

Renal and Cardiovascular Metabolic Impact Caused by Ketogenesis of the SGLT2 Inhibitors

 , ,
, , 
{kind=link}
{kind=link}
Abstract
1. Introduction
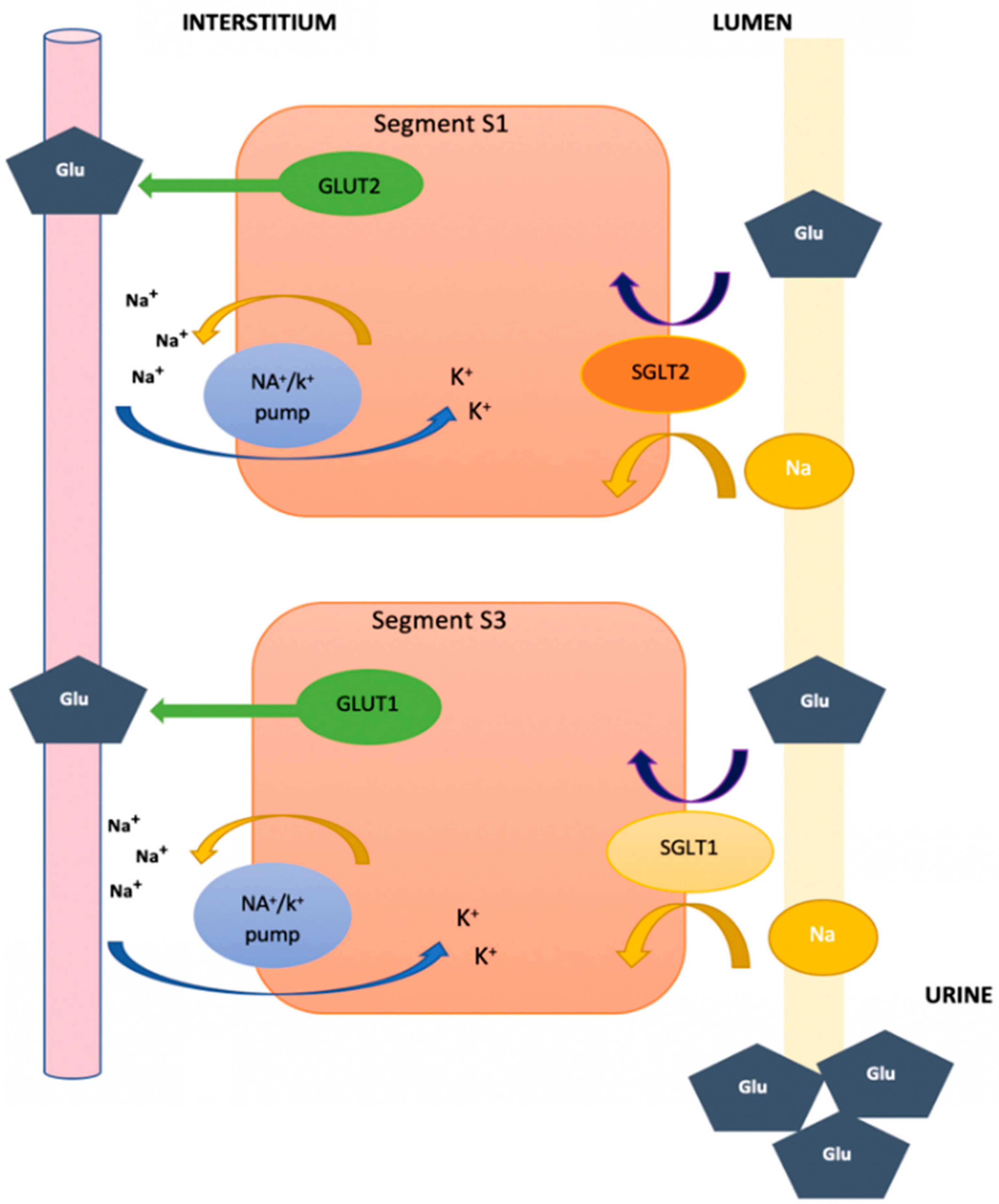
2. Function of Glucose Transporters and Sodium–Glucose Cotransporters at the Renal Level
3. Energy Metabolism in the Heart
4. Change in Cardiac Metabolism during Hypoxia
5. Alternative Fuels used by the Heart
6. Energy Metabolism and Oxidative Stress in T2DM and HF
7. Role of SGLT2i
8. Role of SGLT2i at the Renal Level—Chronic Kidney Disease
9. Hemodynamic Mechanisms of SGLT2 Inhibitors
10. Conclusions
Funding
Conflicts of Interest
References
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Muñiz, R.; Vargas-Guerrero, B.; Flores-Alvarado, L.J.; Gurrola-Díaz, C.M. Glucotransportadores (GLUT): Aspectos clí-nicos, moleculares y genéticos. Gac. Médica México 2016, 152, 547–557. Available online: https://www.medigraphic.com/cgi-bin/new/resumen.cgi?IDARTICULO=68005 (accessed on 30 November 2022).
- Castro, P.; Gabrielli, L.; Verdejo, H.; Greig, D.; Mellado, R.; Concepción, R.; Sepulveda, L.; Vukasovic, J.L.; García, L.; Pizarro, M.; et al. Metabolismo energético del corazón y sus proyecciones en el tratamiento de la insuficiencia cardíaca. Rev. Méd. Chile 2010, 138, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- García-Arias, M.R.; Gonzaga-López, T.I.; del Carmen González-Fernández, N.; Guzmán-Ramírez, P.M.; Ángeles-Acuña, A.; Enríquez-Peregrino, K.G.; Hintze-de León, J.C.; Marín-Reyes, A.H.; Cedillo-Rivera, E.A. Efecto cardiometabólico de los inhibidores del cotransportador sodio glucosa tipo 2 (SGLT2). Med. Interna México 2019, 34, 924–932. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Braunwald, E. Mechanisms of Cardiorenal Effects of Sodium-Glucose Cotransporter 2 Inhibitors: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2020, 75, 422–434. [Google Scholar] [CrossRef]
- Morales-Olvera, D.; Obregón-Aguilar, A.; Pérez-Mendoza, M.T.; Zanabria-Giles, P.; Fanghänel-Salmón, G.; Sánchez-Reyes, L. iS-GLT2 y su potencial efecto nefroprotector en pacientes con diabetes mellitus 2. Med. Interna México 2017, 33, 503–510. Available online: https://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S0186-48662017000400503 (accessed on 30 November 2022).
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef]
- Bertrand, L.; Auquier, J.; Renguet, E.; Angé, M.; Cumps, J.; Horman, S.; Beauloye, C. Glucose transporters in cardiovascular system in health and disease. Pflugers Arch. 2020, 472, 1385–1399. [Google Scholar] [CrossRef]
- Luptak, I.; Sverdlov, A.L.; Panagia, M.; Qin, F.; Pimentel, D.R.; Croteau, D.; Siwik, D.A.; Ingwall, J.S.; Bachschmid, M.M.; Balschi, J.A.; et al. Decreased ATP production and myocardial contractile reserve in metabolic heart disease. J. Mol. Cell. Cardiol. 2018, 116, 106–114. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG outcome Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef]
- Köhler, F.B.; Bevacqua, R.J.; Perrone, S.V. Inhibidores del Cotransportador de Sodio-Glucosa Tipo 2 en la Insuficiencia Cardíaca. Available online: http://www.insuficienciacardiaca.org/pdf/v16n4_21/v16n4a02.pdf (accessed on 30 November 2022).
- García-Ropero, Á.; Vargas-Delgado, A.P.; Santos-Gallego, C.G.; Badimon, J.J. Inhibition of Sodium Glucose Cotransporters Improves Cardiac Performance. Int. J. Mol. Sci. 2019, 20, 3289. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sadoshima, J. Ketone body can be a fuel substrate for failing heart. Cardiovasc. Res. 2019, 115, 1567–1569. [Google Scholar] [CrossRef] [PubMed]
- Gabr, R.E.; El-Sharkawy, A.-M.M.; Schär, M.; Panjrath, G.S.; Gerstenblith, G.; Weiss, R.G.; Bottomley, P.A. Cardiac work is related to creatine kinase energy supply in human heart failure: A cardiovascular magnetic resonance spectroscopy study. J. Cardiovasc. Magn. Reson. 2018, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Carbó, R.; Guarner, V. Cambios en el metabolismo cardíaco y su posible aprovechamiento en la terapéutica (Parte I). Arch. Cardiol. México 2003, 73, 218–229. Available online: http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S1405-99402003000300008&lng=es (accessed on 10 February 2023).
- Hewitson, K.S.; Lienard, B.M.; McDonough, M.A.; Clifton, I.J.; Butler, D.; Soares, A.S.; Oldham, N.J.; McNeill, L.A.; Schofield, C.J. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricar-boxylic acid cycle intermediates. J. Biol. Chem. 2007, 282, 3293–3301. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Hirsila, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Dalgard, C.L.; Mohyeldin, A.; McFate, T.; Tait, A.S.; Verma, A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J. Biol. Chem. 2005, 280, 41928–41939. [Google Scholar] [CrossRef]
- Dupuy, F.; Tabaries, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015, 22, 577–589. [Google Scholar] [CrossRef]
- Semba, H.; Takeda, N.; Isagawa, T.; Sugiura, Y.; Honda, K.; Wake, M.; Miyazawa, H.; Yamaguchi, Y.; Miura, M.; Jenkins, D.M.; et al. HIF-1alpha-PDK1 axis-induced active glycolysis plays an essential role in macrophage mi-gratory capacity. Nat. Commun. 2016, 7, 11635. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef]
- Van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future: Treating oxidative stress in heart failure. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. Guía ESC 2021 sobre el diagnóstico y tratamiento de la insuficiencia cardiaca aguda y crónica. Rev. Esp. Cardiol. 2022, 75, 523.e1–523.e114. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canvas. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Garcia-Ropero, A.; Mancini, D.; Pinney, S.P.; Contreras, J.P.; Fergus, I.; Abascal, V.; Moreno, P.; Atallah-Lajam, F.; Tamler, R.; et al. Rationale and Design of the EMPA-TROPISM Trial (ATRU-4): Are the “Cardiac Benefits” of Empagliflozin Independent of its Hypoglycemic Activity? Cardiovasc. Drugs Ther. 2019, 33, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Bethel, M.A.; McMurray, J.J. Class Effect for Sodium Glucose-Cotransporter-2 Inhibitors in Cardiovascular Outcomes. Circulation 2018, 137, 1218–1220. [Google Scholar] [CrossRef]
- Perkovic, V.; de Zeeuw, D.; Mahaffey, K.W.; Fulcher, G.; Erondu, N.; Shaw, W.; Barrett, T.D.; Weidner-Wells, M.; Deng, H.; Matthews, D.R.; et al. Canagliflozin and renal outcomes in type 2 diabetes: Results from the CANVAS Program randomised clinical trials. Lancet Diabetes Endocrinol. 2018, 6, 691–704. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Vargas-Delgado, A.P.; Requena-Ibanez, J.A.; Garcia-Ropero, A.; Mancini, D.; Pinney, S.; Macaluso, F.; Sartori, S.; Roque, M.; Sabatel-Perez, F.; et al. Randomized Trial of Empagliflozin in Nondiabetic Patients With Heart Failure and Reduced Ejection Fraction. J. Am. Coll. Cardiol. 2020, 77, 243–255. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Butler, J.; Filippatos, G.; Siddiqi, T.J.; Ferreira, J.P.; Brueckmann, M.; Bocchi, E.; Böhm, M.; Chopra, V.K.; Giannetti, N.; Iwata, T.; et al. Effects of Empagliflozin in Women and Men with Heart Failure and Preserved Ejection Fraction. Circulation 2022, 146, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Granata, A.; Pesce, F.; Iacoviello, M.; Anzaldi, M.; Amico, F.; Catalano, M.; Leonardi, G.; Gatta, C.; Costanza, G.; Corrao, S.; et al. SGLT2 Inhibitors: A Broad Impact Therapeutic Option for the Nephrologist. Front. Nephrol. 2022, 2, 4. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Gómez, L.S.; Negrete, J.P.G. Evidencia molecular y clínica del beneficio cardiovascular de los inhibidores SGLT2: Estado del arte. Med. UPB 2022, 41, 145–156. [Google Scholar] [CrossRef]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef]
- Zanchi, A.; Burnier, M.; Muller, M.; Ghajarzadeh-Wurzner, A.; Maillard, M.; Loncle, N.; Milani, B.; Dufour, N.; Bonny, O.; Pruijm, M. Acute and Chronic Effects of SGLT2 Inhibitor Empagliflozin on Renal Oxygenation and Blood Pressure Control in Nondiabetic Normotensive Subjects: A Randomized, Placebo-Controlled Trial. J. Am. Heart Assoc. 2020, 9, e016173. [Google Scholar] [CrossRef]
- Raza, S.; Osasan, S.; Sethia, S.; Batool, T.; Bambhroliya, Z.; Sandrugu, J.; Lowe, M.; Okunlola, O.; Hamid, P. A Systematic Review of Sodium-Glucose Cotransporter 2 (SGLT2) Inhibitors and Sympathetic Nervous System Inhibition: An Underrated Mechanism of Cardiorenal Protection. Cureus 2022, 14, e26313. [Google Scholar] [CrossRef]
- Bragagni, A.; Piani, F.; Borghi, C. Surprises in cardiology: Efficacy of gliflozines in heart failure even in the absence of diabetes. Eur. Heart J. Suppl. 2021, 23, E40–E44. [Google Scholar] [CrossRef]
- Uthman, L.; Li, X.; Baartscheer, A.; Schumacher, C.A.; Baumgart, P.; Hermanides, J.; Preckel, B.; Hollmann, M.W.; Coronel, R.; Zuurbier, C.J.; et al. Empagliflozin reduces oxidative stress through inhibition of the novel inflammation/NHE/[Na+]c/ROS-pathway in human endothelial cells. Biomed. Pharmacother. 2022, 146, 112515. [Google Scholar] [CrossRef]
- Santilli, F.; D’Ardes, D.; Guagnano, M.T.; Davi, G. Metabolic Syndrome: Sex-Related Cardiovascular Risk and Therapeutic Approach. Curr. Med. Chem. 2017, 24, 2602–2627. [Google Scholar] [CrossRef]
- Piani, F.; Melena, I.; Tommerdahl, K.L.; Nokoff, N.; Nelson, R.G.; Pavkov, M.E.; van Raalte, D.H.; Cherney, D.Z.; Johnson, R.J.; Nadeau, K.J.; et al. Sex-related differences in diabetic kidney disease: A review on the mechanisms and potential therapeutic implications. J. Diabetes Complicat. 2021, 35, 107841. [Google Scholar] [CrossRef] [PubMed]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab. 2020, 32, 404–419.e6. [Google Scholar] [CrossRef] [PubMed]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.; McMurray, J.J.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Rossing, P.; Caramori, M.L.; Chan, J.C.; Heerspink, H.J.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A.; et al. KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2022, 102, S1–S127. [Google Scholar] [CrossRef]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Mayr, M.; Badimon, J. SGLT2 Inhibitors in Heart Failure: Targeted Metabolomics and Energetic Metabolism. Circulation 2022, 146, 819–821. [Google Scholar] [CrossRef]
- Ho, K.L.; Zhang, L.; Wagg, C.; Al Batran, R.; Gopal, K.; Levasseur, J.; Leone, T.; Dyck, J.R.B.; Ussher, J.R.; Muoio, D.M.; et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 2019, 115, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E. Sodium-Glucose Co-transporters and Their Inhibition: Clinical Physiology. Cell Metab. 2017, 26, 27–38. [Google Scholar] [CrossRef]
- Buitrago Sandoval, A.F.; Sánchez Vallejo, C.A. Mecanismos de acción de los inhibidores de cotransportador de sodio y glucosa tipo 2 —SGLT2—: Más allá del control de la glicemia. Rev. Colomb. Cardiol. 2020, 27, 22–25. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; Antonio, R.S.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empagliflozin Ameliorates Adverse Left Ventricular Remodeling in Nondiabetic Heart Failure by Enhancing Myocardial Energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef]
- Verma, S.; Rawat, S.; Ho, K.L.; Wagg, C.S.; Zhang, L.; Teoh, H.; Dyck, J.E.; Uddin, G.M.; Oudit, G.Y.; Mayoux, E.; et al. Empagliflozin Increases Cardiac Energy Production in Diabetes: Novel translational insights into the heart failure benefits of SGLT2 inhibitors. JACC Basic Transl. Sci. 2018, 3, 575–587. [Google Scholar] [CrossRef]
- López-Hernández, M.A. Inhibidores del cotransportador de sodio y glucosa tipo 2 (SGLT2), el riñón como objetivo en el control glucémico de la diabetes mellitus tipo 2. Med. Interna México 2017, 33, 363–371. Available online: https://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S0186-48662017000300363 (accessed on 10 February 2023).
- Garnica-Cuéllar, J.C.; Lavalle-González, F.J.; Magaña-Serrano, J.A.; Almeda-Valdés, P.; Cetina-Canto, J.A.; Chávez-Iñiguez, J.; Garza-García, C.A.; González-Chávez, A.; González-Gálvez G, G.; Medina-Chávez, J.H.; et al. Documento de consenso sobre el uso de los iSGLT2 en el tratamiento de pacientes con diabetes mellitus tipo 2. Gac. Med. Mex. 2022, 158, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.S.; Singh, T.; Newby, D.E.; Singh, J. Sodium-glucose co-transporter 2 inhibitor therapy: Mechanisms of action in heart failure. Heart 2021, 107, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Pérez López, G.; González Albarrán, O.; Cano Megías, M. Inhibidores del cotransportador sodio-glucosa tipo 2 (SGLT2): De la glucosuria renal familiar al tratamiento de la diabetes mellitus tipo 2. Nefrología 2010, 30, 618–625. [Google Scholar] [CrossRef]
- Gliflozinas: Más Que Antidiabéticos Orales. Una Breve Revisión de la Literatura. Rev. Urug. Cardiol. 2021, 36, e3611. [Google Scholar] [CrossRef]
- Hidalgo Santiago, J.C.; Maraver Delgado, J.; Cayón Blanco, M.; López Saez, J.B.; Gómez-Fernández, P. Efecto de dapagliflozina sobre la rigidez arterial en pacientes con diabetes mellitus tipo 2. Med. Clínica 2020, 154, 171–174. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vargas-Delgado, A.P.; Arteaga Herrera, E.; Tumbaco Mite, C.; Delgado Cedeno, P.; Van Loon, M.C.; Badimon, J.J. Renal and Cardiovascular Metabolic Impact Caused by Ketogenesis of the SGLT2 Inhibitors. Int. J. Mol. Sci. 2023, 24, 4144. https://doi.org/10.3390/ijms24044144
Vargas-Delgado AP, Arteaga Herrera E, Tumbaco Mite C, Delgado Cedeno P, Van Loon MC, Badimon JJ. Renal and Cardiovascular Metabolic Impact Caused by Ketogenesis of the SGLT2 Inhibitors. International Journal of Molecular Sciences. 2023; 24(4):4144. https://doi.org/10.3390/ijms24044144
Chicago/Turabian StyleVargas-Delgado, Ariana P., Estefania Arteaga Herrera, Cesar Tumbaco Mite, Patricia Delgado Cedeno, Maria Cristina Van Loon, and Juan J. Badimon. 2023. "Renal and Cardiovascular Metabolic Impact Caused by Ketogenesis of the SGLT2 Inhibitors" International Journal of Molecular Sciences 24, no. 4: 4144. https://doi.org/10.3390/ijms24044144
APA StyleVargas-Delgado, A. P., Arteaga Herrera, E., Tumbaco Mite, C., Delgado Cedeno, P., Van Loon, M. C., & Badimon, J. J. (2023). Renal and Cardiovascular Metabolic Impact Caused by Ketogenesis of the SGLT2 Inhibitors. International Journal of Molecular Sciences, 24(4), 4144. https://doi.org/10.3390/ijms24044144