3.1. Synthesis
General Chemical Procedures. Commercial reagents and solvents were used as received from the suppliers without further purification unless otherwise stated. The solvents employed in some reactions were dried prior to use. DMF dry was commercially available (Aldrich, St. Louis, MO, USA).
A microwave reactor EmrysTM Synthesizer (Biotage AB, Uppsala, Sweden) was used for the reactions which needed microwave irradiation (synthesis of intermediate 2).
Analytical thin-layer chromatography (TLC) was performed on aluminum plates precoated with silica gel 60 (F254, 0.20 mm). Products were visualized using an ultraviolet lamp (254 nm and 365 nm) or by heating after treatment with a 5% solution of phosphomolybdic acid (PMA) or vanillin in ethanol.
The compounds were purified by (a) high-performance flash chromatography (HPFC) with an “Isolera One” (Biotage) system in reverse phase using water/acetonitrile (100:0 to 0:100) as eluent, (b) flash column chromatography on silica gel (60 Merck 230–400 mesh), (c) preparative centrifugal circular thin layer chromatography (CCTLC) on a Chromatotron® (Kiesegel 60 PF254 gipshaltig, Merck, Rahway, NJ, USA) with layer thickness 1 mm and a flow rate of 2–4 mL/min.
For HPLC analysis, an Agilent Technologies 1120 Compact LC with a reverse-phase column ACE 5 C18-300 (4.6 mm × 150 mm, 3.5 μm) equipped with a PDA (photo diode array) detector was used. Acetonitrile was used as mobile phase A, and water with 0.05% of TFA was used as mobile phase B with at a flow rate of 1 mL·min−1. All retention times are quoted in minutes and the gradients are specified for each compound in the experimental data.
For high-resolution mass spectrometry (HRMS), an Agilent 6520 Accurate Mass QTOF (quadrupole time of flight) coupled with LC/MS and equipped with an electrospray interface (ESI) working in the positive-ion (ESI+) and negative-ion (ESI−) mode was used.
NMR spectra (1H, 13C NMR) were recorded on Varian UNIT INOVA-300 (300 MHz), Bruker AVANCE 300 (300 and 75 MHz), Varian INOVA-400 (400 and 100 MHz), Varian MERCURY-400 (400 and 100 MHz), and Varian-500 (500 and 125 MHz) spectrometers, using DMSO-d6 or CDCl3 as solvents. Chemical shift (δ) values are reported in parts per million (ppm) relative to tetramethylsilane (TMS) in 1H and CDCl3 (δ = 77.0) in 13C NMR. Coupling constant (J values) are reported in hertz (Hz), and multiplicities of signals are indicated by the following symbol: s (singlet), d (doublet), t (triplet), q (quadruplet), m (multiplet), and bs (broad singlet). Some two-dimensional spectra (COSY, HSQC, and HMBC) were performed to identify the structure.
The values of specific rotation were determined in a Perkin Elmer Polarimeter (Model 141).
The final compounds were lyophilized using a Telstar 6–80 system. Their purity was at least 95% based on HPLC, LC/MS, and 1HNMR analyses. All compounds are >95% pure by HPLC analysis.
3.1.1. General Procedure for C2 Alkylation of Trp Derivatives
To a mixture of commercially available Nα-Boc-L-tryptophan methyl ester 7 (0.50 mmol, 1.00 equiv), norbornene (1.00 mmol, 2.00 equiv), K2CO3 (3.00 mmol, 4.00 equiv), and PdCl2 (10 mol%), 2.5 mL of DMF (containing 0.5 M H2O) and the corresponding primary alkyl bromide (2.00 mmol, 4.00 equiv) were added. The resulting suspension was stirred at 60 °C for 24 h. After being cooled to room temperature, the reaction mixture was concentrated under reduce pressure and then diluted with EtOAc (20 mL) and washed with water (3 × 20 mL). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated. The crude product was purified by flash column chromatography on silica gel or CCTLC to afford the corresponding 2-alkyltryptophan intermediate.
3.1.2. General Coupling Procedure for the Synthesis of OMe-Protected Tetrapodal Trp Derivatives
To a solution of the tetrapodal polyacid
4 [
27] (1.00 equiv), in anhydrous DMF (10 mL), HATU (1.20 equiv each carboxylic acid group), the corresponding OMe protected C-2 alkyl tryptophan derivative (1.20 equiv each carboxylic acid group), and DIPEA (2.40 equiv each carboxylic acid group) were added. The reaction mixture was stirred under argon atmosphere at 30 °C for 12 h and then evaporated to dryness. The residue was dissolved in ethyl acetate (20 mL) and washed successively with aqueous solutions of citric acid (10%) (3 × 20 mL), saturated NaHCO
3 (3 × 20 mL), and brine (3 × 20 mL). The organic phase was dried over anhydrous Na
2SO
4, filtered, and evaporated to dryness. The crude product was purified by Biotage HPFC (high-performance flash chromatography) purification system on reverse phase using water/acetonitrile (100:0 to 0:100) as eluent to afford the corresponding products.
3.1.3. General Procedure for Methyl Ester Deprotection
To a solution containing the corresponding methyl ester derivative (1.00 equiv) in THF (10 mL) at 0 °C (ice bath), a solution of LiOH∙H2O (2.00 equiv for each methyl ester group) in water (2 mL) was added, and the mixture was stirred at 30 °C overnight. Then 1 M hydrochloric acid aqueous solution was added to reach pH~2, and volatiles were evaporated to dryness. The residue was dissolved in iso-butanol (15 mL) and washed with brine (3 × 10 mL) and water (3 × 10 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and evaporated to dryness. The residue was purified with a Biotage HPFC (high-performance flash chromatography) purification system on reverse phase using water/acetonitrile (100:0 to 0:100) as eluent, frozen and lyophilized, yielding the product as a fluffy powder.
3.1.4. Dimethyl (R)-5-[3-(2-{[(Benzyloxy)carbonyl]amino}-3-methoxy-3-oxopropyl)-1H-indol-2-yl]isophthalate 2
Commercially available Nα-benzyloxycarbonyl-d-tryptophan methyl ester 1 (680 mg, 1.93 mmol, 1.00 equiv), dimethyl 5-iodoisophthalate (927 mg, 2.90 mmol, 1.50 equiv), Pd(OAc)2 (22 mg, 0.10 mmol, 5 mol%), AgBF4 (751 mg, 3.86 mmol, 2.00 equiv), and TFA (148 μL, 1.93 mmol, 1.00 equiv) were placed in an MW reactor vessel under argon atmosphere in anhydrous DMF (12 mL). The mixture was heated under MW irradiation (250 W) at 120 °C for 30 min. The resulting suspension was filtered through Whatman® filter paper 42, and the solvent was removed under vacuum. The residue was dissolved in ethyl acetate (20 mL) and washed successively with saturated NaHCO3 (3 × 20 mL) and brine (3 × 20 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and evaporated to dryness. The residue was purified by flash column chromatography using hexane:ethyl acetate (7:3) to afford 2 (575 mg, 55%) as an amorphous solid of cream color. 1H NMR (300 MHz, CDCl3) δ: 8.59 (p, J = 1.6 Hz, 1H, NH-1iTrp), 8.34 (d, J = 7.5 Hz, 3H, Ar), 7.59 (d, J = 7.9 Hz, 1H, Ar), 7.38–7.18 (m, 5H, Ar), 7.10 (t, J = 7.3 Hz, 1H, Ar), 5.16 (d, J = 8.3 Hz, 1H, NHCO), 4.97–4.76 (m, CH2Ph), 4.70–4.55 (m, 1H, α-CHTrp), 3.91 (s, 6H, OCH3), 3.46 (m, 2H, β-CH2Trp), 3.36 (s, 3H, OCH3). HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 9.327 min.
3.1.5. Dimethyl (R)-5-[3-(2-Amino-3-methoxy-3-oxopropyl)-1H-indol-2-yl]isophthalate 3
To a solution of compound 2 (570 mg, 1.05 mmol, 1.00 equiv) in DMF (10 mL), ammonium formate (219 mg, 3.45 mmol, 3.30 equiv) and Pd/C (10% on C; 30 wt %) were added. After 3 h under argon atmosphere, the residue was filtered through a Whatman® filter paper 42 and the solvent was removed under reduced pressure to give 356 mg (83%) of 3 as an amorphous white solid. The crude was used in the next step without purification. HPLC (gradient: H2O:MeCN, 10 min: 6.277 min.
3.1.6. Tetramer 5
According to the general coupling procedure, compound
3 (340 mg, 0.83 mmol, 4.80 equiv), was added to a mixture of
4 [
27] (73 mg, 0.17 mmol, 1.00 equiv), HATU (315 mg, 0.83 mmol, 4.80 equiv), and DIPEA (281 µL, 1.73 mmol, 10.00 equiv). Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water:acetonitrile (100:0 to 0:100) yielded
5 (254 mg, 74%).
1H NMR (400 MHz, DMSO-
d6)
δ: 11.52 (s, 3H, NH-1
iTrp), 8.44 (t,
J = 1.6 Hz, 3H, Ar), 8.39 (d,
J = 1.7 Hz, 8H, Ar), 8.32 (d,
J = 7.7 Hz, 4H, NHCO), 7.59 (d,
J = 7.5 Hz, 4H, Ar), 7.37 (d,
J = 7.4 Hz, 4H, Ar), 7.13 (t,
J = 7.5 Hz, 4H, Ar), 7.01 (t,
J = 7.4 Hz, 4H, Ar), 4.62–4.48 (m, 4H,
α-CHTrp), 3.90 (s, 24H, OCH
3), 3.38–3.29 (m, 26H, OCH
3 +
β-CH
2Trp + CH
2), 3.20 (dd,
J = 14.5, 6.6 Hz, 4H,
β-CH
2Trp), 3.06 (m, 8H, CH
2), 2.20 (m, 4H, CH
2), 2.16–2.02 (m, 4H, CH
2). HPLC (gradient: H
2O:MeCN, 50–100% of MeCN in 10 min): 7.682 min.
3.1.7. Tetramer 6
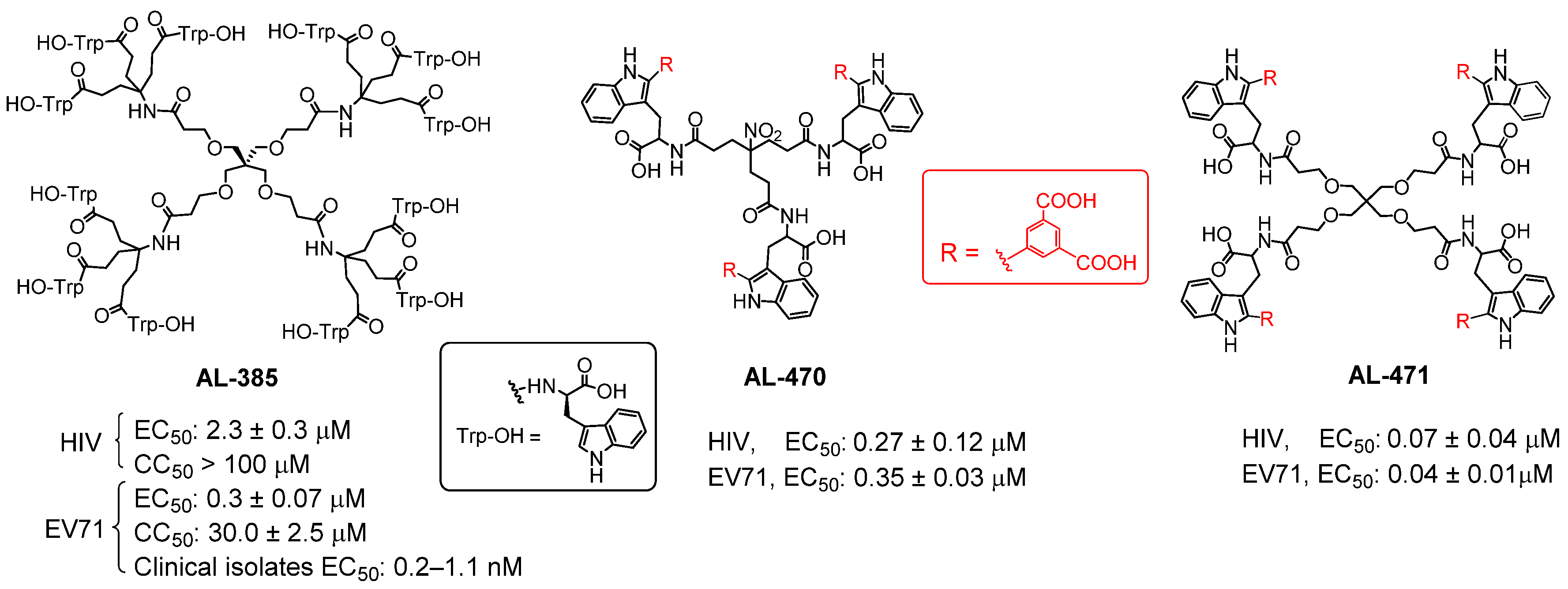
According to the general procedure for deprotection of methyl esters, compound 5 (100 mg, 0.05 mmol, 1.00 equiv) was treated with LiOH·H2O (51 mg, 1.20 mmol, 24.00 equiv). Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water/acetonitrile (100:0 to 0:100) yielded 6 (75 mg, 82%). 1H NMR (500 MHz, DMSO-d6) δ: 11.43 (s, 4H, NH-1iTrp), 8.45 (t, J = 1.6 Hz, 4H, Ar), 8.41–8.32 (m, 8H, Ar), 8.18 (d, J = 8.0 Hz, 4H, NHCO), 7.66 (d, J = 8.0 Hz, 4H, Ar), 7.35 (d, J = 8.1 Hz, 4H, Ar), 7.11 (t, J = 8.1, 4H, Ar), 7.03–6.96 (m, 4H, Ar), 4.66–4.33 (m, 4H, α-CHTrp), 3.40–3.33 (m, 4H, β-CH2Trp), 3.29 (m, 8H, CH2), 3.17 (dd, J = 14.3, 6.9 Hz, 4H, β-CH2Trp), 3.08 (s, 8H, CH2), 2.21 (dt, J = 13.9, 6.7 Hz, 4H, CH2), 2.11 (dq, J = 16.1, 9.5, 8.0 Hz, 4H, CH2). 13C NMR (126 MHz, DMSO-d6) δ: 173.0, 169.9, 166.6, 136.2, 133.7, 133.6, 132.6, 132.0, 128.6, 128.6, 122.0, 119.2, 119.0, 111.3, 108.5, 68.8, 67.0, 52.8, 44.8, 35.6, 27.2. HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 6.814 min. [α]D = −15.50° (c1, MeOH). The specific rotation observed for 6 is opposite in sign to those found for the prototype AL-471, [α]D = +16.10° (c1, MeOH), confirming that both compounds are enantiomers.
3.1.8. Methyl 2-[(tert-Butoxycarbonyl)amino]-3-{2-[3-(1,3-dioxoisoindolin-2-yl)propyl]-1H-indol-3-yl}propanoate 8
According to the general procedure for C2 alkylation, commercially available Nα-Boc-(S)-tryptophan methyl ester 7 (800 mg, 2.51 mmol, 1.00 equiv) was treated with norbornene (473 mg, 5.03 mmol, 2.00 equiv), K2CO3 (1.78 g, 10.05 mmol, 4.00 equiv), PdCl2 (35 mg, 0.25 mmol, 10 mol%), and N-(3-bromopropyl)phthalimide (2.70 g, 10.05 mmol, 4.00 equiv). Purification by flash column chromatography using hexane:ethyl acetate (6:4) yielded 8 (744 mg, 60%) as a pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ: 9.09 (s, 1H, NH-1iTrp), 7.83 (m, 2H, Ar), 7.71 (m, 2H, Ar), 7.43 (d, J = 7.9 Hz, 1H, Ar), 7.34 (d, J = 7.9 Hz, 1H, Ar), 7.10 (t, J = 7.5 Hz, 1H, Ar), 7.04 (t, J = 7.5 Hz, 1H, Ar), 5.06 (d, J = 8.5 Hz, 1H, NHCO), 4.58 (m, 1H, α-CHTrp), 3.78 (t, J = 6.3 Hz, 2H, CH2), 3.62 (s, 3H, OCH3), 3.21 (m, 2H, β-CH2Trp), 2.74 (t, J = 8.0 Hz, 2H, CH2), 2.04 (m, 2H, CH2), 1.39 (s, 9H, CH3). 13C NMR (101 MHz, CDCl3) δ: 173.0, 169.1, 155.2, 136.0, 135.5, 134.3, 132.0, 128.8, 123.5, 121.5, 119.5, 118.3, 110.8, 105.9, 79.9, 54.3, 52.4, 37.3, 28.7, 28.4, 27.5, 22.6. HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 9.472 min.
3.1.9. Methyl 2-[(tert-Butoxycarbonyl)amino[-3-{2-[5-(1,3-dioxoisoindolin-2-yl)pentyl]-1H-indol-3-yl}propanoate 9
According to the general procedure for C2 alkylation, Nα-Boc-(S)-tryptophan methyl ester 7 (800 mg, 2.51 mmol, 1.00 equiv) was treated with norbornene (473 mg, 5.03 mmol, 2.00 equiv), K2CO3 (1.78 mg, 10.05 mmol, 4.00 equiv), PdCl2 (35 mg, 0.25 mmol, 10 mol%), and N-(5-bromopentyl)phthalimide (3.00 g, 10.05 mmol, 4.00 equiv). Purification by flash column chromatography using hexane:ethyl acetate (6:4) yielded 9 (1.19 g, 85%) as a pale-yellow oil. 1H NMR (300 MHz, CDCl3) δ: 8.13 (s, 1H, NH-1iTrp), 7.82 (m, 2H, Ar), 7.70 (m, 2H, Ar), 7.43 (d, J = 7.5 Hz, 1H, Ar), 7.29 (d, J = 7.5 Hz, 1H, Ar), 7.08 (m, 2H, Ar), 5.06 (d, J = 8.3 Hz, 1H, NHCO), 4.56 (m, 1H, α-CHTrp), 3.70 (m, 2H, NCH2), 3.61 (s, 3H, OCH3), 3.20 (d, J = 5.8 Hz, 2H, β-CH2Trp), 2.72 (t, J = 7.6 Hz, 2H, CH2), 1.74 (m, 4H, CH2), 1.41 (m, 11H, CH2 + CH3). 13C NMR (75 MHz, CDCl3) δ: 173.0, 168.7, 155.2, 137.0, 135.5, 134.1, 132.2, 128.8, 123.4, 119.5, 118.3, 110.5, 105.8, 79.8, 54.3, 52.4, 37.5, 28.7, 28.4, 28.3, 27.4, 26.3, 25.8. HPLC (gradient: H2O:MeCN, 40–100% of MeCN in 10 min): 8.287 min. HRMS (ESI+) m/z: calculated for C30H35N3O6 533.25259; found 533.25164.
3.1.10. Methyl 2-[(tert-Butoxycarbonyl)amino]-3-{2-[8-(1,3-dioxoisoindolin-2-yl)octyl]-1H-indol-3-yl}propanoate 10
According to the general procedure for C2 alkylation, Nα-Boc-(S)-tryptophan methyl ester 7 (500 mg, 1.57 mmol, 1.00 equiv) was treated with norbornene (296 mg, 3.14 mmol, 2.00 equiv), K2CO3 (1.11 g, 6.28 mmol, 4.00 equiv), PdCl2 (22 mg, 0.16 mmol, 10 mol%), and N-(8-bromooctyl)phthalimide (2.12 g, 6.28 mmol, 4.00 equiv). Purification by flash column chromatography using hexane:ethyl acetate (6:4) yielded 10 (723 mg, 80%) as a pale-yellow oil. 1H NMR (300 MHz, CDCl3) δ: 8.14 (s, 1H, NH-1iTrp), 7.83 (m, 2H, Ar), 7.70 (m, 2H, Ar), 7.45 (d, J = 7.5 Hz, 1H, Ar), 7.26 (m, 1H, Ar), 7.08 (m, 2H, Ar), 5.05 (d, J = 8.3 Hz, 1H, NHCO), 4.58 (m, 1H, α-CHTrp), 3.68 (m, 2H, CH2), 3.63 (s, 3H, OCH3), 3.21 (d, J = 5.7 Hz, 2H, β-CH2Trp), 2.68 (m, 2H, CH2), 1.65 (m, 4H, CH2), 1.40 (s, 9H, CH3), 1.33 (s, 8H, CH2). 13C NMR (75 MHz, CDCl3) δ: 173.0, 168.7, 155.2, 137.6, 135.4, 134.0, 134.0, 132.3, 128.8, 123.3, 121.3, 119.5, 118.3, 110.4, 105.5, 79.8, 54.3, 52.3, 38.0, 29.6, 29.3, 29.2, 28.8, 28.6, 28.4, 27.4, 26.7, 26.1. HPLC (gradient: H2O:MeCN, 40–100% of MeCN in 10 min): 9.640 min. HRMS (ESI+) m/z: calculated for C33H41N3O6 575.29954; found 575.30022.
3.1.11. Dimethyl 5-{[3-(3-{2-[(tert-Butoxycarbonyl)amino]-3-methoxy-3-oxopropyl}-1H-indol-2-yl)propyl]amino}isophthalate 14
To a solution of compound 8 (24 mg, 0.11 mmol, 1.00 equiv) in EtOH (5 mL), hydrazine monohydrate (64%) (14 µL, 0.21 mmol, 2.00 equiv) was added. The resulting solution was stirred at 30 °C for 5 h. After being cooled to room temperature, the reaction mixture was concentrated under reduced pressure and then diluted with H2O (10 mL) and extracted with dichloromethane (3 × 15 mL). The combined organic extracts were washed with brine (3 × 15 mL), dried over anhydrous Na2SO4, and concentrated. The crude product 11 (30 mg, 0.08 mmol, 1.00 equiv) was dissolved under argon atmosphere in anhydrous DMF (5 mL), and dimethyl 5-isocyanatoisophthalate (23 mg, 0.10 mmol, 1.20 equiv) and DIPEA (16 µL, 0.10 mmol, 1.20 equiv) were added. The reaction mixture was stirred at 30 °C for 4 h and the crude product was purified by flash column chromatography on silica gel using hexane:ethyl acetate (3:7) to afford 14 (36 mg, 74%) as a pale-yellow oil. 1H NMR (400 MHz, CDCl3) δ: 9.07 (s, 1H, NH-1iTrp), 8.25 (t, J = 1.5 Hz, 1H, Ar), 8.18 (s, 2H, Ar), 7.66 (s, 1H), 7.41 (d, J = 6.2 Hz, 1H, Ar), 7.23 (m, 1H, Ar), 7.04 (m, 2H, Ar), 5.36 (d, J = 8.3 Hz, 1H, NHCO), 4.44 (m, 1H, α-CHTrp), 3.88 (s, 7H, CH3 + CH2), 3.68 (m, 1H, CH2) 3.57 (m, 3H, OCH3), 3.41 (m, 1H, β-CH2Trp), 3.22 (m, 3H, β-CH2Trp + CH2), 2.81 (m, 2H, CH2), 1.92 (m, 2H, CH2), 1.38 (s, 9H, CH3). 13C NMR (101 MHz, CDCl3) δ: 173.0, 166.4, 156.2, 155.8, 140.1, 136.5, 135.6, 131.1, 128.7, 124.5, 124.2, 121.3, 119.4, 117.8, 110.8, 105.7, 55.3, 52.6, 52.5, 39.2, 29.6, 28.4, 23.0. HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 9.305 min.
3.1.12. Dimethyl 5-{3-[5-(3-{2-[(tert-Butoxycarbonyl)amino]-3-methoxy-3-oxopropyl}-1H-indol-2-yl)pentyl]ureido}isophthalate 15
To a solution of compound 9 (463 mg, 2.13 mmol, 1.00 equiv) in EtOH (5 mL), hydrazine monohydrate (64%) (158 µL, 2.09 mmol, 2.50 equiv) was added. The resulting solution was stirred at 30 °C for 5 h. After being cooled to room temperature, the reaction mixture was concentrated under reduced pressure and then diluted with H2O (10 mL) and extracted with dichloromethane (3 × 15 mL). The combined organic extracts were washed with brine (3 × 15 mL), dried over anhydrous Na2SO4, and concentrated. The crude product 12 (230 mg, 0.57 mmol, 1.00 equiv) was dissolved in anhydrous DMF (5 mL), and dimethyl 5-isocyanatoisophthalate (161 mg, 0.68 mmol, 1.20 equiv) and DIPEA (111 µL, 0.68 mmol, 1.20 equiv) were added. The reaction was stirred at 30 °C for 4 h and the crude product was purified by flash column chromatography on silica gel using hexane:ethyl acetate (6:4) to afford 15 (151 mg, 40%) as a pale-yellow oil. 1H NMR (300 MHz, CDCl3) δ: 8.75 (s, 1H, NH-1iTrp), 8.22 (s, 1H, Ar), 8.20 (s, 2H, Ar), 7.86 (m, 1H, NHCO), 7.40 (d, J = 7.2 Hz, 1H, Ar), 7.26 (m, 1H, Ar), 7.02 (m, 2H, Ar), 5.79 (s, 1H, NHCO), 5.20 (d, J = 8.1 Hz, 1H, NHCO), 4.50 (m, 1H, α-CHTrp), 3.82 (s, 8H, OCH3 + CH2), 3.57 (s, 3H, OCH3), 3.17 (m, 4H, β-CH2Trp + CH2), 2.63 (m, 2H, CH2), 1.62 (m, 2H, CH2), 1.36 (s, 11H, CH3 + CH2). 13C NMR (75 MHz, CDCl3) δ: 173.1, 166.3, 156.0, 155.6, 140.3, 137.3, 135.5, 131.1, 128.6, 124.3, 124.0, 121.1, 119.3, 117.9, 110.7, 105.2, 80.2, 54.8, 52.4, 39.7, 29.0, 28.4, 28.1, 26.5, 25.9. HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 9.343 min.
3.1.13. Dimethyl 5-{3-[8-(3-{2-[(tert-Butoxycarbonyl)amino]-3-methoxy-3-oxopropyl}-1H-indol-2-yl)octyl]ureido}isophthalate 16
To a solution of compound 10 (700 mg, 1.22 mmol, 1.00 equiv) in EtOH (5 mL), hydrazine monohydrate (64%) (178 µL, 3.65 mmol, 3.00 equiv) was added. The resulting solution was stirred at 30 °C for 5 h. After being cooled to room temperature, the reaction mixture was concentrated under reduced pressure and then diluted with H2O (10 mL) and extracted with dichloromethane (3 × 15 mL). The organic layers were washed with brine (3 × 15 mL), dried over anhydrous Na2SO4, and concentrated. The crude product 13 (500 mg, 1.12 mmol, 1.00 equiv) was dissolved in anhydrous DMF (5 mL), and dimethyl 5-isocyanatoisophthalate (317 mg, 1.35 mmol, 1.20 equiv) and DIPEA (219 µL, 1.35 mmol, 1.20 equiv) were added. The reaction was stirred at 30 °C for 4 h and the crude product was purified by flash column chromatography on silica gel using hexane:ethyl acetate (5:5) to afford 16 (621 mg, 81%) as a pale-yellow oil. 1H NMR (300 MHz, CDCl3) δ: 8.50 (s, 1H, NH-1iTrp), 8.27 (t, J = 1.5 Hz, 1H Ar), 8.24 (d, J = 1.5 Hz, 2H, Ar), 7.43 (d, J = 7.0 Hz, 1H, Ar), 7.28 (m, 1H, Ar), 7.06 (m, 2H, Ar), 5.70 (bs, 1H, NHCO), 5.15 (d, J = 8.0 Hz, 1H, NHCO), 4.52 (m, 1H, α-CHTrp), 3.84 (s, 6H, OCH3), 3.64 (s, 3H, OCH3), 3.20 (m, 4H, β-CH2Trp + CH2), 2.66 (t, J = 7.7 Hz, 2H, CH2), 1.54 (m, 2H, CH2), 1.38 (s, 8H, CH2), 1.21 (m, 11H, CH3 + CH2). 13C NMR (75 MHz, CDCl3) δ: 173.2, 166.5, 156.0, 155.6, 140.4, 137.7, 135.6, 131.2, 128.7, 124.4, 123.9, 121.3, 119.4, 118.0, 110.7, 105.2, 80.2, 54.7, 52.5, 52.5, 40.1, 29.9, 29.6, 29.1, 28.9, 28.4, 28.1, 27.4, 26.7, 26.0. HPLC (gradient: H2O:MeCN, 50–100% of MeCN in 10 min): 6.439 min.
3.1.14. Tetramer 20
To a cold (0 °C) solution of compound
14 (150 mg, 0.25 mmol) in dichloromethane (8 mL), TFA (0.4 mL) was added. The mixture was stirred at room temperature for 5 h, then volatiles were evaporated to dryness and the residue was coevaporated successively with methanol and dichloromethane. The residue (compound
17) (100 mg, 0.20 mmol, 4.80 equiv) was next added to a mixture of
4 [27] (18 mg, 0.04 mmol, 1.00 equiv), HATU (77 mg, 0.20 mmol, 4.80 equiv), and DIPEA (68 µL, 0.42 mmol, 10.00 equiv). The mixture was treated as described in the general coupling procedure. Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water:acetonitrile (100:0 to 0:100) yielded
20 (21 mg, 20%).
1H NMR (500 MHz, DMSO-
d6)
δ: 10.78 (s, 4H, NH-1
iTrp), 9.08 (s, 4H, NHCO), 8.34 (d,
J = 7.8 Hz, 4H, Ar), 8.28 (m, 8H, Ar), 8.00 (t,
J = 1.6 Hz, 4H, Ar), 7.39 (d,
J = 7.8 Hz, 4H, Ar), 7.22 (d,
J = 7.8 Hz, 4H, Ar), 6.96 (t,
J = 7.5 Hz, 4H, Ar), 6.90 (t,
J = 7.5 Hz, 4H, Ar), 6.38 (t,
J = 5.9 Hz, 4H, NHCO), 4.50 (m, 4H, α-CHTrp), 3.85 (s, 24H, OCH
3), 3.46 (m, 20H, OCH
3 + CH
2), 3.10 (m, 20H,
β-CH
2Trp + CH
2), 2.97 (dd,
J = 14.4, 6.9 Hz, 4H,
β-CH
2Trp), 2.68 (m, 8H, CH
2), 2.33 (m, 4H, CH
2), 2.23 (m, 4H, CH
2), 1.80 (quint,
J = 7.6 Hz, 8H, CH
2).
13C NMR (126 MHz, DMSO-
d6)
δ: 172.4, 170.3, 165.5, 155.0, 141.7, 136.8, 135.4, 130.5, 128.0, 122.1, 121.8, 120.2, 118.2, 117.4, 110.6, 105.2, 68.8, 66.9, 53.3, 52.4, 51.7, 35.6, 30.0, 26.7, 22.9. HPLC (gradient: H
2O:MeCN, 10–100% of MeCN in 10 min): 10.357 min.
3.1.15. Tetramer 21
To a cold (0 °C) solution of compound
15 (130 mg, 0.20 mmol) in dichloromethane (8 mL), TFA (0.4 mL) was added. The mixture was stirred at room temperature for 5 h, then volatiles were evaporated to dryness and the residue was coevaporated successively with methanol and dichloromethane. The residue (compound
18) (110 mg, 0.17 mmol, 4.80 equiv) was next added to a mixture of
4 [27] (15 mg, 0.04 mmol, 1.00 equiv), HATU (64 mg, 0.17 mmol, 4.80 equiv), and DIPEA (57 µL, 0.35 mmol, 10.00 equiv). The mixture was treated as described in the general coupling procedure. Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water:acetonitrile (100:0 to 0:100) yielded
21 (64 mg, 73%).
1H NMR (300 MHz, MeOD-
d4)
δ: 10.16 (s, 4H, NH-1
iTrp), 8.22 (d,
J = 1.5 Hz, 8H, Ar), 8.14 (t,
J = 1.5 Hz, 4H, Ar), 7.38 (d,
J = 6.9 Hz, 4H, Ar), 7.19 (d,
J = 6.9 Hz, 4H, Ar), 6.93 (m, 8H, Ar), 6.16 (t,
J = 5.7 Hz, 4H, NHCO), 4.70 (m, 4H,
α-CHTrp), 3.87 (s, 24H, OCH
3), 3.52 (m, 12H, OCH
3), 3.32 (m, 16H, CH
2), 3.18 (m, 12H,
β-CH
2Trp + CH
2) 3.02 (m, 12H, CH
2), 2.70 (qd,
J = 7.6, 4.1 Hz, 8H, CH
2), 2.31 (quint,
J = 5.6 Hz, 8H, CH
2), 1.71 (m, 8H, CH
2), 1.53 (m, 8H, CH
2), 1.39 (m, 8H, CH
2).
13C NMR (126 MHz, CDCl
3)
δ: 166.4, 156.6, 140.4, 137.8, 135.9, 131.2, 128.2, 124.4, 124.1, 121.0, 119.1, 117.6, 111.2, 104.6, 52.7, 52.5, 44.4, 40.0, 38.1, 29.7, 29.5, 27.0, 26.2, 7.8. HPLC (gradient: H
2O:MeCN, 60–100% of MeCN in 10 min): 5.615 min. HRMS (ESI
+)
m/z: calculated for C
128H
151N
16O
36 2488.04769; found 2488.04916.
3.1.16. Tetramer 22
To a cold (0 °C) solution of compound
16 (400 mg, 0.59 mmol) in dichloromethane (10 mL), TFA (0.5 mL) was added. The mixture was stirred at room temperature for 5 h, then volatiles were evaporated to dryness and the residue was coevaporated successively with methanol and dichloromethane. The residue (compound
19) (350 mg, 0.50 mmol, 4.80 equiv), was next added to a mixture of
4 [27] (44 mg, 0.10 mmol, 1.00 equiv), HATU (191 mg, 0.50 mmol, 4.80 equiv), and DIPEA (126 µL, 1.05 mmol, 10.00 equiv). The mixture was treated as described in the general coupling procedure. Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water:acetonitrile (100:0 to 0:100) yielded
22 (219 mg, 78%).
1H NMR (500 MHz, DMSO-
d6)
δ: 10.69 (s, 4H, NHCO), 8.97 (s, 4H, NH-1
iTrp), 8.31 (d,
J = 7.7 Hz, 4H, NHCO), 8.27 (d,
J = 1.6 Hz, 8H, Ar), 8.00 (t,
J = 1.6 Hz, 4H, Ar), 7.38 (d,
J = 7.8 Hz, 4H, Ar), 7.20 (d,
J = 7.8 Hz, 4H, Ar), 6.95 (t,
J = 7.3 Hz, 4H, Ar), 6.89 (t,
J = 7.3 Hz, 4H, Ar), 6.21 (t,
J = 5.6 Hz, 4H, NHCO), 4.50 (m, 4H,
α-CHTrp), 3.86 (s, 24H, OCH
3), 3.46 (s, 12H, OCH
3), 3.41 (m, 4H, CH
2), 3.12 (m, 8H, CH
2), 3.07 (m, 12H,
β-CH
2Trp + CH
2), 2.95 (dd,
J = 14.3, 6.8 Hz, 4H,
β-CH
2Trp), 2.63 (m, 8H, CH
2), 2.31 (m, 4H, CH
2), 2.23 (m, 4H, CH
2), 1.59 (m, 8H, CH
2), 1.41 (m, 8H, CH
2), 1.27 (m, 36H, CH
2).
13C NMR (126 MHz, DMSO-
d6)
δ: 172.4, 170.2, 165.5, 154.9, 141.7, 137.4, 135.3, 130.5, 128.0, 122.0, 121.8, 120.0, 118.1, 117.3, 110.5, 105.0, 68.7, 66.9, 53.3, 52.4, 51.6, 44.9, 35.6, 29.7, 29.3, 28.9, 28.9 28.8, 26.8, 26.4, 25.3. HPLC (gradient: H
2O:MeCN, 10–100% of MeCN in 10 min): 11.256 min. HRMS (ESI
+): calculated for C
140H
175N
16O
36 2656.23549; found 2656.24812.
3.1.17. Tetramer 23
According to the general procedure for deprotection of methyl esters, compound 20 (20 mg, 0.01 mmol, 1.00 equiv) was treated with LiOH·H2O (9 mg, 0.20 mmol, 24.00 equiv). Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water:acetonitrile (100:0 to 0:100) yielded 23 (15 mg, 79%). 1H NMR (500 MHz, DMSO-d6) δ: 10.72 (s, 4H, NH-1iTrp), 8.29 (s, 8H, Ar), 8.15 (s, 4H, NHCO), 8.01 (s, 4H, Ar), 7.51 (d, J = 7.7 Hz, 4H, Ar), 7.18 (d, J = 7.7 Hz, 4H, Ar), 6.93 (t, J = 7.5 Hz, 4H, Ar), 6.88 (t, J = 7.5 Hz, 4H, Ar), 4.48 (m, 4H, α-CHTrp), 3.32 (m, 8H, CH2), 3.13 (m, 12H, β-CH2Trp + CH2), 3.03 (s, 8H, CH2), 2.95 (m, 4H, β-CH2Trp), 2.73 (t, J = 7.4 Hz, 8H, CH2), 2.30 (dt, J = 14.2, 6.5 Hz, 4H, CH2), 2.17 (dt, J = 14.2, 6.5 Hz, 4H, CH2), 1.79 (m, 8H, CH2). 13C NMR (126 MHz, DMSO-d6) δ: 170.0, 167.3, 155.5, 141.6, 136.7, 135.3, 128.4, 122.2, 121.8, 119.9, 118.1, 117.9, 110.4, 106.4, 68.7, 66.8, 64.9, 54.2, 44.7, 35.9, 29.8, 22.9, 15.2. HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 6.997 min. HRMS (ESI+) m/z: calculated for C109H116N16O36 2224.77381; found 2224.76703.
3.1.18. Tetramer 24
According to the general procedure for deprotection of methyl esters, compound 21 (63 mg, 0.02 mmol, 1.00 equiv) was treated with LiOH·H2O (25 mg, 0.60 mmol, 24.00 equiv). Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water:acetonitrile (100:0 to 0:100) yielded 24 (20 mg, 34%). 1H NMR (500 MHz, DMSO-d6) δ: 10.65 (s, 4H, NHCO), 9.33 (s, 4H, NH-1iTrp), 8.23 (s, 8H, Ar), 8.11 (d, J = 8.2 Hz, 4H, NHCO), 8.00 (s, 4H, Ar), 7.46 (d, J = 7.8 Hz, 4H, Ar), 7.18 (d, J = 7.8 Hz, 4H, Ar), 6.92 (t, J = 7.4 Hz, 4H, Ar), 6.87 (t, J = 7.4 Hz, 4H, Ar), 6.72 (m, 4H, NHCO), 4.47 (m, 4H, α-CHTrp), 3.32 (m, 8H, CH2), 3.09 (m, 20H, β-CH2Trp + CH2), 2.94 (dd, J = 14.3, 6.9 Hz, 4H, β-CH2Trp), 2.66 (m, 8H, CH2), 2.28 (m, 4H, CH2), 2.20 (m, 4H, CH2), 1.63 (quint, J = 7.3 Hz, 8H), 1.47 (quint, J = 7.3 Hz, 8H, CH2), 1.33 (quint, J = 7.3, 8H, CH2). 13C NMR (126 MHz, DMSO-d6) δ: 174.0, 170.0, 167.2, 155.2, 141.4, 137.4, 135.3, 132.1, 128.4, 122.3, 121.9, 119.9, 118.0, 117.8, 110.4, 105.9, 68.8, 66.9, 53.8, 44.7, 40.4, 35.8, 29.4, 29.1, 27.1, 26.4, 25.4. HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 7.043 min.
3.1.19. Tetramer 25
According to the general procedure for deprotection of methyl esters, compound 22 (117 mg, 0.04 mmol, 1.00 equiv) was treated with LiOH·H2O (44 mg, 1.06 mmol, 24.00 equiv). Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water/acetonitrile (100:0 to 0:100) yielded 25 (88 mg, 80%). 1H NMR (500 MHz, DMSO-d6) δ: 8.99 (s, 4H, NH-1iTrp), 8.22 (s, 8H, Ar), 8.13 (d, J = 8.1 Hz, 4H, NHCO), 8.01 (s, 4H, Ar), 7.45 (d, J = 7.7 Hz, 4H, Ar), 7.19 (d, J = 7.9 Hz, 4H, Ar), 6.94 (t, J = 7.5 Hz, 4H, Ar), 6.88 (t, J = 7.5 Hz, 4H, Ar), 6.33 (s, 4H, NHCO), 4.46 (m, 4H, α-CHTrp), 3.38 (m, 8H), 3.08 (s, 16H, β-CH2Trp + CH2), 2.93 (s, 4H, β-CH2Trp), 2.65 (m, 8H, CH2), 2.29 (m, 4H, CH2), 2.21 (m, 4H, CH2), 1.61 (m, 8H, CH2), 1.41 (m, 8H, CH2), 1.27 (m, 36H, CH2). 13C NMR (126 MHz, DMSO-d6) δ: 173.6, 170.1, 166.9, 155.0, 141.3, 137.5, 135.3, 131.8, 128.3, 122.4, 122.0, 119.9, 118.1, 117.7, 110.4, 105.6, 68.8, 66.9, 53.5, 44.8, 35.8, 29.7, 29.4, 28.9, 28.8, 26.5, 25.5. HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 7.876 min. HRMS (ESI-) m/z: calculated for C129H156N16O36 2505.08682; found 2505.08471.
3.1.20. Methyl 4-(3-{2-[(tert-Butoxycarbonyl)amino]-3-methoxy-3-oxopropyl}-1H-indol-2-yl)butanoate 26
According to the general C2 alkylation procedure, Nα-Boc-(S)-tryptophan methyl ester 7 (100 mg, 0.31 mmol, 1.00 equiv) was treated with norbornene (59 mg, 0.63 mmol, 2.00 equiv), K2CO3 (177 mg, 1.26 mmol, 4.00 equiv), PdCl2 (6 mg, 0.03 mmol, 10 mol%), and methyl 4-bromobutyrate (227 mg, 1.26 mmol, 4.00 equiv). Purification by CCTLC using dichloromethane:methanol (20:1) yielded 26 (126 mg, 96%) as a pale-yellow oil. 1H NMR (300 MHz, Acetone-d6) δ: 10.05 (s, 1H, NH-1iTrp), 7.49 (d, J = 7.9 Hz, 1H, Ar), 7.28 (d, J = 7.3 Hz, 1H, Ar), 7.00 (m, 2H, Ar), 6.00 (d, J = 8.5 Hz, 1H, NHCO), 4.43 (m, J = 7.3 Hz, 1H, α-CHTrp), 3.61 (m, 6H, OCH3), 3.23 (dd, J = 14.6, 7.1 Hz, 1H, β-CH2Trp), 3.12 (dd, J = 14.6, 7.1 Hz, 1H, β-CH2Trp), 2.83 (t, J = 7.6 Hz, 2H, CH2), 2.38 (t, J = 7.6 Hz, 2H, CH2), 1.95 (m, 2H, CH2), 1.34 (s, 9H, CH3). 13C NMR (101 MHz, Acetone-d6) δ: 173.9, 173.6, 156.0, 137.3, 136.8, 129.5, 121.5, 119.5, 118.7, 111.4, 107.1, 79.3, 55.5, 52.2, 51.6, 44.4, 35.6, 33.8, 28.5, 27.7, 25.8, 24.9. HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 8.829 min. HRMS (ESI+) m/z: calculated for C22H30N2O6 418.21039; found 418.21162.
3.1.21. Ethyl 7-(3-{2-[(tert-Butoxycarbonyl)amino]-3-methoxy-3-oxopropyl}-1H-indol-2-yl)heptanoate 27
According to the general C2 alkylation procedure, Nα-Boc-(S)-tryptophan ethyl ester 7 (800 mg, 2.51 mmol, 1.00 equiv) was treated with norbornene (473 mg, 5.03 mmol, 2.00 equiv), K2CO3 (1.78 g, 10.05 mmol, 4.00 equiv), PdCl2 (35 mg, 0.25 mmol, 10 mol%), and ethyl 7-bromoheptanoate (1.96 mL, 10.05 mmol, 4.00 equiv). Purification by flash column chromatography using dichloromethane:methanol (20:1) yielded 27 (872 mg, 77%) as a pale-yellow oil. 1H NMR (300 MHz, CDCl3) δ: 8.02 (s, 1H, NH-1iTrp), 7.44 (d, J = 7.5 Hz, 1H, Ar), 7.26 (m, 1H, Ar), 7.08 (m, 2H, Ar), 5.06 (d, J = 8.3 Hz, 1H, NHCO), 4.57 (m, 1H, α-CHTrp), 3.63 (s, 3H, OCH3), 3.21 (d, J = 5.7 Hz, 2H, β-CH2Trp), 2.70 (t, J = 7.7 Hz, 2H, CH2), 2.29 (t, J = 7.4 Hz, 2H, CH2), 1.65 (m, 6H, CH2), 1.38 (m, 13H, CH3 + CH2). 13C NMR (75 MHz, CDCl3) δ: 174.0, 173.0, 155.2, 137.3, 135.4, 128.8, 121.4, 119.5, 118.3, 110.5, 105.7, 60.4, 54.3, 52.4, 34.3, 29.6, 29.1, 28.9, 28.5, 27.5, 26.0, 24.9, 14.4. HPLC (gradient: H2O:MeCN, 60–100% of MeCN in 10 min): 4.706 min. HRMS (ESI+) m/z: calculated for C26H38N2O6 474.27299; found 474.27158.
3.1.22. Tetramer 30
To a cold (0 °C) solution of compound
26 (169 mg, 0.40 mmol) in dichloromethane (15 mL), TFA (1 mL) was added. The mixture was stirred at room temperature for 5 h, then volatiles were evaporated to dryness and the residue (compound
28) was coevaporated successively with methanol and dichloromethane. Intermediate
28 (150 mg, 0.35 mmol, 4.80 equiv) was added under argon atmosphere to a solution of
4 [27] (31 mg, 0.07 mmol, 1.00 equiv), HATU (132 mg, 0.35 mmol, 4.80 equiv), and DIPEA (117 µL, 0.72 mmol, 10.00 equiv) in anhydrous DMF (15 mL). The reaction mixture was treated as described in the general coupling procedure. Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water:acetonitrile (100:0 to 0:100) yielded
30 (32 mg, 26%).
1H NMR (500 MHz, CDCl
3)
δ: 9.06 (s, 4H, NH-1
iTrp), 7.39 (m, 4H, Ar), 7.10 (m, 4H, Ar), 7.00 (m, 8H, Ar), 6.77 (d,
J = 8.2 Hz, 4H, NHCO), 4.90 (m, 4H,
α-CHTrp), 3.65 (s, 24H, OCH
3), 3.26 (dd,
J = 14.8, 5.6 Hz, 4H,
β-CH
2Trp), 3.19 (dd,
J = 14.8, 5.6 Hz, 4H,
β-CH
2Trp), 3.06 (m, 4H, OCH
2), 3.02 (m, 4H, OCH
2), 2.70 (m, 10H, CH
2), 2.45 (d,
J = 9.3 Hz, 6H, CH
2), 2.30 (t,
J = 7.2 Hz, 8H, CH
2), 2.20 (m, 8H, CH
2), 1.95 (quint,
J = 7.2 Hz, 8H, CH
2).
13C NMR (126 MHz, CDCl
3)
δ: 174.1, 172.7, 172.2, 136.3, 135.7, 128.6, 121.4, 119.4, 117.9, 111.0, 105.7, 70.0, 66.6, 53.2, 52.5, 51.8, 51.8, 44.1, 36.6, 33.1, 26.8, 25.2, 25.0. HPLC (gradient: H
2O:MeCN, 30–95% of MeCN in 10 min): 9.230 min.
3.1.23. Tetramer 31
To a cold (0 °C) solution of compound
27 (850 mg, 1.79 mmol) in dichloromethane (15 mL), TFA (1 mL) was added. The mixture was stirred at room temperature for 5 h, then volatiles were evaporated to dryness and the residue (compound
29) was coevaporated successively with methanol and dichloromethane. Intermediate
29 (800 mg, 1.64 mmol, 4.80 equiv) was added under argon atmosphere to a solution of
4 [27] (144 mg, 0.34 mmol, 1.00 equiv), HATU (622 mg, 1.64 mmol, 4.80 equiv), and DIPEA (554 µL, 3.41 mmol, 10.00 equiv) in anhydrous DMF (15 mL) and treated as described in the general coupling procedure. Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water:acetonitrile (100:0 to 0:100) yielded
31 (304 mg, 48%).
1H NMR (400 MHz, CDCl
3)
δ: 8.90 (s, 4H, NH-1
iTrp), 7.37 (d,
J = 6.6, 4H, Ar), 7.13 (d,
J = 6.6, 4H, Ar), 7.00 (m, 8H, Ar), 6.73 (d,
J = 8.0 Hz, 4H, NHCO), 4.90 (m, 4H,
α-CHTrp), 4.11 (q,
J = 7.1 Hz, 8H, OC
H2CH
3), 3.63 (s, 12H, CH
3), 3.25 (d,
J = 5.5 Hz, 8H,
β-CH
2Trp), 3.06 (m, 8H, OCH
2), 2.74 (d,
J = 9.3 Hz, 4H, CH
2), 2.64 (t,
J = 7.5 Hz, 8H, CH
2), 2.54 (d,
J = 9.2 Hz, 4H, CH
2), 2.26 (m, 16H, CH
2), 1.59 (m, 16H, CH
2), 1.30 (m, 16H, CH
2), 1.24 (t,
J = 7.1 Hz, 12H, OCH
2C
H3).
13C NMR (101 MHz, CDCl
3)
δ: 174.0, 172.7 172.1, 137.6, 135.6, 128.8, 121.2, 119.3, 117.8, 110.9, 105.0, 70.0, 66.6, 60.4, 53.2, 52.5, 44.2, 36.7, 34.3, 29.5, 29.0, 29.0, 26.9, 26.2, 24.9, 14.4. HPLC (gradient: H
2O:MeCN, 60–100% of MeCN in 10 min): 8.092 min. HRMS (ESI
+)
m/z: calculated for C
101H
140N
8O
24 1849.00616; found 1848.99805.
3.1.24. Tetramer 32
According to the general procedure for deprotection of methyl esters, compound 30 (56 mg, 0.04 mmol, 1.00 equiv) was treated with LiOH·H2O (24 mg, 0.57 mmol, 16.00 equiv). Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water:acetonitrile (100:0 to 0:100) yielded 32 (33 mg, 66%). 1H NMR (500 MHz, DMSO-d6) δ: 10.70 (s, 4H, NH-1iTrp), 8.17 (m, 4H, NHCO), 7.47 (d, J = 7.8 Hz, 4H, Ar), 7.20 (d, J = 7.8 Hz, 4H, Ar), 6.96 (t, J = 7.5 Hz, 4H, Ar), 6.90 (t, J = 7.5 Hz, 4H, Ar), 4.45 (m, 4H, α-CHTrp), 3.46–3.29 (m, 8H, CH2), 3.12–3.05 (m, 12H, β-CH2Trp + CH2), 2.92 (dd, J = 14.3, 7.3 Hz, 4H, β-CH2Trp), 2.70 (h, J = 7.1 Hz, 8H, CH2), 2.35–2.25 (m, 4H, CH2), 2.25–2.14 (m, 12H, CH2), 1.86 (quint, J = 7.3 Hz, 8H, CH2). 13C NMR (126 MHz, DMSO-d6) δ: 174.4, 173.8, 170.1, 136.6, 135.4, 128.2, 120.1, 118.1, 117.8, 110.5, 106.3, 68.9, 67.0, 53.7, 44.7, 35.8, 33.4, 26.9, 24.8. HPLC (gradient: H2O:MeCN, 10–100% of MeCN, in 10 min): 7.123 min. HRMS (ESI+) m/z: calculated for C77H92N8O24 1512.62245; found 1512.62337.
3.1.25. Tetramer 33
According to the general procedure for deprotection of methyl esters, compound 31 (97 mg, 0.05 mmol, 1.00 equiv) was treated with LiOH·H2O (35 mg, 0.84 mmol, 16.00 equiv). Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water:acetonitrile (100:0 to 0:100) yielded 33 (25 mg, 28%). 1H NMR (500 MHz, DMSO-d6) δ: 10.65 (s, 4H, NH-1iTrp), 8.17 (d, J = 8.0 Hz, 4H, NHCO), 7.46 (d, J = 7.8 Hz, 4H, Ar), 7.19 (d, J = 7.8 Hz, 4H, Ar), 6.94 (t, J = 7.5 Hz, 4H, Ar), 6.89 (t, J = 7.5 Hz, 4H, Ar), 4.45 (m, 4H, α-CHTrp), 3.39 (m, 16H, CH2), 3.10 (m, 12H, β-CH2Trp + OCH2), 2.93 (dd, J = 14.3, 7.0 Hz, 4H, β-CH2Trp), 2.63 (m, 8H, CH2), 2.18 (t, J = 7.4 Hz, 8H, CH2), 1.61 (q, J = 7.1 Hz, 8H, CH2), 1.48 (m, 8H, CH2), 1.30 (m, 16H, CH2). 13C NMR (126 MHz, DMSO-d6) δ: 174.5, 173.7, 170.0, 137.4, 135.3, 128.3, 119.9, 118.0, 117.7, 110.4, 105.7, 68.9, 67.0, 53.6, 44.7, 35.8, 33.7, 29.2, 28.7, 28.4, 25.4, 24.5. HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 7.873 min. HRMS (ESI+) m/z: calculated for C89H116N8O24 1680.81025; found 1680.80782.
3.1.26. Tetramer 34
Commercially available dimethyl 5-aminobenzene-1,3-dicarboxylate (236 mg, 1.13 mmol, 4.80 equiv) was added under argon atmosphere to a mixture of
4 [27] (100 mg, 0.24 mmol, 1.00 equiv), HATU (430 mg, 1.13 mmol, 4.80 equiv), and DIPEA (383 µL, 2.36 mmol, 10.00 equiv) in anhydrous DMF (15 mL). The reaction mixture was treated as described in the general coupling procedure. The crude product was purified by CCTLC using hexane:ethyl acetate (1:9) to afford
34 (211 mg, 75%).
1H NMR (300 MHz, CDCl
3)
δ: 9.00 (s, 4H, NHCO), 8.42 (d,
J = 1.6 Hz, 8H, Ar), 8.34 (t,
J = 1.4 Hz, 4H, Ar), 3.90 (s, 24H, OCH
3), 3.74 (t,
J = 5.5 Hz, 8H, OC
H2CH
2), 3.42 (s, 8H, CH
2O), 2.61 (t,
J = 5.5 Hz, 8H, OCH
2C
H2).
13C NMR (126 MHz, DMSO-
d6)
δ: 170.8, 166.2, 139.0, 131.3, 126.2, 125.2, 69.6, 67.4, 52.7, 45.6, 37.9. HPLC (gradient: H
2O:MeCN, 10–100% of MeCN in 10 min): 9.029 min.
3.1.27. Tetramer 35
According to the general procedure for deprotection of methyl esters, compound 34 (125 mg, 0.11 mmol, 1.00 equiv) was treated with LiOH·H2O (71 mg, 1.68 mmol, 16.00 equiv). Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water/acetonitrile (100:0 to 0:100) yielded 35 (89 mg, 78%).1H NMR (500 MHz, DMSO-d6) δ: 10.23 (s, 4H, NHCO), 8.41 (d, J = 1.6 Hz, 8H, Ar), 8.12 (d, J = 1.6 Hz, 4H, Ar), 3.55 (t, J = 6.3 Hz, 8H, OCH2CH2), 3.27 (s, 8H, CH2O), 2.46 (t, J = 6.3 Hz, 8H, OCH2CH2). 13C NMR (126 MHz, DMSO-d6) δ: 169.7, 166.5, 139.7, 131.7, 124.5, 123.5, 69.1, 67.1, 45.2, 37.2. HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 4.989 min. HRMS (ESI-) m/z: calculated for C49H48N4O24 1076.26585; found 1076.26573.
3.1.28. Dimethyl 5-[3-(2-{[(Benzyloxy)carbonyl]amino}ethyl)ureido]isophthalate 37
A solution of N-Cbz-ethylenediamine hydrochloride 36 (500 mg, 2.17 mmol, 1.00 equiv) in anhydrous DMF (5 mL) was treated with dimethyl 5-isocyanatoisophthalate (765 mg, 3.25 mmol, 1.50 equiv) and DIPEA (1.13 mL, 6.50 mmol, 3.00 equiv). The reaction was stirred at 30 °C overnight under argon atmosphere and the crude product was purified by flash column chromatography on silica gel using hexane:ethyl acetate (7:3) to afford 37 (500 mg, 54%) as a white amorphous solid. 1H NMR (400 MHz, DMSO-d6) δ: 9.13 (s, 1H, Ar), 8.30 (d, J = 1.4 Hz, 2H, Ar), 8.02 (s, 1H, NHCO), 7.42–7.20 (m, 6H, Ar + NHCO), 6.30 (t, J = 5.7 Hz, 1H, NHCO), 5.02 (s, 2H, -CH2Ph), 3.87 (s, 6H, OCH3), 3.22–3.15 (m, 2H, CH2), 3.15–3.07 (m, 2H, CH2). HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 7.609 min.
3.1.29. Dimethyl 5-[3-(2-Aminoethyl)ureido]isophthalate 38
A solution of 37 (264 mg, 0.61 mmol, 1.00 equiv) in DMF (10 mL) was treated with ammonium formate (128 mg, 2.03 mmol, 3.30 equiv) and Pd/C (10% on C; 30 wt %) under argon atmosphere. After 3 h, the residue was filtered through a Whatman® filter paper 42 and the solvent was removed under reduced pressure to give 137 mg of 38 as a brown oil. The crude was used for the next step without purification. 1H NMR (300 MHz, CDCl3) δ: 8.08 (d, J = 1.6 Hz, 2H, Ar), 8.05 (t, J = 1.5 Hz, 1H, Ar), 7.98 (m, NH), 6.52 (s, 2H, NH2), 3.79 (s, 6H, OCH3), 3.55–3.35 (m, 4H, CH2). HPLC (gradient: H2O:MeCN, 15–95% of MeCN in 10 min): 4.981 min.
3.1.30. Tetramer 39
Compound
38 (130 mg, 0.44 mmol, 4.80 equiv) was added under argon atmosphere to a mixture of
4 [
27] (39 mg, 0.09 mmol, 1.00 equiv), HATU (167 mg, 0.44 mmol, 4.80 equiv), and DIPEA (150 µL, 0.92 mmol, 10.00 equiv) in anhydrous DMF (10 mL). The reaction mixture was treated as described in the general coupling procedure. The crude product was purified by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water:acetonitrile (100:0 to 0:100) to afford
39 (48 mg, 34%).
1H NMR (500 MHz, CD
3OD)
δ: 8.15 (d,
J = 1.6 Hz, 8H, Ar), 8.07 (t,
J = 1.6 Hz, 4H, Ar), 3.88 (s, 24H, OCH
3), 3.58 (t,
J = 6.0 Hz, 8H, -OC
H2CH
2CO-), 3.39–3.32 (m, 16H, CH
2), 3.25 (s, 8H, CH
2), 2.40 (
t,
J = 6.0 Hz, 8H, -OCH
2C
H2CO-). HPLC (gradient: H
2O:MeCN, 10–100% of MeCN in 10 min): 7.866 min.
3.1.31. Tetramer 40
According to the general procedure for deprotection of methyl esters, compound 39 (25 mg, 0.02 mmol, 1.00 equiv) was treated with LiOH·H2O (42 mg, 0.26 mmol, 16.00 equiv). Purification by Biotage HPFC (high-performance flash chromatography) system on reverse phase using water/acetonitrile (100:0 to 0:100) yielded product 40 (15 mg, quant.). 1H NMR (500 MHz, DMSO-d6) δ: 9.78 (s, 4H, NHPh), 8.25 (s, 8H, Ar), 8.04 (d, J = 1.6 Hz, 4H, Ar), 8.00 (d, J = 6.2 Hz, 3H, NHCO), 7.34 (s, 3H, NHCO), 3.45 (t, J = 6.4 Hz, 8H, CH2), 3.17 (s, 16H, CH2), 3.04 (m, 8H, CH2), 2.26 (q, J = 6.3 Hz, 8H, CH2). 13C NMR (126 MHz, DMSO-d6) δ: 170.6, 168.0, 155.6, 141.1, 133.3, 122.5, 121.6, 68.3, 67.2, 44.9, 36.3, 31.3, 29.0. HPLC (gradient: H2O:MeCN, 10–100% of MeCN in 10 min): 4.781 min. HRMS (ESI+) m/z: calculated for C61H72N12O28 1420.4579; found 1420.45595.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
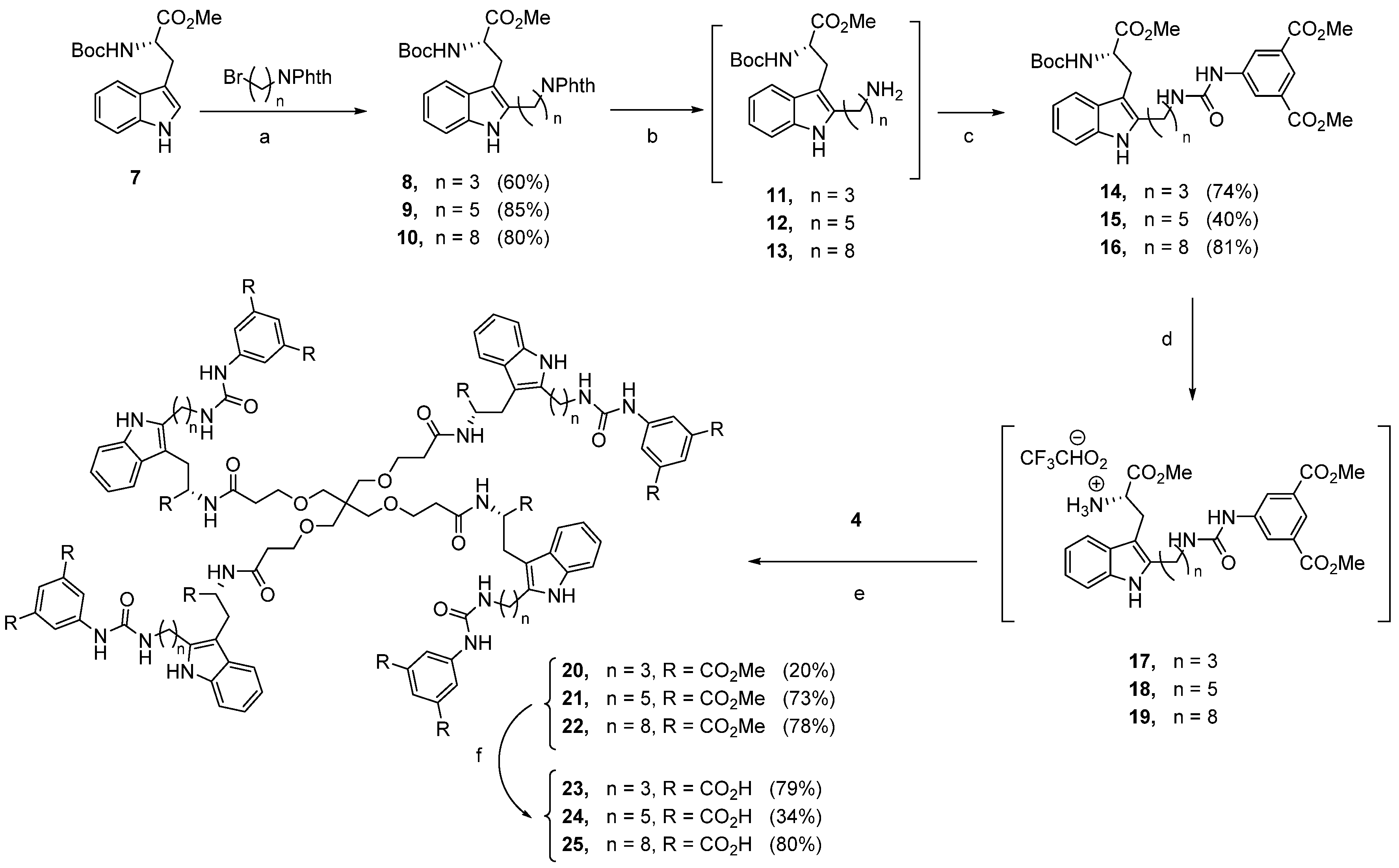{kind=link}
{kind=link}
{kind=link}
{kind=link}