

Malonyl-CoA Accumulation as a Compensatory Cytoprotective Mechanism in Cardiac Cells in Response to 7-Ketocholesterol-Induced Growth Retardation

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
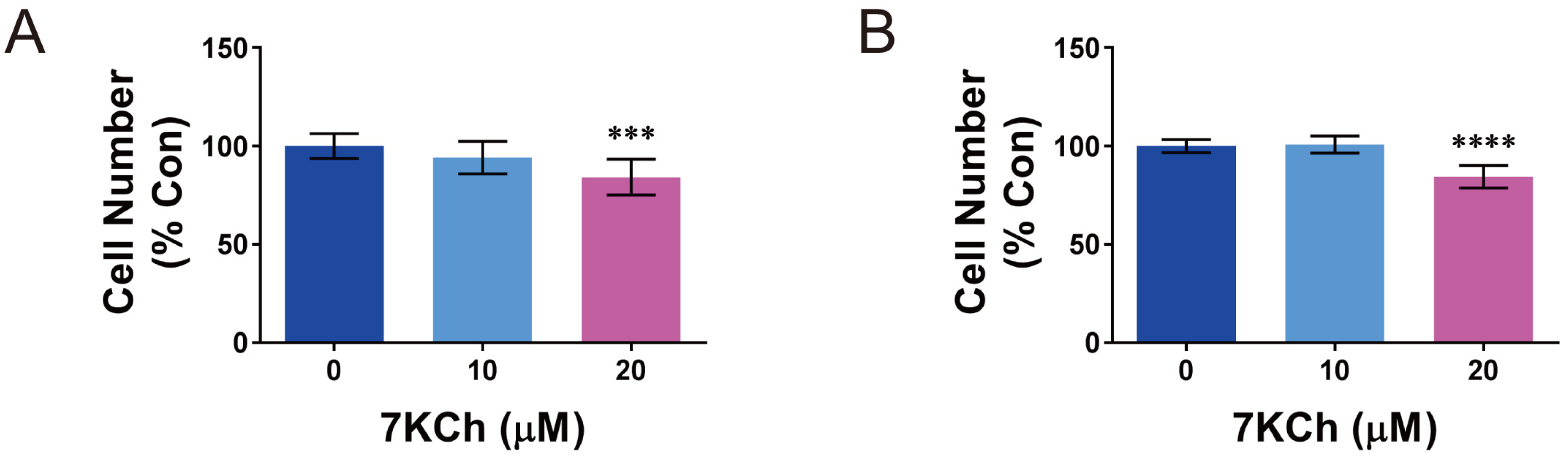
2.1. The Growth of Cardiomyocytes Is Inhbited by 7KCh
2.2. Mitochondrial Dysfunction and ROS Formation in Cardiomyocytes Are Induced by 7KCh
2.3. Compensatory Biogenesis of Mitochondria in 7KCh-Treated Cardiomyocytes
2.4. Energy Metabolic Profiling and the Changes in Metabolic Fluxes in the 7KCh-Treated Cardiomyocytes
2.5. Fatty Acid Oxidation in Cardiomyocytes Is Inhibited by 7KCh
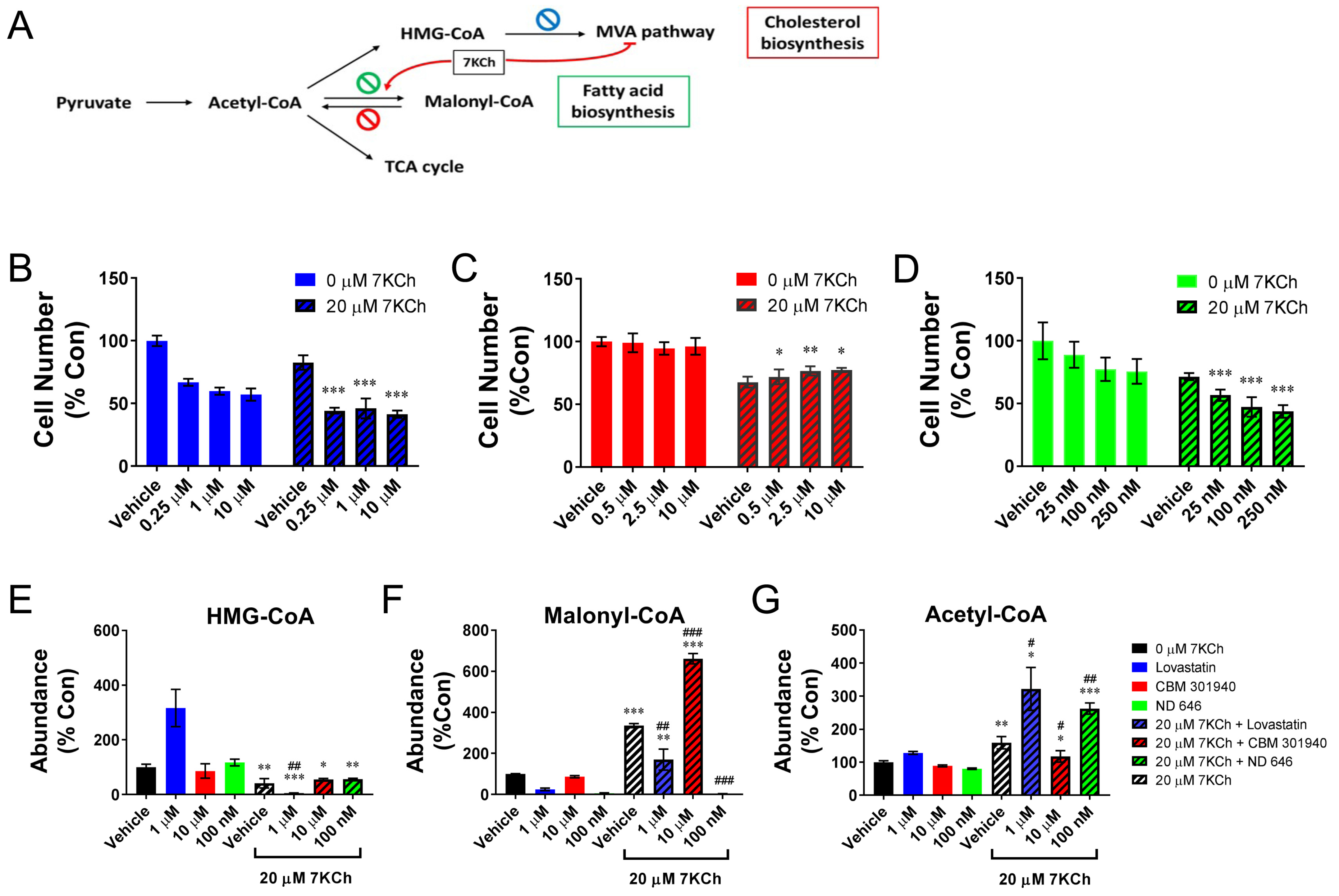
2.6. Inhibition of the Mevalonic Acid (MVA) Pathway Contributes to the Growth Retardation Effect of 7KCh
2.7. Malonyl-CoA Production Is Cytoprotective
2.8. The Expression of the Genes Involved in Malonyl-CoA Metabolism in Cardiomyocytes Is Differentially Modulated by 7KCh
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Viability Assay
4.3. Assessment of Mitochondrial Function Assays
4.3.1. Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) Determination and Mitochondrial Function Tests
4.3.2. Fatty Acid Oxidation (FAO) Assay
4.4. Metabolite Analysis Using Liquid-Chromatography Coupled with Tandem Mass Spectrometry (LC-MS/MS)
4.5. [U-13C] Glucose Labeling and Isotopologue Analysis
4.6. Isolation of Mitochondria and CPT-1 Activity Assay
4.7. Western Blotting and Immunofluorescence
4.8. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, A.J.; Jessup, W. Oxysterols and atherosclerosis. Atherosclerosis 1999, 142, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, R.A.; Zurcher, M.; Balazs, Z.; Dick, B.; Odermatt, A. Rapid hepatic metabolism of 7-ketocholesterol by 11beta-hydroxysteroid dehydrogenase type 1: Species-specific differences between the rat, human, and hamster enzyme. J. Biol. Chem. 2004, 279, 18415–18424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, M.A.; Samman, S.; Gatto, L.; Brown, A.J. Rapid hepatic metabolism of 7-ketocholesterol in vivo: Implications for dietary oxysterols. J. Lipid Res. 1999, 40, 1846–1857. [Google Scholar] [CrossRef]
- Lee, J.W.; Huang, J.D.; Rodriguez, I.R. Extra-hepatic metabolism of 7-ketocholesterol occurs by esterification to fatty acids via cPLA2alpha and SOAT1 followed by selective efflux to HDL. Biochim. Biophys. Acta 2015, 1851, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.Y.; Wang, C.H.; Ho, H.Y.; Wu, P.T.; Hung, C.L.; Huang, C.Y.; Wu, P.R.; Yeh, Y.H.; Cheng, M.L. Lipidomics reveals accumulation of the oxidized cholesterol in erythrocytes of heart failure patients. Redox Biol. 2018, 14, 499–508. [Google Scholar] [CrossRef]
- Kolwicz, S.C., Jr.; Tian, R. Glucose metabolism and cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 194–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Pulinilkunnil, T.; Kienesberger, P.; Nagendran, J. Editorial: Novel Concepts in Cardiac Energy Metabolism: From Biology to Disease. Front. Cardiovasc. Med. 2019, 6, 97. [Google Scholar] [CrossRef]
- Luchetti, F.; Canonico, B.; Cesarini, E.; Betti, M.; Galluzzi, L.; Galli, L.; Tippins, J.; Zerbinati, C.; Papa, S.; Iuliano, L. 7-Ketocholesterol and 5,6-secosterol induce human endothelial cell dysfunction by differential mechanisms. Steroids 2015, 99, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Kahn, E.; Baarine, M.; Pelloux, S.; Riedinger, J.M.; Frouin, F.; Tourneur, Y.; Lizard, G. Iron nanoparticles increase 7-ketocholesterol-induced cell death, inflammation, and oxidation on murine cardiac HL1-NB cells. Int. J. Nanomed. 2010, 5, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Lutjohann, D.; Papassotiropoulos, A.; Bjorkhem, I.; Locatelli, S.; Bagli, M.; Oehring, R.D.; Schlegel, U.; Jessen, F.; Rao, M.L.; von Bergmann, K.; et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J. Lipid Res. 2000, 41, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Girao, H.; Mota, M.C.; Ramalho, J.; Pereira, P. Cholesterol oxides accumulate in human cataracts. Exp. Eye Res. 1998, 66, 645–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Musman, J.; Pons, S.; Barau, C.; Caccia, C.; Leoni, V.; Berdeaux, A.; Ghaleh, B.; Morin, D. Regular treadmill exercise inhibits mitochondrial accumulation of cholesterol and oxysterols during myocardial ischemia-reperfusion in wild-type and ob/ob mice. Free Radic. Biol. Med. 2016, 101, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Paradis, S.; Leoni, V.; Caccia, C.; Berdeaux, A.; Morin, D. Cardioprotection by the TSPO ligand 4′-chlorodiazepam is associated with inhibition of mitochondrial accumulation of cholesterol at reperfusion. Cardiovasc. Res. 2013, 98, 420–427. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.L.; Tang, H.Y.; Wu, P.T.; Yang, C.H.; Lo, C.J.; Lin, J.F.; Ho, H.Y. 7-Ketocholesterol Induces Lipid Metabolic Reprogramming and Enhances Cholesterol Ester Accumulation in Cardiac Cells. Cells 2021, 10, 3597. [Google Scholar] [CrossRef]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [Green Version]
- Pataj, Z.; Liebisch, G.; Schmitz, G.; Matysik, S. Quantification of oxysterols in human plasma and red blood cells by liquid chromatography high-resolution tandem mass spectrometry. J. Chromatogr. A 2016, 1439, 82–88. [Google Scholar] [CrossRef]
- Folmes, C.D.; Lopaschuk, G.D. Role of malonyl-CoA in heart disease and the hypothalamic control of obesity. Cardiovasc. Res. 2007, 73, 278–287. [Google Scholar] [CrossRef]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef]
- Alberts, A.W. Discovery, biochemistry and biology of lovastatin. Am. J. Cardiol. 1988, 62, 10J–15J. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, W.; Li, S.; Guo, D.; He, J.; Wang, Y. Acetyl-CoA Carboxylases and Diseases. Front. Oncol. 2022, 12, 836058. [Google Scholar] [CrossRef] [PubMed]
- Gramajo, A.L.; Zacharias, L.C.; Neekhra, A.; Luthra, S.; Atilano, S.R.; Chwa, M.; Brown, D.J.; Kuppermann, B.D.; Kenney, M.C. Mitochondrial DNA damage induced by 7-ketocholesterol in human retinal pigment epithelial cells in vitro. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.J.; Kahl, A.; Qian, L.; Stepanova, A.; Starkov, A.; Manfredi, G.; Iadecola, C.; Zhou, P. Prohibitin is a positive modulator of mitochondrial function in PC12 cells under oxidative stress. J. Neurochem. 2018, 146, 235–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, C.; Xu, F.; Hou, T.; Sun, T.; Li, J.; Cheng, H.; Wang, X. Deficiency of PHB complex impairs respiratory supercomplex formation and activates mitochondrial flashes. J. Cell Sci. 2017, 130, 2620–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vartak, R.; Porras, C.A.; Bai, Y. Respiratory supercomplexes: Structure, function and assembly. Protein Cell 2013, 4, 582–590. [Google Scholar] [CrossRef] [Green Version]
- Timon-Gomez, A.; Garlich, J.; Stuart, R.A.; Ugalde, C.; Barrientos, A. Distinct Roles of Mitochondrial HIGD1A and HIGD2A in Respiratory Complex and Supercomplex Biogenesis. Cell Rep. 2020, 31, 107607. [Google Scholar] [CrossRef]
- Dyck, J.R.; Cheng, J.F.; Stanley, W.C.; Barr, R.; Chandler, M.P.; Brown, S.; Wallace, D.; Arrhenius, T.; Harmon, C.; Yang, G.; et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ. Res. 2004, 94, e78–e84. [Google Scholar] [CrossRef] [Green Version]
- Dyck, J.R.; Hopkins, T.A.; Bonnet, S.; Michelakis, E.D.; Young, M.E.; Watanabe, M.; Kawase, Y.; Jishage, K.; Lopaschuk, G.D. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation 2006, 114, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Niizuma, S.; Inuzuka, Y.; Kawashima, T.; Okuda, J.; Tamaki, Y.; Iwanaga, Y.; Narazaki, M.; Matsuda, T.; Soga, T.; et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ. Heart Fail. 2010, 3, 420–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimben, L.; Ingwall, J.S.; Lorell, B.H.; Pinz, I.; Schultz, V.; Tornheim, K.; Tian, R. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension 2004, 44, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillmore, N.; Levasseur, J.L.; Fukushima, A.; Wagg, C.S.; Wang, W.; Dyck, J.R.B.; Lopaschuk, G.D. Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol. Med. 2018, 24, 3. [Google Scholar] [CrossRef] [Green Version]
- Lundgrin, E.L.; Park, M.M.; Sharp, J.; Tang, W.H.; Thomas, J.D.; Asosingh, K.; Comhair, S.A.; DiFilippo, F.P.; Neumann, D.R.; Davis, L.; et al. Fasting 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann. Am. Thorac. Soc. 2013, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Pu, W.; He, L.; Li, Y.; Zhao, H.; Li, Y.; Liu, K.; Huang, X.; Weng, W.; Wang, Q.D.; et al. Cell proliferation fate mapping reveals regional cardiomyocyte cell-cycle activity in subendocardial muscle of left ventricle. Nat. Commun. 2021, 12, 5784. [Google Scholar] [CrossRef]
- Fukuda, R.; Marin-Juez, R.; El-Sammak, H.; Beisaw, A.; Ramadass, R.; Kuenne, C.; Guenther, S.; Konzer, A.; Bhagwat, A.M.; Graumann, J.; et al. Stimulation of glycolysis promotes cardiomyocyte proliferation after injury in adult zebrafish. EMBO Rep. 2020, 21, e49752. [Google Scholar] [CrossRef]
- Mohamed, T.M.A.; Abouleisa, R.; Hill, B.G. Metabolic Determinants of Cardiomyocyte Proliferation. Stem Cells 2022, 40, 458–467. [Google Scholar] [CrossRef]
- Uchikawa, T.; Matoba, T.; Kawahara, T.; Baba, I.; Katsuki, S.; Koga, J.I.; Hashimoto, Y.; Yamasaki, R.; Ichi, I.; Akita, H.; et al. Dietary 7-ketocholesterol exacerbates myocardial ischemia-reperfusion injury in mice through monocyte/macrophage-mediated inflammation. Sci. Rep. 2022, 12, 14902. [Google Scholar] [CrossRef]
- Cuillerier, A.; Ruiz, M.; Daneault, C.; Forest, A.; Rossi, J.; Vasam, G.; Cairns, G.; Cadete, V.; Consortium, L.; Des Rosiers, C.; et al. Adaptive optimization of the OXPHOS assembly line partially compensates lrpprc-dependent mitochondrial translation defects in mice. Commun. Biol. 2021, 4, 989. [Google Scholar] [CrossRef]
- Wu, Y.T.; Wu, S.B.; Wei, Y.H. Metabolic reprogramming of human cells in response to oxidative stress: Implications in the pathophysiology and therapy of mitochondrial diseases. Curr. Pharm. Des. 2014, 20, 5510–5526. [Google Scholar] [CrossRef]
- Giordano, C.; Iommarini, L.; Giordano, L.; Maresca, A.; Pisano, A.; Valentino, M.L.; Caporali, L.; Liguori, R.; Deceglie, S.; Roberti, M.; et al. Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain 2014, 137, 335–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vercauteren, K.; Gleyzer, N.; Scarpulla, R.C. Short hairpin RNA-mediated silencing of PRC (PGC-1-related coactivator) results in a severe respiratory chain deficiency associated with the proliferation of aberrant mitochondria. J. Biol. Chem. 2009, 284, 2307–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Elheiga, L.; Matzuk, M.M.; Abo-Hashema, K.A.; Wakil, S.J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 2001, 291, 2613–2616. [Google Scholar] [CrossRef]
- Bowman, C.E.; Wolfgang, M.J. Role of the malonyl-CoA synthetase ACSF3 in mitochondrial metabolism. Adv. Biol. Regul. 2019, 71, 34–40. [Google Scholar] [CrossRef]
- Bowman, C.E.; Rodriguez, S.; Selen Alpergin, E.S.; Acoba, M.G.; Zhao, L.; Hartung, T.; Claypool, S.M.; Watkins, P.A.; Wolfgang, M.J. The Mammalian Malonyl-CoA Synthetase ACSF3 Is Required for Mitochondrial Protein Malonylation and Metabolic Efficiency. Cell Chem. Biol. 2017, 24, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [Green Version]
- Ho, H.Y.; Lin, Y.T.; Lin, G.; Wu, P.R.; Cheng, M.L. Nicotinamide nucleotide transhydrogenase (NNT) deficiency dysregulates mitochondrial retrograde signaling and impedes proliferation. Redox Biol. 2017, 12, 916–928. [Google Scholar] [CrossRef]
- Zhang, J.; Nuebel, E.; Wisidagama, D.R.; Setoguchi, K.; Hong, J.S.; Van Horn, C.M.; Imam, S.S.; Vergnes, L.; Malone, C.S.; Koehler, C.M.; et al. Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat. Protoc. 2012, 7, 1068–1085. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.L.; Wu, C.H.; Chien, K.Y.; Lai, C.H.; Li, G.J.; Liu, Y.Y.; Lin, G.; Ho, H.Y. Enteroviral 2B Interacts with VDAC3 to Regulate Reactive Oxygen Species Generation That Is Essential to Viral Replication. Viruses 2022, 14, 1717. [Google Scholar] [CrossRef]
- Cheng, M.L.; Weng, S.F.; Kuo, C.H.; Ho, H.Y. Enterovirus 71 induces mitochondrial reactive oxygen species generation that is required for efficient replication. PLoS ONE 2014, 9, e113234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, H.Y.; Cheng, M.L.; Cheng, P.F.; Chiu, D.T. Low oxygen tension alleviates oxidative damage and delays cellular senescence in G6PD-deficient cells. Free Radic. Res. 2007, 41, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.Y.; Cheng, M.L.; Shiao, M.S.; Chiu, D.T. Characterization of global metabolic responses of glucose-6-phosphate dehydrogenase-deficient hepatoma cells to diamide-induced oxidative stress. Free Radic. Biol. Med. 2013, 54, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.L.; Shiao, M.S.; Chiu, D.T.; Weng, S.F.; Tang, H.Y.; Ho, H.Y. Biochemical disorders associated with antiproliferative effect of dehydroepiandrosterone in hepatoma cells as revealed by LC-based metabolomics. Biochem. Pharmacol. 2011, 82, 1549–1561. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Zhang, J.; Zhou, L.; Wen, S.; Tang, H.Y.; Jiang, B.; Zhang, F.; Suleman, M.; Sun, D.; Chen, A.; et al. c-Src Promotes Tumorigenesis and Tumor Progression by Activating PFKFB3. Cell Rep. 2020, 30, 4235–4249. [Google Scholar] [CrossRef]
- Setoyama, D.; Fujimura, Y.; Miura, D. Metabolomics reveals that carnitine palmitoyltransferase-1 is a novel target for oxidative inactivation in human cells. Genes Cells 2013, 18, 1107–1119. [Google Scholar] [CrossRef]
- Dai, J.; Liang, K.; Zhao, S.; Jia, W.; Liu, Y.; Wu, H.; Lv, J.; Cao, C.; Chen, T.; Zhuang, S.; et al. Chemoproteomics reveals baicalin activates hepatic CPT1 to ameliorate diet-induced obesity and hepatic steatosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5896–E5905. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, M.-L.; Yang, C.-H.; Wu, P.-T.; Li, Y.-C.; Sun, H.-W.; Lin, G.; Ho, H.-Y. Malonyl-CoA Accumulation as a Compensatory Cytoprotective Mechanism in Cardiac Cells in Response to 7-Ketocholesterol-Induced Growth Retardation. Int. J. Mol. Sci. 2023, 24, 4418. https://doi.org/10.3390/ijms24054418
Cheng M-L, Yang C-H, Wu P-T, Li Y-C, Sun H-W, Lin G, Ho H-Y. Malonyl-CoA Accumulation as a Compensatory Cytoprotective Mechanism in Cardiac Cells in Response to 7-Ketocholesterol-Induced Growth Retardation. International Journal of Molecular Sciences. 2023; 24(5):4418. https://doi.org/10.3390/ijms24054418
Chicago/Turabian StyleCheng, Mei-Ling, Cheng-Hung Yang, Pei-Ting Wu, Yi-Chin Li, Hao-Wei Sun, Gigin Lin, and Hung-Yao Ho. 2023. "Malonyl-CoA Accumulation as a Compensatory Cytoprotective Mechanism in Cardiac Cells in Response to 7-Ketocholesterol-Induced Growth Retardation" International Journal of Molecular Sciences 24, no. 5: 4418. https://doi.org/10.3390/ijms24054418
APA StyleCheng, M.-L., Yang, C.-H., Wu, P.-T., Li, Y.-C., Sun, H.-W., Lin, G., & Ho, H.-Y. (2023). Malonyl-CoA Accumulation as a Compensatory Cytoprotective Mechanism in Cardiac Cells in Response to 7-Ketocholesterol-Induced Growth Retardation. International Journal of Molecular Sciences, 24(5), 4418. https://doi.org/10.3390/ijms24054418