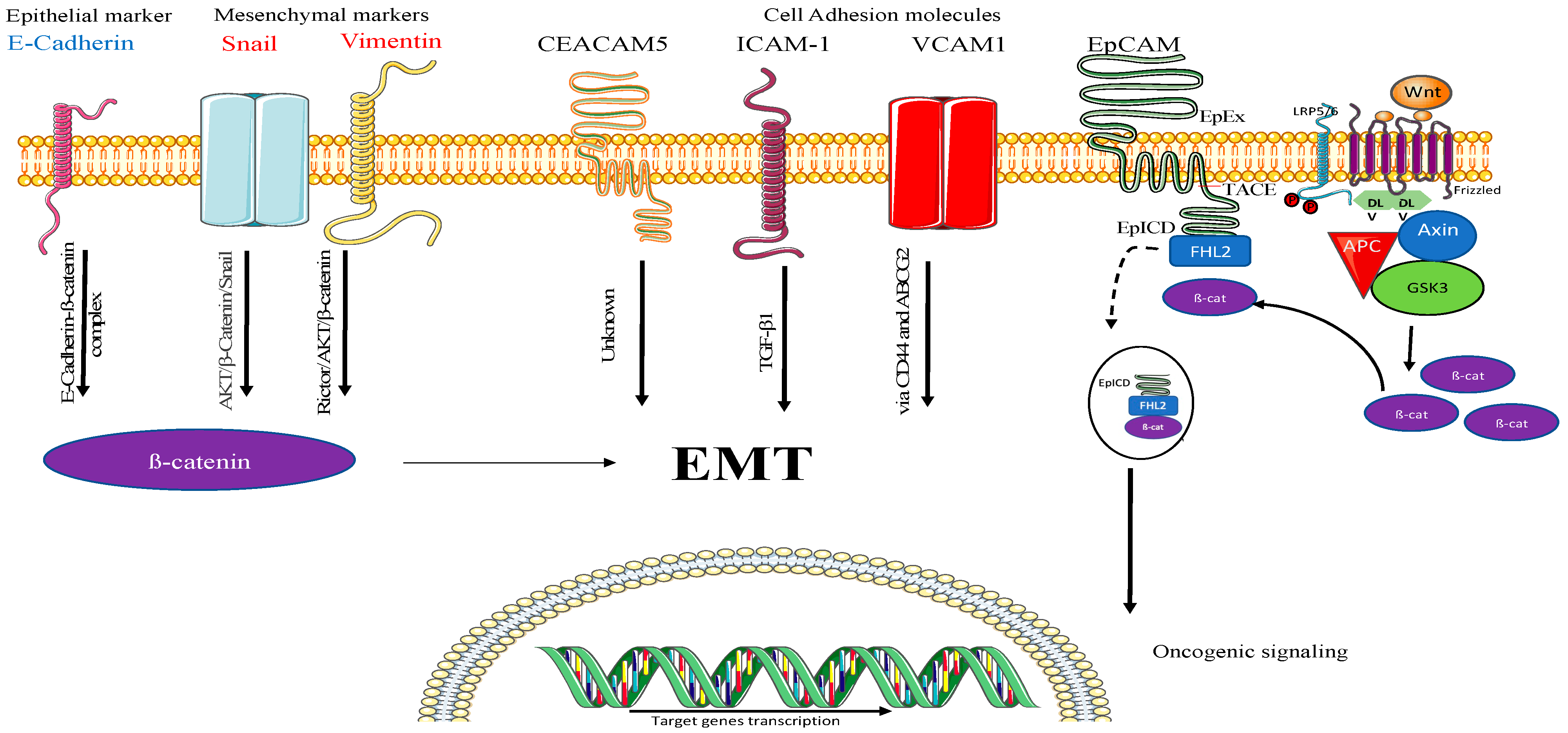
2.3. Understanding the Mechanism of A549-EV-Induced Tumorigenesis
2.4. Understating the Location, Encapsulation, and Uptake of EpCAM
3. Materials and Methods
3.1. Cell Culture
3.2. Collection and Isolation of EVs
3.3. Particle Size Distribution by Dynamic Light Scattering
3.4. Protein Extraction from EVs, Whole Cell Lysate, and Protein Quantification
3.5. Western Blotting
3.6. Immunofluorescence
3.7. Flow Cytometry
3.8. Protein Array of EVs and Cells
3.9. Migration (Scratch Wound Healing Assay)
3.10. MTT Cell Proliferation Assay
3.11. Cell Invasion Assay
3.12. Statistical Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; Bebawy, M. Proteins regulating microvesicle biogenesis and multidrug resistance in cancer. Proteomics 2019, 19, 1800165. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Rabinowits, G.; Gerçel-Taylor, C.; Day, J.M.; Taylor, D.D.; Kloecker, G.H. Exosomal microRNA: A diagnostic marker for lung cancer. Clin. Lung Cancer 2009, 10, 42–46. [Google Scholar] [CrossRef]
- Jakobsen, K.R.; Paulsen, B.S.; Bæk, R.; Varming, K.; Sorensen, B.S.; Jørgensen, M.M. Exosomal proteins as potential diagnostic markers in advanced non-small cell lung carcinoma. J. Extracell. Vesicles 2015, 4, 26659. [Google Scholar] [CrossRef]
- Kalra, H.; Gangoda, L.; Fonseka, P.; Chitti, S.V.; Liem, M.; Keerthikumar, S.; Samuel, M.; Boukouris, S.; Al Saffar, H.; Collins, C. Extracellular vesicles containing oncogenic mutant β-catenin activate Wnt signalling pathway in the recipient cells. J. Extracell. Vesicles 2019, 8, 1690217. [Google Scholar] [CrossRef]
- Krishn, S.R.; Salem, I.; Quaglia, F.; Naranjo, N.M.; Agarwal, E.; Liu, Q.; Sarker, S.; Kopenhaver, J.; McCue, P.A.; Weinreb, P.H. The αvβ6 integrin in cancer cell-derived small extracellular vesicles enhances angiogenesis. J. Extracell. Vesicles 2020, 9, 1763594. [Google Scholar] [CrossRef]
- Malyla, V.; Paudel, K.R.; Shukla, S.D.; Donovan, C.; Wadhwa, R.; Pickles, S.; Chimankar, V.; Sahu, P.; Bielefeldt-Ohmann, H.; Bebawy, M. Recent advances in experimental animal models of lung cancer. Future Med. Chem. 2020, 12, 567–570. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Brozos-Vázquez, E.M.; Díaz-Peña, R.; García-González, J.; León-Mateos, L.; Mondelo-Macía, P.; Peña-Chilet, M.; López-López, R. Immunotherapy in nonsmall-cell lung cancer: Current status and future prospects for liquid biopsy. Cancer Immunol. Immunother. 2021, 70, 1177–1188. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Engholm, G.; Ferlay, J.; Christensen, N.; Bray, F.; Gjerstorff, M.L.; Klint, Å.; Køtlum, J.E.; Ólafsdóttir, E.; Pukkala, E.; Storm, H.H. NORDCAN–a Nordic tool for cancer information, planning, quality control and research. Acta Oncol. 2010, 49, 725–736. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Kerbel, R.S.; Allison, A.C.; Rak, J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 3794–3799. [Google Scholar] [CrossRef]
- Baj-Krzyworzeka, M.; Szatanek, R.; Węglarczyk, K.; Baran, J.; Zembala, M. Tumour-derived microvesicles modulate biological activity of human monocytes. Immunol. Lett. 2007, 113, 76–82. [Google Scholar] [CrossRef]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes Released by Melanoma Cells Prepare Sentinel Lymph Nodes for Tumor MetastasisMelanoma Exosome Preparation of Lymph Nodes for Metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Chen, T.; You, Y.; Jiang, H.; Wang, Z.Z. Epithelial–mesenchymal transition (EMT): A biological process in the development, stem cell differentiation, and tumorigenesis. J. Cell. Physiol. 2017, 232, 3261–3272. [Google Scholar] [CrossRef]
- Xu, S.; Zhan, M.; Wang, J. Epithelial-to-mesenchymal transition in gallbladder cancer: From clinical evidence to cellular regulatory networks. Cell Death Discov. 2017, 3, 17069. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, R.; Luk, F.; Dalla, P.V.; Grau, G.E.R.; Bebawy, M. Breast cancer-derived microparticles display tissue selectivity in the transfer of resistance proteins to cells. PLoS ONE 2013, 8, e61515. [Google Scholar] [CrossRef] [PubMed]
- Jankovičová, J.; Sečová, P.; Michalková, K.; Antalíková, J. Tetraspanins, More than Markers of Extracellular Vesicles in Reproduction. Int. J. Mol. Sci. 2020, 21, 7568. [Google Scholar] [CrossRef] [PubMed]
- Morishita, Y.; Watanabe, M.; Nakazawa, E.; Ishibashi, K.; Kusano, E. The interaction of LFA-1 on mononuclear cells and ICAM-1 on tubular epithelial cells accelerates TGF-β1-induced renal epithelial-mesenchymal transition. PLoS ONE 2011, 6, e23267. [Google Scholar] [CrossRef] [PubMed]
- Schmalhofer, O.; Brabletz, S.; Brabletz, T. E-cadherin, β-catenin, and ZEB1 in malignant progression of cancer. Cancer Metast. Rev. 2009, 28, 151–166. [Google Scholar] [CrossRef]
- Liu, T.; Yu, J.; Ge, C.; Zhao, F.; Miao, C.; Jin, W.; Su, Y.; Geng, Q.; Chen, T.; Xie, H. B-Cell Receptor-Associated Protein 31 Promotes Metastasis via AKT/β-Catenin/Snail Pathway in Hepatocellular Carcinoma. Front. Mol. Biosci. 2021, 8, 278. [Google Scholar] [CrossRef]
- Ding, Y.; Lv, C.; Zhou, Y.; Zhang, H.; Zhao, L.; Xu, Y.; Fan, X. Vimentin loss promotes cancer proliferation through up-regulating Rictor/AKT/β-catenin signaling pathway. Exp. Cell Res. 2021, 405, 112666. [Google Scholar] [CrossRef]
- Shelke, G.V.; Lässer, C.; Gho, Y.S.; Lötvall, J. Importance of exosome depletion protocols to eliminate functional and RNA-containing extracellular vesicles from fetal bovine serum. J. Extracell. Vesicles 2014, 3, 24783. [Google Scholar] [CrossRef]
- Paudel, K.R.; Kim, D.-W. Microparticles-mediated vascular inflammation and its amelioration by antioxidant activity of baicalin. Antioxidants 2020, 9, 890. [Google Scholar] [CrossRef]
- Paudel, K.R.; Mehta, M.; Yin, G.H.S.; Yen, L.L.; Malyla, V.; Patel, V.K.; Panneerselvam, J.; Madheswaran, T.; MacLoughlin, R.; Jha, N.K. Berberine-loaded liquid crystalline nanoparticles inhibit non-small cell lung cancer proliferation and migration in vitro. Environ. Sci. Pollut. Res. 2022, 29, 46830–46847. [Google Scholar] [CrossRef]
- Wu, F.; Yin, Z.; Yang, L.; Fan, J.; Xu, J.; Jin, Y.; Yu, J.; Zhang, D.; Yang, G. Smoking Induced Extracellular Vesicles Release and Their Distinct Properties in Non-Small Cell Lung Cancer. J. Cancer 2019, 10, 3435–3443. [Google Scholar] [CrossRef]
- Jaiswal, R.; Johnson, M.S.; Pokharel, D.; Krishnan, S.R.; Bebawy, M. Microparticles shed from multidrug resistant breast cancer cells provide a parallel survival pathway through immune evasion. BMC Cancer 2017, 17, 104. [Google Scholar] [CrossRef]
- Zhou, F.Q.; Qi, Y.M.; Xu, H.; Wang, Q.Y.; Gao, X.S.; Guo, H.G. Expression of EpCAM and Wnt/β-catenin in human colon cancer. Genet. Mol. Res. 2015, 14, 4485–4494. [Google Scholar] [CrossRef]
- Singh, G.; Hossain, M.M.; Bhat, A.Q.; Ayaz, M.O.; Bano, N.; Eachkoti, R.; Dar, M.J. Identification of a cross-talk between EGFR and Wnt/beta-catenin signaling pathways in HepG2 liver cancer cells. Cell Signal 2021, 79, 109885. [Google Scholar] [CrossRef]
- Zhou, J.; Jiang, J.; Wang, S.; Xia, X. DKK1 inhibits proliferation and migration in human retinal pigment epithelial cells via the Wnt/β-catenin signaling pathway. Exp. Ther. Med. 2016, 12, 859–863. [Google Scholar] [CrossRef]
- Sant’ana, J.M.; Chammas, R.; Liu, F.T.; Nonogaki, S.; Cardoso, S.V.; Loyola, A.M.; de Faria, P.R. Activation of the Wnt/beta-catenin signaling pathway during oral carcinogenesis process is not influenced by the absence of galectin-3 in mice. Anticancer Res. 2011, 31, 2805–2811. [Google Scholar]
- Zhang, X.; Yang, M.; Shi, H.; Hu, J.; Wang, Y.; Sun, Z.; Xu, S. Reduced E-cadherin facilitates renal cell carcinoma progression by WNT/β-catenin signaling activation. Oncotarget 2017, 8, 19566–19576. [Google Scholar] [CrossRef]
- Wu, T.; Duan, X.; Hu, T.; Mu, X.; Jiang, G.; Cui, S. Effect of endostatin on Wnt pathway of stem-like cells in bladder cancer in tumor microenvironment. Mol. Biol. Rep. 2020, 47, 3937–3948. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, Y.; Sun, C.; Wang, T.; Shen, Y.; Cai, W.; Sun, J.; Chi, L.; Wang, H.; Song, N. Feedback activation of basic fibroblast growth factor signaling via the Wnt/β-catenin pathway in skin fibroblasts. Front. Pharmacol. 2017, 8, 32. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Y.; Li, M.-Y.; Ng, C.S.; Yang, S.-l.; Wang, S.; Zou, C.; Dong, Y.; Du, J.; Long, X. FOXP3 promotes tumor growth and metastasis by activating Wnt/β-catenin signaling pathway and EMT in non-small cell lung cancer. Mol. Cancer 2017, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Li, Y.; Yao, X. PGRN Suppresses Inflammation and Promotes Autophagy in Keratinocytes Through the Wnt/β-Catenin Signaling Pathway. Inflammation 2016, 39, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Jachin, S.; Bae, J.S.; Sung, J.J.; Park, H.S.; Jang, K.Y.; Chung, M.J.; Kim, D.G.; Moon, W.S. The role of nuclear EpICD in extrahepatic cholangiocarcinoma: Association with β-catenin. Int. J. Oncol. 2014, 45, 691–698. [Google Scholar] [CrossRef]
- Warneke, V.; Behrens, H.; Haag, J.; Krüger, S.; Simon, E.; Mathiak, M.; Ebert, M.; Röcken, C. Members of the EpCAM signalling pathway are expressed in gastric cancer tissue and are correlated with patient prognosis. Br. J. Cancer 2013, 109, 2217–2227. [Google Scholar] [CrossRef]
- Denzel, S.; Maetzel, D.; Mack, B.; Eggert, C.; Bärr, G.; Gires, O. Initial activation of EpCAM cleavage viacell-to-cell contact. BMC Cancer 2009, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Bi, J.; Liang, Q.; Wang, S.; Zhang, L.; Han, F.; Li, S.; Qiu, B.; Fan, X.; Chen, W.; et al. VCAM1 Promotes Tumor Cell Invasion and Metastasis by Inducing EMT and Transendothelial Migration in Colorectal Cancer. Front. Oncol. 2020, 10, 1066. [Google Scholar] [CrossRef]
- Wegwitz, F.; Lenfert, E.; Gerstel, D.; von Ehrenstein, L.; Einhoff, J.; Schmidt, G.; Logsdon, M.; Brandner, J.; Tiegs, G.; Beauchemin, N.; et al. CEACAM1 controls the EMT switch in murine mammary carcinoma in vitro and in vivo. Oncotarget 2016, 7, 63730–63746. [Google Scholar] [CrossRef]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef]
- Basu, S.; Cheriyamundath, S.; Ben-Ze’ev, A. Cell-cell adhesion: Linking Wnt/β-catenin signaling with partial EMT and stemness traits in tumorigenesis. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Wong, S.H.M.; Fang, C.M.; Chuah, L.H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018, 121, 11–22. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of β-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef]
- Le Bras, G.F.; Taubenslag, K.J.; Andl, C.D. The regulation of cell-cell adhesion during epithelial-mesenchymal transition, motility and tumor progression. Cell Adh. Migr. 2012, 6, 365–373. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malyla, V.; Paudel, K.R.; Rubis, G.D.; Hansbro, N.G.; Hansbro, P.M.; Dua, K. Extracellular Vesicles Released from Cancer Cells Promote Tumorigenesis by Inducing Epithelial to Mesenchymal Transition via β-Catenin Signaling. Int. J. Mol. Sci. 2023, 24, 3500. https://doi.org/10.3390/ijms24043500
Malyla V, Paudel KR, Rubis GD, Hansbro NG, Hansbro PM, Dua K. Extracellular Vesicles Released from Cancer Cells Promote Tumorigenesis by Inducing Epithelial to Mesenchymal Transition via β-Catenin Signaling. International Journal of Molecular Sciences. 2023; 24(4):3500. https://doi.org/10.3390/ijms24043500
Chicago/Turabian StyleMalyla, Vamshikrishna, Keshav Raj Paudel, Gabriele De Rubis, Nicole G. Hansbro, Philip M. Hansbro, and Kamal Dua. 2023. "Extracellular Vesicles Released from Cancer Cells Promote Tumorigenesis by Inducing Epithelial to Mesenchymal Transition via β-Catenin Signaling" International Journal of Molecular Sciences 24, no. 4: 3500. https://doi.org/10.3390/ijms24043500