
Role of SOCS and VHL Proteins in Neuronal Differentiation and Development

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structure of SOCS and VHL Proteins
3. Characteristics of SOCS Proteins and Their Roles in Neuronal Differentiation and Nervous System Development
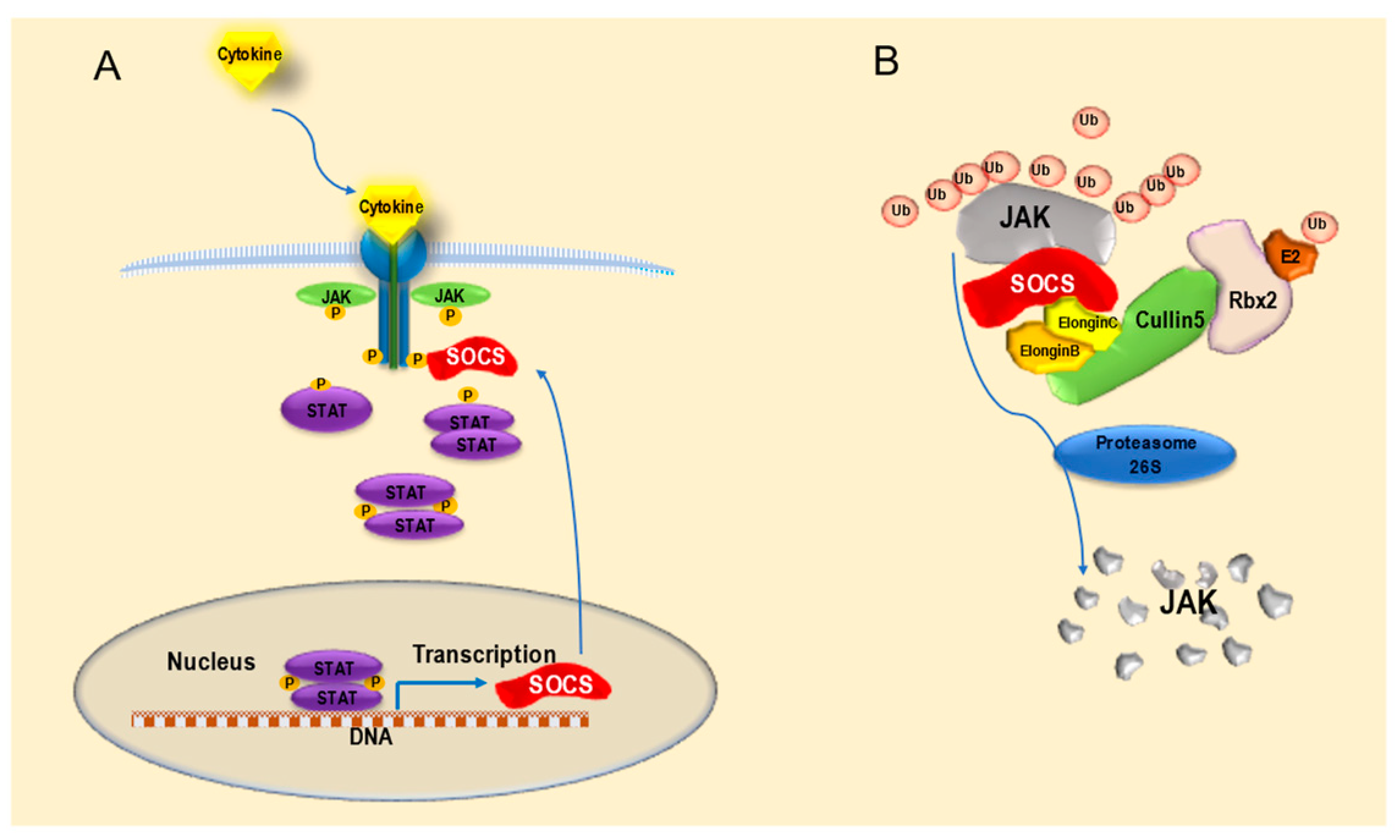
3.1. Features and Functions of SOCS Proteins
3.2. SOCS Gene and Protein Expression in the Nervous System
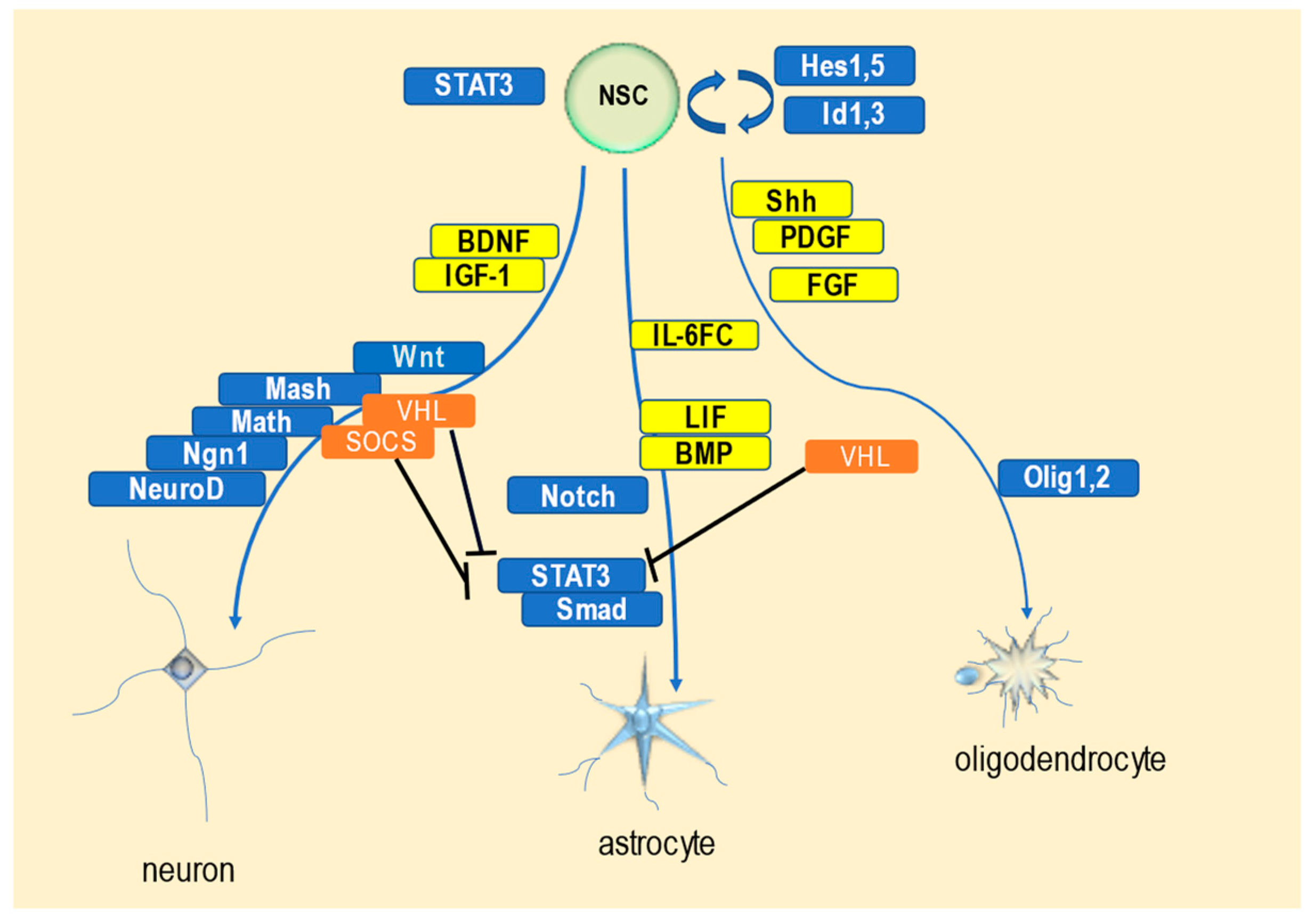
3.3. Role of SOCS Proteins in the Molecular Mechanisms of Neuronal Differentiation and Nervous System Development
4. Characteristics of VHL Proteins and Their Roles in Neuronal Differentiation and Nervous System Development
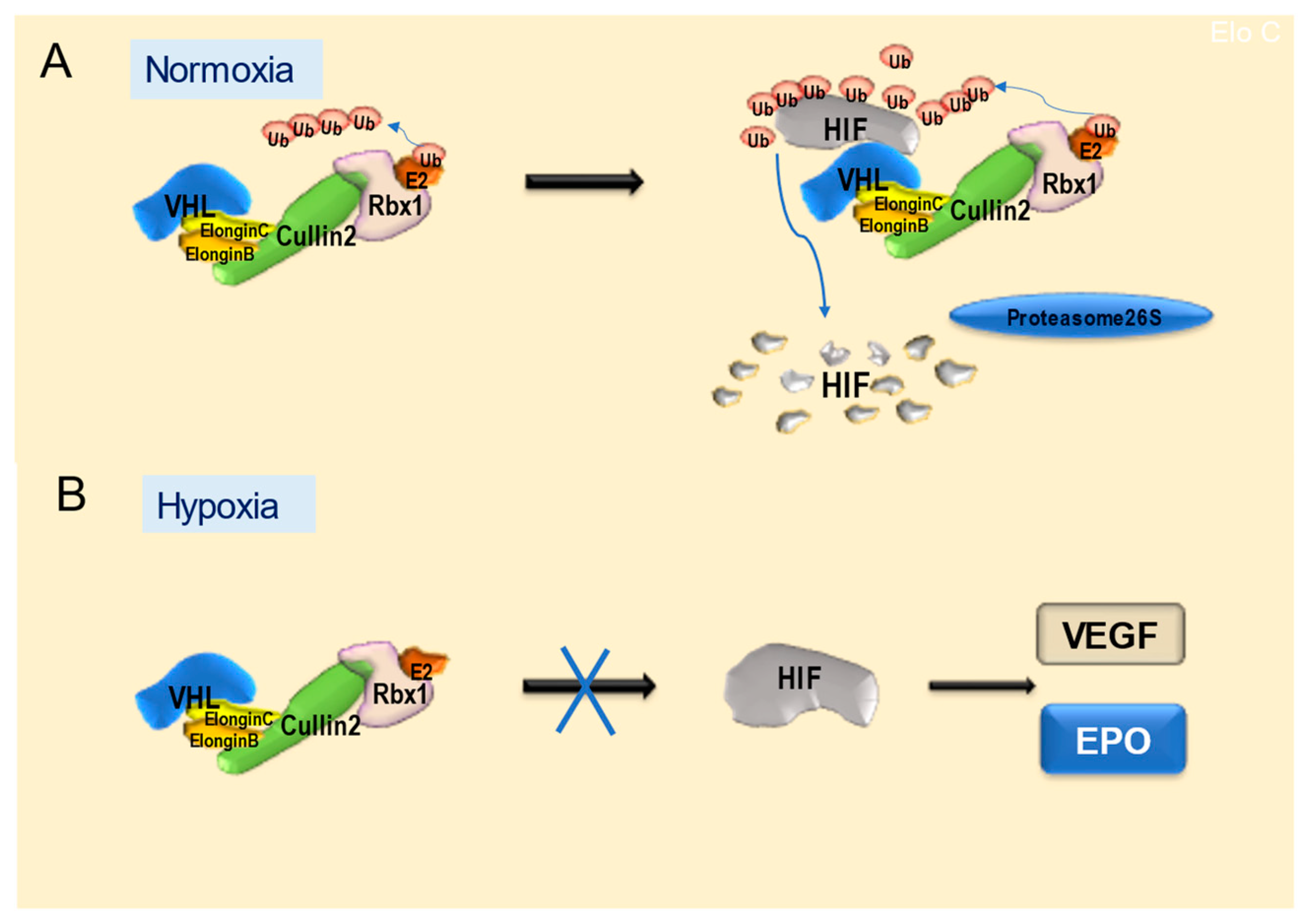
4.1. Features and Functions of VHL Proteins
4.2. Expression of the VHL Gene and Protein in the Nervous System
4.3. Role of VHL Protein in the Molecular Mechanisms of Neuronal Differentiation and Nervous System Development
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Altman, J.; Das, G.D. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol. 1965, 124, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Pincus, D.W.; Keyoung, H.M.; Harrison-Restelli, C.; Goodman, R.R.; Fraser, R.A.; Edgar, M.; Sakakibara, S.; Okano, H.; Nedergaard, M.; Goldman, S.A. Fibroblast growth factor-2/brain-derived neurotrophic factor-associated maturation of new neurons generated from adult human subependymal cells. Ann. Neurol. 1998, 43, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Nadal-Vicens, M.; Misono, S. Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanism. Cell 2001, 104, 365–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, S.; Lim, J.W.; Yellajoshyula, D.; Chang, L.W.; Kroll, K.L. Neurogenin and NeuroD direct transcriptional targets and their regulatory enhancers. EMBO J. 2007, 26, 5093–5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Yuk, D.; Alberta, J.A. Sonic hedgehog-regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron 2000, 25, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Bai, G.; Sheng, N.; Xie, Z.; Bian, W.; Yokota, Y.; Benezra, R.; Kageyama, R.; Guillemot, F.; Jing, N. Id sustains Hes1 expression to inhibit precocious neurogenesis by releasing negative autoregulation of Hes1. Dev. Cell 2007, 13, 283–297. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.J.; Perez, S.E.; Qiao, Z. Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell 2000, 101, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, K.; Takizawa, T.; Ochiai, W.; Yanagisawa, M.; Hisatsune, T.; Nakafuku, M.; Miyazono, K.; Kishimoto, T.; Kageyama, R.; Taga, T. BMP2-mediated alteration in the development pathway of astrocytogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 5868–5873. [Google Scholar] [CrossRef] [Green Version]
- Taga, T.; Fukuda, S. Role of IL-6 in the neural stem cell differentiation. Clin. Rev. Allergy Immunol. 2005, 28, 249–256. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Nakashima, K.; Takizawa, T.; Ochiai, W.; Arakawa, H.; Taga, T. Signaling crosstalk underlying synergistic induction of astrocyte differentiation by BMPs and IL-6 family of cytokines. FEBS Lett. 2001, 489, 139–144. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374 Pt 1, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooney, R.N. Suppressors of cytokine signaling (SOCS): Inhibitors of the JAK/STAT pathway. Shock 2002, 17, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanno, H.; Sato, H.; Yokoyama, T.; Yoshizumi, T.; Yamada, S. The VHL tumor suppressor protein regulates tumorigenicity of U87-derived glioma stem-like cells by inhibiting the JAK/STAT signaling pathway. Int. J. Oncol. 2013, 42, 881–886. [Google Scholar] [CrossRef] [Green Version]
- Kanno, H.; Yoshizumi, T.; Shinonaga, M.; Kubo, A.; Murata, H.; Yao, M. Role of VHL-JAK-STAT signaling pathway in central nervous system hemangioblastoma associated with von Hippel-Lindau disease. J. Neurooncol. 2020, 148, 29–38. [Google Scholar] [CrossRef]
- Kim, H.; Shim, B.Y.; Lee, S.J.; Lee, J.Y.; Lee, H.J.; Kim, I.H. Loss of von Hippel-Lindau (VHL) tumor suppressor gene function: VHL-HIF pathway and advances in treatments for metastatic renal cell carcinoma (RCC). Int. J. Mol. Sci. 2021, 22, 9795. [Google Scholar] [CrossRef]
- Kamura, T.; Maenaka, K.; Kotoshiba, S.; Matsumoto, M.; Kohda, D.; Conaway, R.C.; Conaway, J.W.; Nakayama, K.I. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004, 18, 3055–3065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, A.; Ohkubo, T.; Kiguchi, T.; Jenkins, N.A.; Gilbert, D.J.; Copeland, N.G.; Hara, T.; Miyajima, A. A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J. 1995, 14, 2816–2826. [Google Scholar] [CrossRef]
- Starr, R.; Willson, T.A.; Viney, E.M.; Murray, L.J.; Rayner, J.R.; Jenkins, B.J.; Gonda, T.J.; Alexander, W.S.; Metcalf, D.; Nicola, N.A.; et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997, 26, 917–921. [Google Scholar] [CrossRef]
- Endo, T.A.; Masuhara, M.; Yokouchi, M.; Suzuki, R.; Sakamoto, H.; Mitsui, K.; Matsumoto, A.; Tanimura, S.; Ohtsubo, M.; Misawa, H.; et al. A new protein containing an SH2 domain that inhibits JAK kinase. Nature 1997, 26, 921–924. [Google Scholar] [CrossRef]
- Naka, T.; Narazaki, M.; Hirata, M.; Matsumoto, T.; Minamoto, S.; Aono, A.; Nishimoto, N.; Kajita, T.; Taga, T.; Yoshizaki, K.; et al. Structure and function of a new STAT-induced STAT inhibitor. Nature 1997, 387, 924–929. [Google Scholar] [CrossRef]
- Hilton, D.J.; Richardson, R.T.; Alexander, W.S.; Viney, E.M.; Willson, T.A.; Sprigg, N.S.; Starr, R.; Nicholson, S.E.; Metcalf, D.; Nicola, N.A. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA 1998, 95, 114–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamura, T.; Sato, S.; Haque, D.; Liu, L.; Kaelin, W.G., Jr.; Conaway, R.C.; Conaway, J.W. The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. 1998, 12, 3872–3881. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.G.; Farley, A.; Nicholson, S.E.; Willson, T.A.; Zugaro, L.M.; Simpson, R.J.; Moritz, R.L.; Cary, D.; Richardson, R.; Hausmann, G.; et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. USA 1999, 96, 2071–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasi, W.; Sharma, A.K.; Mokbel, K. The role of suppressors of cytokine signalling in human neoplasms. Mol. Biol. Int. 2014, 2014, 630797. [Google Scholar] [CrossRef]
- Piessevaux, J.; Lavens, D.; Montoye, T.; Wauman, J.; Catteeuw, D.; Vandekerckhove, J.; Belsham, D.; Peelman, F.; Tavernier, J. Functional cross-modulation between SOCS proteins can stimulate cytokine signaling. J. Biol. Chem. 2006, 281, 32953–32966. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, A.; Masuhara, M.; Mitsui, K.; Yokouchi, M.; Ohtsubo, M.; Misawa, H.; Miyajima, A.; Yoshimura, A. CIS, a cytokine inducible SH2 protein, is a target of the JAK-STAT5 pathway and modulates STAT5 activation. Blood 1997, 89, 3148–3154. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, A.; Seki, Y.; Kubo, M.; Ohtsuka, S.; Suzuki, A.; Hayashi, I.; Tsuji, K.; Nakahata, T.; Okabe, M.; Yamada, S.; et al. Suppression of STAT5 functions in liver, mammary glands, and T cells in cytokine inducible SH2-containing protein 1 transgenic mice. Mol. Cell. Biol. 1999, 19, 6396–6407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ram, P.A.; Waxman, D.J. Role of the cytokine-inducible SH2 protein CIS in desensitization of STAT5b signaling by continuous growth hormone. J. Biol. Chem. 2000, 275, 39487–39496. [Google Scholar] [CrossRef] [Green Version]
- Aman, M.J.; Migone, T.S.; Sasaki, A.; Ascherman, D.P.; Zhu, M.; Soldaini, E.; Imada, K.; Miyajima, A.; Yoshimura, A.; Leonard, W.J. CIS associates with the interleukin-2 receptor beta chain and inhibits interleukin-2- dependent signaling. J. Biol. Chem. 1999, 274, 30266–30272. [Google Scholar] [CrossRef] [Green Version]
- Ram, P.A.; Waxman, D.J. SOCS/CIS protein inhibition of growth hormone-stimulated STAT5 signaling by multiple mechanisms. J. Biol. Chem. 1999, 274, 35553–35561. [Google Scholar] [CrossRef]
- Song, M.M.; Shuai, K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon mediated antiviral and antiproliferative activities. J. Biol. Chem. 1998, 273, 35056–35062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losman, J.A.; Chen, X.P.; Hilton, D.; Rothman, P. Cutting edge: SOCS-1 is a potent inhibitor of IL-4 signal transduction. J. Immunol. 1999, 162, 3770–3774. [Google Scholar] [CrossRef] [PubMed]
- Dickensheets, H.L.; Venkataraman, C.; Schindler, U.; Donnelly, R.P. Interferons inhibit activation of STAT6 by interleukin 4 in human monocytes by inducing SOCS-1 gene expression. Proc. Natl. Acad. Sci. USA 1999, 96, 10800–10805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezet, A.; Favre, H.; Kelly, P.A. Inhibition and restoration of prolactin signal transduction by suppressors of cytokine signaling. J. Biol. Chem. 1999, 274, 24497–24502. [Google Scholar] [CrossRef] [Green Version]
- Metcalf, D.; Greenhalgh, C.J.; Viney, E.; Willson, T.A.; Starr, R.; Nicola, N.A.; Hilton, D.J.; Alexander, W.S. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature 2000, 405, 1069–1073. [Google Scholar] [CrossRef]
- Kopchick, J.J.; Bellush, L.L.; Coschigano, K.T. Transgenic models of growth hormone action. Annu. Rev. Nutr. 1999, 19, 437–446. [Google Scholar] [CrossRef]
- Wanke, R.; Milz, S.; Rieger, N.; Ogiolda, L.; Renner-Müller, I.; Brem, G.; Hermanns, W.; Wolf, E. Overgrowth of skin in growth hormone transgenic mice depends on the presence of male gonads. J. Invest. Dermatol. 1999, 113, 967–971. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, A.; Yasukawa, H.; Shouda, T.; Kitamura, T.; Dikic, I.; Yoshimura, A. CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J. Biol. Chem. 2000, 275, 29338–29347. [Google Scholar] [CrossRef] [Green Version]
- Bjørbaek, C.; Elmquist, J.K.; El-Haschimi, K.; Kelly, J.; Ahima, R.S.; Hileman, S.; Flier, J.S. Activation of SOCS-3 messenger ribonucleic acid in the hypothalamus by ciliary neurotrophic factor. Endocrinology 1999, 140, 2035–2043. [Google Scholar] [CrossRef]
- Brender, C.; Nielsen, M.; Röpke, C.; Nissen, M.H.; Svejgaard, A.; Billestrup, N.; Geisler, C.; Ødum, N. Interferon-alpha induces transient suppressors of cytokine signalling expression in human T cells. Exp. Clin. Immunogenet. 2001, 18, 80–85. [Google Scholar] [CrossRef]
- Dogusan, Z.; Hooghe-Peters, E.L.; Berus, D.; Velkeniers, B.; Hooghe, R. Expression of SOCS genes in normal and leukemic human leukocytes stimulated by prolactin, growth hormone and cytokines. J. Neuroimmunol. 2000, 109, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Yun, S.; Piao, Z.H.; Jeong, M.; Kim, D.O.; Jung, H.; Lee, J.; Kim, M.J.; Kim, M.S.; Chung, J.W.; et al. Suppressor of cytokine signaling 2 regulates IL-15-primed human NK cell function via control of phosphorylated Pyk2. J. Immunol. 2010, 185, 917–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadowski, C.L.; Choi, T.S.; Le, M.; Wheeler, T.T.; Wang, L.H.; Sadowski, H.B. Insulin induction of SOCS-2 and SOCS-3 mRNA expression in C2C12 skeletal muscle cells is mediated by Stat5*. J. Biol. Chem. 2001, 276, 20703–20710. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.; Stanford, P.M.; Sutherland, K.; Oakes, S.R.; Naylor, M.J.; Robertson, F.G.; Blazek, K.D.; Kazlauskas, M.; Hilton, H.N.; Wittlin, S.; et al. Socs2 and elf5 mediate prolactin-induced mammary gland development. Mol. Endocrinol. 2006, 20, 1177–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minamoto, S.; Ikegame, K.; Ueno, K.; Narazaki, M.; Naka, T.; Yamamoto, H.; Matsumoto, T.; Saito, H.; Hosoe, S.; Kishimoto, T. Cloning and functional analysis of new members of STAT Induced STAT Inhibitor (SSI) family: SSI-2 and SSI-3. Biochem. Biophys. Res. Comm. 1997, 237, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Elliott, J.; Barry, A.C.; Hibbert, L.; Cacalano, N.A.; Johnston, J.A. SOCS2 can enhance interleukin-2 (IL-2) and IL-3 signaling by accelerating SOCS3 degradation. Mol. Cell. Biol. 2005, 25, 9115–9126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholson, S.E.; Willson, T.A.; Farley, A.; Starr, R.; Zhang, J.G.; Baca, M.; Alexander, W.S.; Metcalf, D.; Hilton, D.J.; Nicola, N.A. Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J. 1999, 18, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Goldshmit, Y.; Walters, C.E.; Scott, H.J.; Greenhalgh, C.J.; Turnley, A.M. SOCS2 induces neurite outgrowth by regulation of epidermal growth factor receptor activation. J. Biol. Chem. 2004, 279, 16349–16355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, B.R.; Spence, S.L.; Nissley, P.; Furlanetto, R.W. Interaction of human suppressor of cytokine signaling (SOCS)-2 with the insulin-like growth factor-I receptor. J. Biol. Chem. 1998, 273, 24095–24101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boisclair, Y.R.; Wang, J.; Shi, J.; Hurst, K.R.; Ooi, G.T. Role of the suppressor of cytokine signaling-3 in mediating the inhibitory effects of interleukin-1beta on the growth hormone-dependent transcription of the acid-labile subunit gene in liver cells. J. Biol. Chem. 2000, 275, 3841–3847. [Google Scholar] [CrossRef]
- Cohney, S.J.; Sanden, D.; Cacalano, N.A.; Yoshimura, A.; Mui, A.; Migone, T.S.; Johnston, J.A. SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol. Cell. Biol. 1999, 19, 4980–4988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrangeas, F.; Boisteau, O.; Denis, S.; Jacques, Y.; Minvielle, S. Negative cross-talk between interleukin-3 and interleukin-11 is mediated by suppressor of cytokine signalling-3 (SOCS-3). Biochem. J. 2001, 353, 223–230. [Google Scholar] [CrossRef]
- Croker, B.A.; Krebs, D.L.; Zhang, J.G.; Wormald, S.; Willson, T.A.; Stanley, E.G.; Robb, L.; Greenhalgh, C.J.; Förster, I.; Clausen, B.E.; et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 2003, 4, 540–545. [Google Scholar] [CrossRef]
- Lejeune, D.; Demoulin, J.B.; Renauld, J.C. Interleukin 9 induces expression of three cytokine signal inhibitors: Cytokine-inducible SH2-containing protein, suppressor of cytokine signalling (SOCS)-2 and SOCS-3, but only SOCS-3 overexpression suppresses interleukin 9 signalling. Biochem. J. 2001, 353, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Hong, F.; Nguyen, V.A.; Gao, B. IL-10 attenuates IFN-alpha-activated STAT1 in the liver: Involvement of SOCS2 and SOCS3. FEBS Lett. 2000, 480, 132–136. [Google Scholar] [CrossRef] [Green Version]
- Auernhammer, C.J.; Melmed, S. Interleukin-11 stimulates proopiomelanocortin gene expression and adrenocorticotropin secretion in corticotroph cells: Evidence for a redundant cytokine network in the hypothalamo-pituitary-adrenal axis. Endocrinology 1999, 140, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Izotova, L.S.; Mirochnitchenko, O.V.; Esterova, E.; Dickensheets, H.; Donnelly, R.P.; Pestka, S. Identification, cloning, and characterization of a novel soluble receptor that binds IL-22 and neutralizes its activity. J. Immunol. 2001, 166, 7096–7103. [Google Scholar] [CrossRef] [Green Version]
- Adams, T.E.; Hansen, J.A.; Starr, R.; Nicola, N.A.; Hilton, D.J.; Billestrup, N. Growth hormone preferentially induces the rapid, transient expression of SOCS-3, a novel inhibitor of cytokine receptor signaling. J. Biol. Chem. 1998, 273, 1285–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjørbaek, C.; Elmquist, J.K.; Frantz, J.D.; Shoelson, S.E.; Flier, J.S. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol. Cell 1998, 1, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Faderl, S.; Harris, D.; Van, Q.; Kantarjian, H.M.; Talpaz, M.; Estrov, Z. Granulocyte-macrophage colony-stimulating factor (GM-CSF) induces antiapoptotic and proapoptotic signals in acute myeloid leukemia. Blood 2003, 102, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Miyakawa, Y.; Fox, N.; Kaushansky, K. Interferon-alpha directly represses megakaryopoiesis by inhibiting thrombopoietin-induced signaling through induction of SOCS-1. Blood 2000, 96, 2093–2099. [Google Scholar] [CrossRef]
- Hong, F.; Nguyen, V.A.; Gao, B. Tumor necrosis factor alpha attenuates interferon alpha signaling in the liver: Involvement of SOCS3 and SHP2 and implication in resistance to interferon therapy. FASEB J. 2001, 15, 1595–1597. [Google Scholar] [CrossRef]
- Hamanaka, I.; Saito, Y.; Yasukawa, H.; Kishimoto, I.; Kuwahara, K.; Miyamoto, Y.; Harada, M.; Ogawa, E.; Kajiyama, N.; Takahashi, N.; et al. Induction of JAB/SOCS-1/SSI-1 and CIS3/SOCS-3/ SSI-3 is involved in gp130 resistance in cardiovascular system in rat treated with cardiotrophin-1 in vivo. Circ. Res. 2001, 88, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Magrangeas, F.; Boisteau, O.; Denis, S.; Jacques, Y.; Minvielle, S. Negative regulation of oncostatin M signaling by suppressor of cytokine signaling (SOCS-3). Eur. Cytokine. Netw. 2001, 12, 309–315. [Google Scholar]
- Cacalano, N.A.; Sanden, D.; Johnston, J.A. Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat. Cell Biol. 2001, 3, 460–465. [Google Scholar] [CrossRef]
- Park, E.S.; Kim, H.; Suh, J.M.; Park, S.J.; Kwon, O.Y.; Kim, Y.K.; Ro, H.K.; Cho, B.Y.; Chung, J.; Shong, M. Thyrotropin induces SOCS-1 (suppressor of cytokine signaling-1) and SOCS-3 in FRTL-5 thyroid cells. Mol. Endocrinol. 2000, 14, 440–448. [Google Scholar] [CrossRef]
- Terstegen, L.; Gatsios, P.; Bode, J.G.; Schaper, F.; Heinrich, P.C.; Graeve, L. The inhibition of interleukin-6-dependent STAT activation by mitogen-activated protein kinases depends on tyrosine 759 in the cytoplasmic tail of glycoprotein 130. J. Biol. Chem. 2000, 275, 18810–18817. [Google Scholar] [CrossRef] [Green Version]
- Sobah, M.L.; Liongue, C.; Ward, A.C. SOCS Proteins in Immunity, Inflammatory Diseases, and Immune-Related Cancer. Front. Med. (Lausanne) 2021, 8, 727987. [Google Scholar] [CrossRef]
- Chen, Z.; Laurence, A.; Kanno, Y.; Pacher-Zavisin, M.; Zhu, B.M.; Tato, C.; Yoshimura, A.; Hennighausen, L.; O’Shea, J.J. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8137–8142. [Google Scholar] [CrossRef] [Green Version]
- Brender, C.; Tannahill, G.M.; Jenkins, B.J.; Fletcher, J.; Columbus, R.; Saris, C.J.M.; Ernst, M.; Nicola, N.A.; Hilton, D.J.; Alexander, W.S.; et al. Suppressor of cytokine signaling 3 regulates CD8 T-cell proliferation by inhibition of interleukins 6 and 27. Blood 2007, 110, 2528–2536. [Google Scholar] [CrossRef]
- Croker, B.A.; Metcalf, D.; Robb, L.; Wei, W.; Mifsud, S.; DiRago, L.; Cluse, L.A.; Sutherland, K.D.; Hartley, L.; Williams, E.; et al. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity 2004, 20, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.A.; Lindberg, K.; Hilton, D.J.; Nielsen, J.H.; Billestrup, N. Mechanism of inhibition of growth hormone receptor signaling by suppressor of cytokine signaling proteins. Mol. Endocrinol. 1999, 13, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, A.E.; Rønn, S.G.; Lindberg, K.; Johannesen, J.; Galsgaard, E.D.; Pociot, F.; Nielsen, J.H.; Mandrup-Poulsen, T.; Nerup, J.; Billestrup, N. Suppressor of cytokine signaling 3 (SOCS-3) protects β-cells against interleukin-1β- and interferon-γ-mediated toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 12191–12196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, C.S.; Chan, J.; Levy, D.E.; Horvath, C.; Sadowski, H.B.; Wang, L.H. Mechanism of STAT3 activation by insulin-like growth factor I receptor. J. Biol. Chem. 2000, 275, 15099–15105. [Google Scholar] [CrossRef] [Green Version]
- Emanuelli, B.; Peraldi, P.; Filloux, C.; Sawka-Verhelle, D.; Hilton, D.; Van Obberghen, E. SOCS-3 is an insulin-induced negative regulator of insulin signaling. J. Biol. Chem. 2000, 275, 15985–15991. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Banks, A.S.; Nawijn, M.C.; Chen, X.P.; Rothman, P.B. Cutting edge: Suppressor of cytokine signaling 3 inhibits activation of NFATp. J. Immunol. 2002, 168, 4277–4281. [Google Scholar] [CrossRef] [Green Version]
- Aziz, N.; Kim, M.Y.; Cho, J.Y. Anti-inflammatory effects of luteolin: A review of in vitro, in vivo, and in silico studies. J. Ethnopharmacol. 2018, 225, 342–358. [Google Scholar] [CrossRef]
- Nicholson, S.E.; Metcalf, D.; Sprigg, N.S.; Columbus, R.; Walker, F.; Silva, A.; Cary, D.; Willson, T.A.; Zhang, J.-G.; Hilton, D.J.; et al. Suppressor of cytokine signaling (SOCS)-5 is a potential negative regulator of epidermal growth factor signaling. Proc. Natl. Acad. Sci. USA 2004, 102, 2328–2333. [Google Scholar] [CrossRef] [Green Version]
- Kario, E.; Marmor, M.D.; Adamsky, K.; Citri, A.; Amit, I.; Amariglio, N.; Rechavi, G.; Yarden, Y. Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J. Biol. Chem. 2005, 280, 7038–7048. [Google Scholar] [CrossRef] [Green Version]
- Bullock, A.N.; Rodriguez, M.C.; Debreczeni, J.E.; Songyang, Z.; Knapp, S. Structure of the SOCS4-ElonginB/C complex reveals a distinct SOCS box interface and the molecular basis for SOCS-dependent EGFR degradation. Structure 2007, 15, 1493–1504. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, J.M.; Keightley, R.A.; Nixon, B.; Roman, S.D.; Robker, R.L.; Russell, D.L.; McLaughlin, E.A. Suppressor of cytokine signaling 4 (SOCS4): Moderator of ovarian primordial follicle activation. J. Cell. Physiol. 2012, 227, 1188–1198. [Google Scholar] [CrossRef]
- Lindemann, C.; Hackmann, O.; Delic, S.; Schmidt, N.; Reifenberger, G.; Riemenschneider, M.J. SOCS3 promoter methylation is mutually exclusive to EGFR amplification in gliomas and promotes glioma cell invasion through STAT3 and FAK activation. Acta Neuropathol. 2011, 122, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Seki, Y.; Hayashi, K.; Matsumoto, A.; Seki, N.; Tsukada, J.; Ransom, J.; Naka, T.; Kishimoto, T.; Yoshimura, A.; Kubo, M. Expression of the suppressor of cytokine signaling-5 (SOCS5) negatively regulates IL-4-dependent STAT6 activation and Th2 differentiation. Proc. Natl. Acad. Sci. USA 2002, 99, 13003–13008. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, A.; Seki, Y.; Fukushima, A.; Kubo, M. The control of allergic conjunctivitis by suppressor of cytokine signaling (SOCS)3 and SOCS5 in a murine model. J. Immunol. 2005, 175, 5489–5497. [Google Scholar] [CrossRef] [Green Version]
- Mooney, R.A.; Senn, J.; Cameron, S.; Inamdar, N.; Boivin, L.M.; Shang, Y.; Furlanetto, R.W. Suppressors of cytokine signaling-1 and -6 associate with and inhibit the insulin receptor. A potential mechanism for cytokine-mediated insulin resistance. J. Biol. Chem. 2001, 276, 25889–25893. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Gronning, L.M.; Anderson, P.O.; Li, S.; Edvardsen, K.; Johnston, J.; Kioussis, D.; Shepherd, P.R.; Wang, P. Insulin induces SOCS-6 expression and its binding to the p85 monomer of phosphoinositide 3-kinase, resulting in improvement in glucose metabolism. J. Biol. Chem. 2004, 279, 34107–34114. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Mishra, K.; Surolia, A.; Banerjee, K. Suppressor of cytokine signalling-6 promotes neurite outgrowth via JAK2/STAT5-mediated signalling pathway, involving negative feedback inhibition. PLoS ONE 2011, 6, e26674. [Google Scholar] [CrossRef] [Green Version]
- Kazi, J.U.; Sun, J.; Phung, B.; Zadjali, F.; Flores-Morales, A.; Rönnstrand, L. Suppressor of cytokine signaling 6 (SOCS6) negatively regulates Flt3 signal transduction through direct binding to phosphorylated tyrosines 591 and 919 of Flt3. J. Biol. Chem. 2012, 287, 36509–36517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayle, J.; Letard, S.; Frank, R.; Dubreuil, P.; De Sepulveda, P. Suppressor of cytokine signaling 6 associates with KIT and regulates KIT receptor signaling. J. Biol. Chem. 2004, 279, 12249–12259. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.B.; Son, M.; Park, M.; Shin, J.; Yun, Y. SOCS-6 negatively regulates T cell activation through targeting p56lck to proteasomal degradation. J. Biol. Chem. 2010, 285, 7271–7280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matuoka, K.; Miki, H.; Takahashi, K.; Takenawa, T. A novel ligand for an SH3 domain of the adaptor protein Nck bears an SH2 domain and nuclear signaling motifs. Biochem. Biophys. Res. Commun. 1997, 239, 488–492. [Google Scholar] [CrossRef]
- Banks, A.S.; Li, J.; McKeag, L.; Hribal, M.L.; Kashiwada, M.; Accili, D.; Rothman, P.B. Deletion of SOCS7 leads to enhanced insulin action and enlarged islets of Langerhans. J. Clin. Invest. 2005, 115, 2462–2471. [Google Scholar] [CrossRef] [Green Version]
- Krebs, D.L.; Uren, R.T.; Metcalf, D.; Rakar, S.; Zhang, J.G.; Starr, R.; De Souza, D.P.; Hanzinikolas, K.; Eyles, J.; Connolly, L.M.; et al. SOCS-6 binds to insulin receptor substrate 4, and mice lacking the SOCS-6 gene exhibit mild growth retardation. Mol. Cell. Biol. 2002, 22, 4567–4578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tellechea, M.L.; Steinhardt, A.P.; Rodriguez, G.; Taverna, M.J.; Poskus, E.; Frechtel, G. Common variants in SOCS7 gene predict obesity, disturbances in lipid metabolism and insulin resistance. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 424–431. [Google Scholar] [CrossRef]
- Polizzoto, M.N.; Bartlett, P.F.; Turnley, A.M. Expression of “suppressor of cytokine signalling” (SOCS) genes in the developing and adult mouse nervous system. J. Comp. Neurol. 2000, 423, 348–358. [Google Scholar] [CrossRef]
- Magrangeas, F.; Apiou, F.; Denis, S.; Weidle, U.; Jacques, Y.; Minvielle, S. Cloning and expression of CIS6, chromosome assignment to 3p22 and 2p21 by in situ hybridization. Cytogenet. Cell. Genet. 2000, 88, 78–81. [Google Scholar] [CrossRef]
- Liu, X.; Mameza, M.G.; Lee, Y.S.; Eseonu, C.I.; Yu, C.R.; Derwent, J.J.K.; Egwuagu, C.E. Suppressors of cytokine-signaling proteins induce insulin resistance in the retina and promote survival of retinal cells. Diabetes 2008, 57, 1651–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Ichiyama, K.; Hashimoto, M.; Yoshida, H.; Takimoto, T.; Takaesu, G.; Torisu, T.; Hanada, T.; Yasukawa, H.; Fukuyama, S.; et al. Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN-gamma on STAT3 and Smads. J. Immunol. 2008, 180, 3746–3756. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zeng, B.; Zhang, Z.; Jiao, G.; Li, H.; Jing, Z.; Ouyang, J.; Yuan, X.; Chai, L.; Che, Y.; et al. Suppressor of cytokine signaling 3 promotes bone marrow cells to differentiate into CD8+ T lymphocytes in lung tissue via up-regulating Notch1 expression. Cancer Res. 2009, 69, 1578–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnley, A.M.; Starr, R.; Bartlett, P.F. SOCS1 regulates interferon-gamma mediated sensory neuron survival. Neuroreport 2001, 12, 3443–3445. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Dai, B.; Xin, J.; He, J.; Feng, S. Overexpression of suppressors of cytokine signaling 1 promotes the neuronal differentiation of C17.2. neural stem cells. Cell. Physiol. Biochem. 2014, 33, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Ma, X.L.; Sun, J.; He, J.Q.; Shen, L.; Li, F.G. Overexpression of suppressors of cytokine signaling 1 regulate the proliferation and differentiation of rat-derived neural stem cells. Acta Histochem. 2017, 119, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Goldshmit, Y.; Greenhalgh, C.J.; Turnley, A.M. Suppressor of cytokine signalling-2 and epidermal growth factor regulate neurite outgrowth of cortical neurons. Eur. J. Neurosci. 2004, 20, 2260–2266. [Google Scholar] [CrossRef] [PubMed]
- Ransome, M.I.; Turnley, A.M. Analysis of neuronal subpopulations in mice over-expressing suppressor of cytokine signaling-2. Neuroscience 2005, 132, 673–687. [Google Scholar] [CrossRef]
- Turnley, A.M.; Faux, C.H.; Rietze, R.L.; Coonan, J.R.; Bartlett, P.F. Suppressor of cytokine signalling 2 regulates neuronal differentiation by inhibiting growth hormone signalling. Nat. Neurosci. 2002, 5, 1155–1162. [Google Scholar] [CrossRef]
- Scott, H.J.; Stebbing, M.J.; Walters, C.E.; McLenachan, S.; Ransome, M.I.; Nichols, N.R.; Turnley, A.M. Differential effects of SOCS2 on neuronal differentiation and morphology. Brain Res. 2006, 1067, 138–145. [Google Scholar] [CrossRef]
- Yadav, A.; Kalita, A.; Dhillon, S.; Banerjee, K. JAK/STAT3 pathway is involved in survival of neurons in response to insulin-like growth factor and negatively regulated by suppressor of cytokine signalling-3. J. Biol. Chem. 2005, 280, 31830–31840. [Google Scholar] [CrossRef] [Green Version]
- Mishra, K.K.; Gupta, S.; Banerjee, K. SOCS3 induces neurite differentiation and promotes neuronal cell survival. IUBMB Life 2016, 68, 468–476. [Google Scholar] [CrossRef] [Green Version]
- Yoshizumi, T.; Kubo, A.; Murata, H.; Shinonaga, M.; Kanno, H. BC-box motif in SOCS6 induces differentiation of epidermal stem cells into GABAnergic neurons. Int. J. Mol. Sci. 2020, 21, 4947. [Google Scholar] [CrossRef]
- Bauer, S. Cytokine control of adult neural stem cells. Ann. N. Y. Acad. Sci. 2009, 1153, 48–56. [Google Scholar] [CrossRef]
- Uren, R.T.; Turnley, A.M. Regulation of neurotrophin receptor (Trk) signaling: Suppressor of cytokine signaling 2 (SOCS2) is a new player. Front. Mol. Neurosci. 2014, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Li, Z.; Liang, W.; Hu, W.; Zhou, S.; Yang, Z.; Tao, Y.; Hou, X.; Xing, Z.; Mao, J.; et al. SOCS proteins and their roles in the development of glioblastoma. Oncol Lett. 2022, 23, 5. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.J.; Akhtar, L.N.; Benveniste, E.N. SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol. 2009, 30, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphreys, L.M.; Smith, P.; Chen, Z.; Fouad, S.; D’Angiolella, V. The role of E3 ubiquitin ligases in the development and progression of glioblastoma. Cell Death. Differ. 2021, 28, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. Von Hipple-Lindau disease. Annu. Rev. Pathol. Mech. Dis. 2007, 2, 145–173. [Google Scholar] [CrossRef]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Pack, S.D.; Zbar, B.; Pak, E.; Ault, D.O.; Humphrey, J.S.; Pham, T.; Hurley, K.; Weil, R.J.; Park, W.S.; Kuzmin, I.; et al. Constitutional von Hippel-Lindau (VHL) gene deletions detected in VHL families by fluorescence in situ hybridization. Cancer Res. 1999, 59, 5560–5564. [Google Scholar] [PubMed]
- Iliopoulos, O.; Kibel, A.; Gray, S.; Kaelin, W.G. Tumor suppression by the human von Hippel-Lindau gene product. Nat. Med. 1995, 1, 822–826. [Google Scholar] [CrossRef]
- Groulx, I.; Bonicalzi, M.E.; Lee, S. Ran-mediated nuclear export of the von Hippel-Lindau tumor suppressor protein occurs independently of its assembly with cullin-2*. J. Biol. Chem. 2000, 275, 8991–9000. [Google Scholar] [CrossRef] [Green Version]
- Groulx, I.; Lee, S. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol. Cell. Biol. 2002, 22, 5319–5336. [Google Scholar] [CrossRef] [Green Version]
- Corless, C.L.; Kibel, A.S.; Iliopoulos, O.; Kaelin, W.G. Immunostaining of the von Hippel-Lindau gene product in normal and neoplastic human tissues. Hum. Pathol. 1997, 28, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Los, M.; Jansen, G.H.; Kaelin, W.G.; Lips, C.J.; Blijham, G.H.; Voest, E.E. Expression pattern of the von Hippel-Lindau protein in human tissues. Lab. Invest. 1996, 75, 231–238. [Google Scholar] [PubMed]
- Ye, Y.; Vasavada, S.; Kuzmin, I.; Stackhouse, T.; Zbar, B.; Williams, B.R. Subcellular localization of the von Hippel-Lindau disease gene product is cell cycle-dependent. Int. J. Cancer 1998, 78, 62–69. [Google Scholar] [CrossRef]
- Duan, D.R.; Humphrey, J.S.; Chen, D.Y.; Weng, Y.; Sukegawa, J.; Lee, S.; Gnarra, J.R.; Linehan, W.M.; Klausner, R.D. Characterization of the VHL tumor suppressor gene product: Localization, complex formation, and the effect of natural inactivating mutations. Proc. Natl. Acad. Sci. USA 1995, 92, 6459–6463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Neumann, M.; Stearman, R.; Stauber, R.; Pause, A.; Pavlakis, G.N.; Klausner, R.D. Transcription-dependent nuclear-cytoplasmic trafficking is required for the function of the von Hippel-Lindau tumor suppressor protein. Mol. Cell. Biol. 1999, 19, 1486–1497. [Google Scholar] [CrossRef] [Green Version]
- Bonicalzi, M.E.; Groulx, I.; de Paulsen, N.; Lee, S. Role of exon 2-encoded β-domain of the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2001, 276, 1407–1416. [Google Scholar] [CrossRef] [Green Version]
- Shiao, Y.H.; Resau, J.H.; Nagashima, K.; Anderson, L.M.; Ramakrishna, G. The von Hippel-Lindau tumor suppressor targets to mitochondria. Cancer Res. 2000, 60, 2816–2819. [Google Scholar]
- Schoenfeld, A.R.; Davidowitz, E.J.; Burk, R.D. Endoplasmic reticulum/cytosolic localization of von Hippel-Lindau gene products is mediated by a 64-amino acid region. Int. J. Cancer 2001, 91, 457–467. [Google Scholar] [CrossRef]
- Stebbins, C.E.; Kaelin, W.G.; Pavletich, N.P. Structure of the VHL-ElonginC-ElonginB complex: Implications for VHL tumor suppressor function. Science 1999, 284, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Kibel, A.; Iliopoulos, O.; DeCaprio, J.A.; Kaelin, W.G. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 1995, 269, 1444–1446. [Google Scholar] [CrossRef]
- Lonergan, K.M.; Iliopoulos, O.; Ohh, M.; Kamura, T.; Conaway, R.C.; Kaelin, W.G., Jr. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol. Cell. Biol. 1998, 18, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Duan, D.R.; Pause, A.; Burgress, W.H.; Aso, T.; Chen, D.Y.; Garrett, K.P.; Conaway, R.C.; Conaway, J.W.; Linehan, W.M.; Klausner, R.D. Inhibition of transcriptional elongation by the VHL tumor suppressor protein. Science 1995, 269, 1402–1406. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Koepp, D.M.; Conrad, M.N.; Skowyra, D.; Moreland, R.J.; Iliopoulos, O.; Lane, W.S.; Kaelin, W.G., Jr. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 1999, 284, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Conrad, M.N.; Yan, Q.; Conaway, R.C.; Conaway, J.W. The Rbx1 subunit of SCF and VHL E3 ubiquitin ligase activates Rub1 modification of cullins Cdc53 and Cul2. Genes Dev. 1999, 13, 2928–2933. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Yamanaka, K.; Kamura, T.; Minato, N.; Conaway, R.C.; Conaway, J.W.; Klausner, R.D.; Pause, A. Identification of the von Hippel-Lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc. Natl. Acad. Sci. USA 1999, 96, 12436–12441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisztwan, J.; Imbert, G.; Wirbelauer, C.; Gstaiger, M.; Krek, W. The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev. 1999, 13, 1822–1833. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Richards, F.M.; Schofield, P.N.; Fleming, S.; Maher, E.R. Expression of the von Hippel-Lindau disease tumour suppressor gene during human embryogenesis. Hum. Mol. Genet. 1996, 5, 639–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagashima, Y.; Miyagi, Y.; Udagawa, K.; Taki, A.; Misugi, K.; Sakai, N.; Kondo, K.; Kaneko, S.; Yao, M.; Shuin, T. Von Hippel-Lindau tumour suppressor gene. Localization of expression by in situ hybridization. J. Pathol. 1996, 180, 271–274. [Google Scholar] [CrossRef]
- Sakashita, N.; Takeya, M.; Kishida, T.; Stackhouse, T.M.; Zbar, B.; Takahashi, K. Expression of von Hippel-Lindau protein in normal and pathological human tissues. Histochem. J. 1999, 31, 133–144. [Google Scholar] [CrossRef]
- Kanno, H.; Saljooque, F.; Yamamoto, I.; Hattori, S.; Yao, M.; Shuin, T.; U, H.S. Role of the von Hippel-Lindau tumor suppressor protein during neuronal differentiation. Cancer Res. 2000, 60, 2820–2824. [Google Scholar]
- Kanno, H.; Nakano, S.; Kubo, A.; Mimura, T.; Tajima, N.; Sugimoto, N. Neuronal differentiation of neural progenitor cells by intracellular delivery of synthetic oligopeptide derived from Von Hippel-Lindau protein. Protein Pept. Lett. 2009, 16, 1291–1296. [Google Scholar] [CrossRef]
- Yamada, H.; Dezawa, M.; Shimazu, S.; Baba, M.; Sawada, H.; Kuroiwa, Y.; Yamamoto, I.; Kanno, H. Transfer of the von Hippel-Lindau gene to neuronal progenitor cells in treatment for Parkinson’s disease. Ann. Neurol. 2003, 54, 352–359. [Google Scholar] [CrossRef]
- Josten, F.; Fuss, B.; Feix, M.; Meissner, T.; Hoch, M. Cooperation of JAK/STAT and Notch signaling in the Drosophila foregut. Dev. Biol. 2004, 267, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanno, H.; Xu, Y.; Miyakawa, T.; Kubo, A.; Higashida, T.; Kobayashi, N.B.; Yoshida, T.; Tanokura, M. BC-Box motif-mediated neuronal differentiation of somatic stem cells. Int. J. Mol. Sci. 2018, 19, 466. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhou, B.; Yan, T.; Wu, H.; Feng, J.; Chen, H.; Gao, C.; Peng, T.; Yang, D.; Shen, J. Peroxynitrite enhances self-renewal, proliferation and neuronal differentiation of neural stem/progenitor cells through activating HIF-1α and Wnt/β-catenin signaling pathway. Free Radic. Biol. Med. 2018, 117, 158–167. [Google Scholar] [CrossRef]
- Vriend, J.; Reiter, R.J. Melatonin and the von Hippel-Lindau/HIF-1 oxygen sensing mechanism: A review. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2016, 1865, 176–183. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kanno, H.; Dezawa, M.; Mimura, T.; Kubo, A.; Yamamoto, I. The role of von Hippel-Lindau protein in the differentiation of neural progenitor cells under normoxic and anoxic conditions. Neurosci. Lett. 2005, 383, 28–32. [Google Scholar] [CrossRef]
- Chen, L.; Han, L.; Zhang, K.; Shi, Z.; Zhang, J.; Zhang, A.; Wang, Y.; Song, Y.; Li, Y.; Jiang, T.; et al. VHL regulates the effects of miR-23b on glioma survival and invasion via suppression of HIF-1α/VEGF and β-catenin/Tcf-4 signaling. Neuro Oncol. 2012, 14, 1026–1036. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Liu, M.; Wei, Y.; Liu, F.; Zhi, X.; Xu, R.; Krissansen, G.W. Overexpression of von Hippel-Lindau tumor suppressor protein and antisense HIF-1alpha eradicates gliomas. Cancer Gene Ther. 2006, 13, 428–435. [Google Scholar] [CrossRef]
- Higashida, T.; Jitsuki, S.; Kubo, A.; Mitsushima, D.; Kamiya, Y.; Kanno, H. Skin-derived precursors differentiating into dopaminergic neuronal cells in the brains of Parkinson disease model rats. J. Neurosurg. 2010, 113, 648–655. [Google Scholar] [CrossRef]
- Kubo, A.; Yoshida, T.; Kobayashi, N.; Yokoyama, T.; Mimura, T.; Nishiguchi, T.; Higashida, T.; Yamamoto, I.; Kanno, H. Efficient generation of dopamine neuron-like cells from skin-derived precursors with a synthetic peptide derived from von Hippel-Lindau protein. Stem Cells Dev. 2009, 18, 1523–1532. [Google Scholar] [CrossRef]
- Maeda, K.; Kanno, H.; Yamazaki, Y.; Kubo, A.; Sato, F.; Yamaguchi, Y.; Saito, T. Transplantation of Von Hippel-Lindau peptide delivered neural stem cells promotes recovery in the injured rat spinal cord. Neuroreport 2009, 20, 1559–1563. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Kanno, H.; Maeda, K.; Yoshida, T.; Kobayashi, N.; Kubo, A.; Yamaguchi, Y.; Saito, T. Engrafted VHL peptide-delivered bone marrow stromal cells promote spinal cord repair in rats. Neuroreport 2010, 21, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Kubo, A.; Yoshizumi, T.; Mikami, T.; Maegawa, J. Isolation of multipotent nestin-expressing stem cells derived from the epidermis of elderly humans and TAT-VHL peptide-mediated neuronal differentiation of these cells. Int. J. Mol. Sci. 2013, 14, 9604–9617. [Google Scholar] [CrossRef]
- Ding, X.; Jo, J.; Wang, C.Y.; Cristobal, C.D.; Zuo, Z.; Ye, Q.; Wirianto, M.; Lindeke-Myers, A.; Choi, J.M.; Mohila, C.A.; et al. The Daam2-VHL-Nedd4 axis governs developmental and regenerative oligodendrocyte differentiation. Genes Dev. 2020, 34, 1177–1189. [Google Scholar] [CrossRef]
- Cunningham, L.A.; Candelario, K.; Li, L. Roles for HIF-1α in neural stem cell function and the regenerative response to stroke. Behav. Brain Res. 2012, 227, 410–417. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, H.; Matsumoto, S.; Yoshizumi, T.; Nakahara, K.; Kubo, A.; Murata, H.; Shuin, T.; U, H.-S. Role of SOCS and VHL Proteins in Neuronal Differentiation and Development. Int. J. Mol. Sci. 2023, 24, 3880. https://doi.org/10.3390/ijms24043880
Kanno H, Matsumoto S, Yoshizumi T, Nakahara K, Kubo A, Murata H, Shuin T, U H-S. Role of SOCS and VHL Proteins in Neuronal Differentiation and Development. International Journal of Molecular Sciences. 2023; 24(4):3880. https://doi.org/10.3390/ijms24043880
Chicago/Turabian StyleKanno, Hiroshi, Shutaro Matsumoto, Tetsuya Yoshizumi, Kimihiro Nakahara, Atsuhiko Kubo, Hidetoshi Murata, Taro Shuin, and Hoi-Sang U. 2023. "Role of SOCS and VHL Proteins in Neuronal Differentiation and Development" International Journal of Molecular Sciences 24, no. 4: 3880. https://doi.org/10.3390/ijms24043880