
Cell-Type-Specific Gene Regulatory Networks of Pro-Inflammatory and Pro-Resolving Lipid Mediator Biosynthesis in the Immune System
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
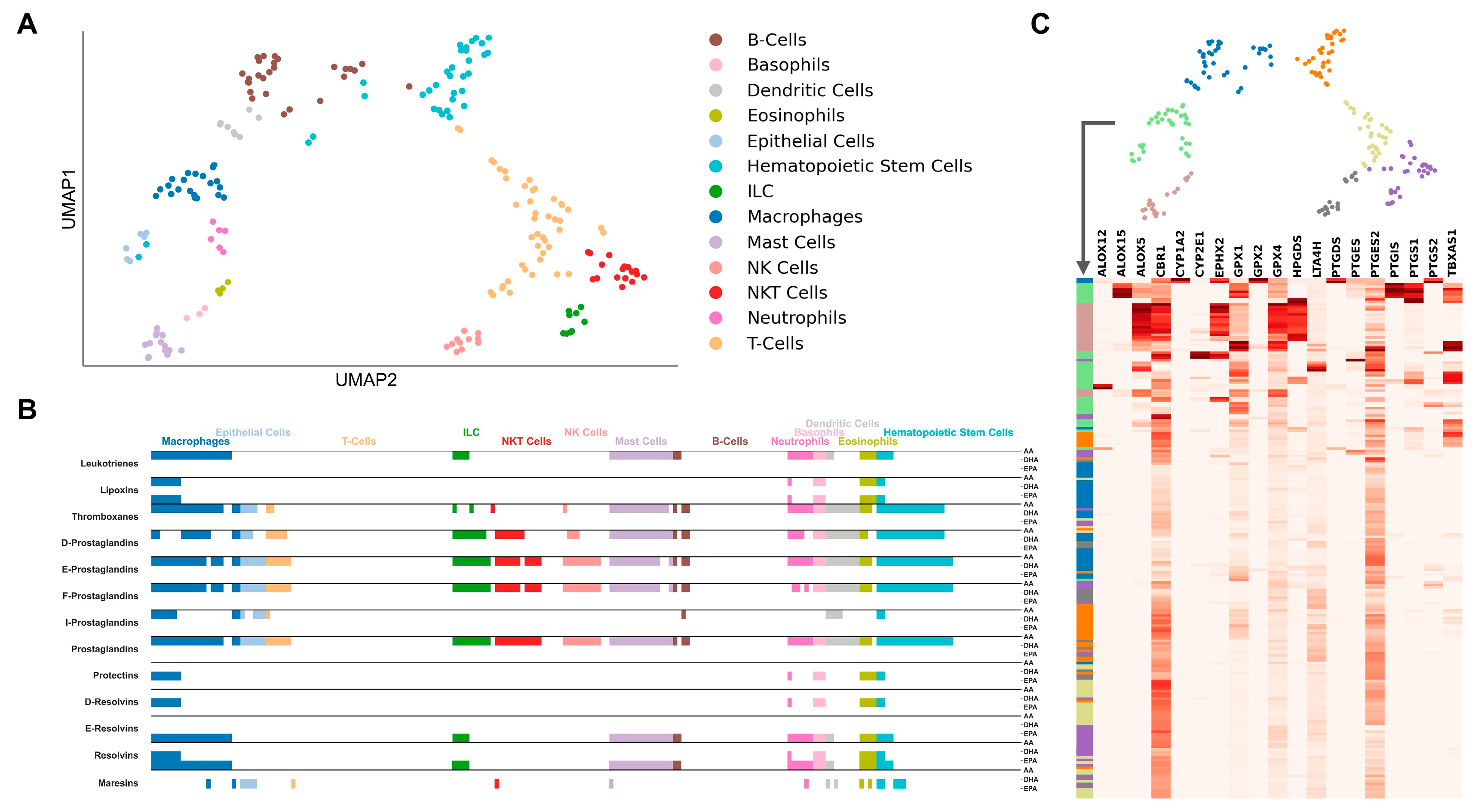
2.1. Cell-Type-Specific LM Pathways
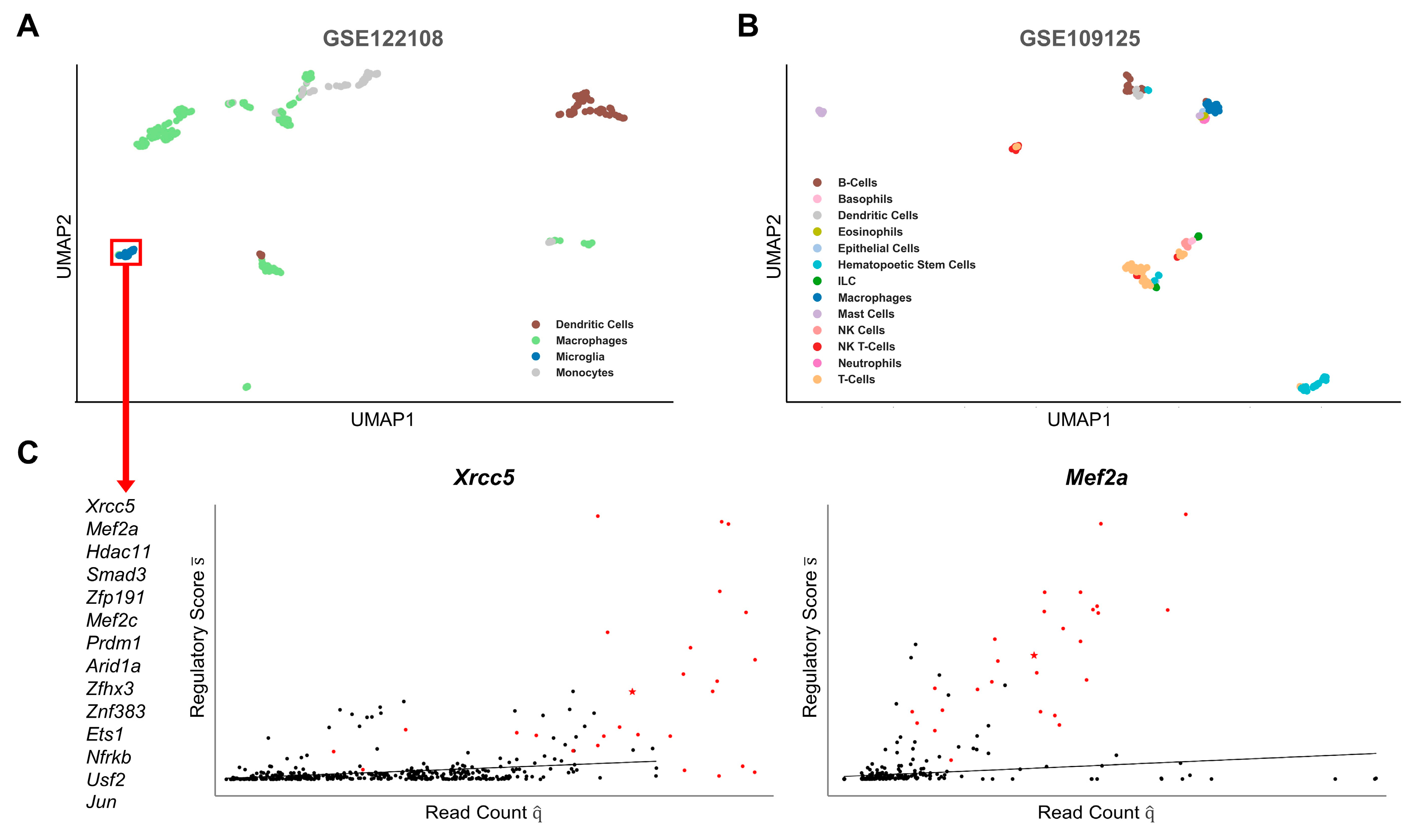
2.2. Cell-Type-Specific Gene Regulation
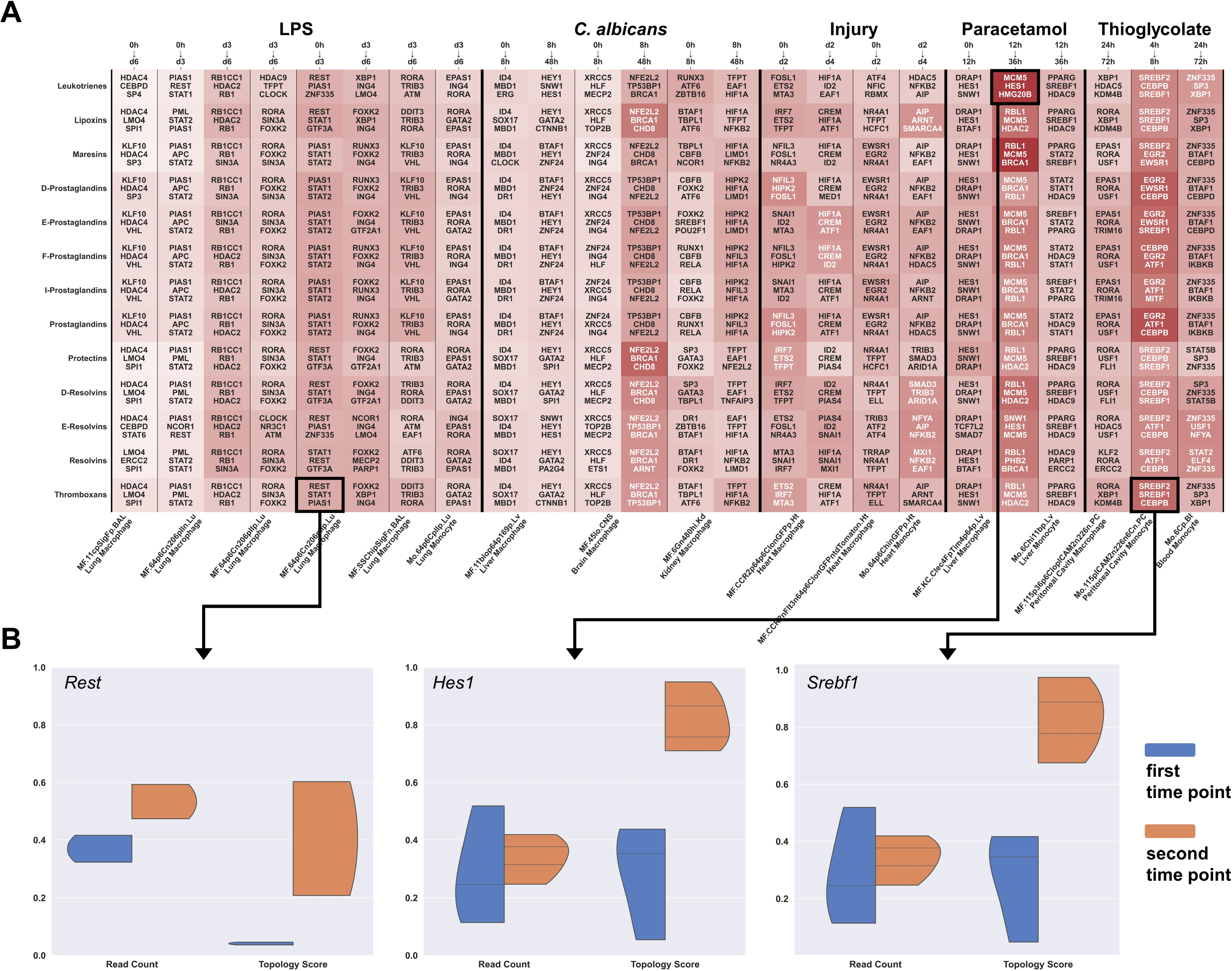
2.3. Immune Cell Activation Modulates Gene Regulatory Networks of Lipid Mediators
2.4. LM Gene Regulation Shows Substantial Differences in Related Cell Types
3. Discussion
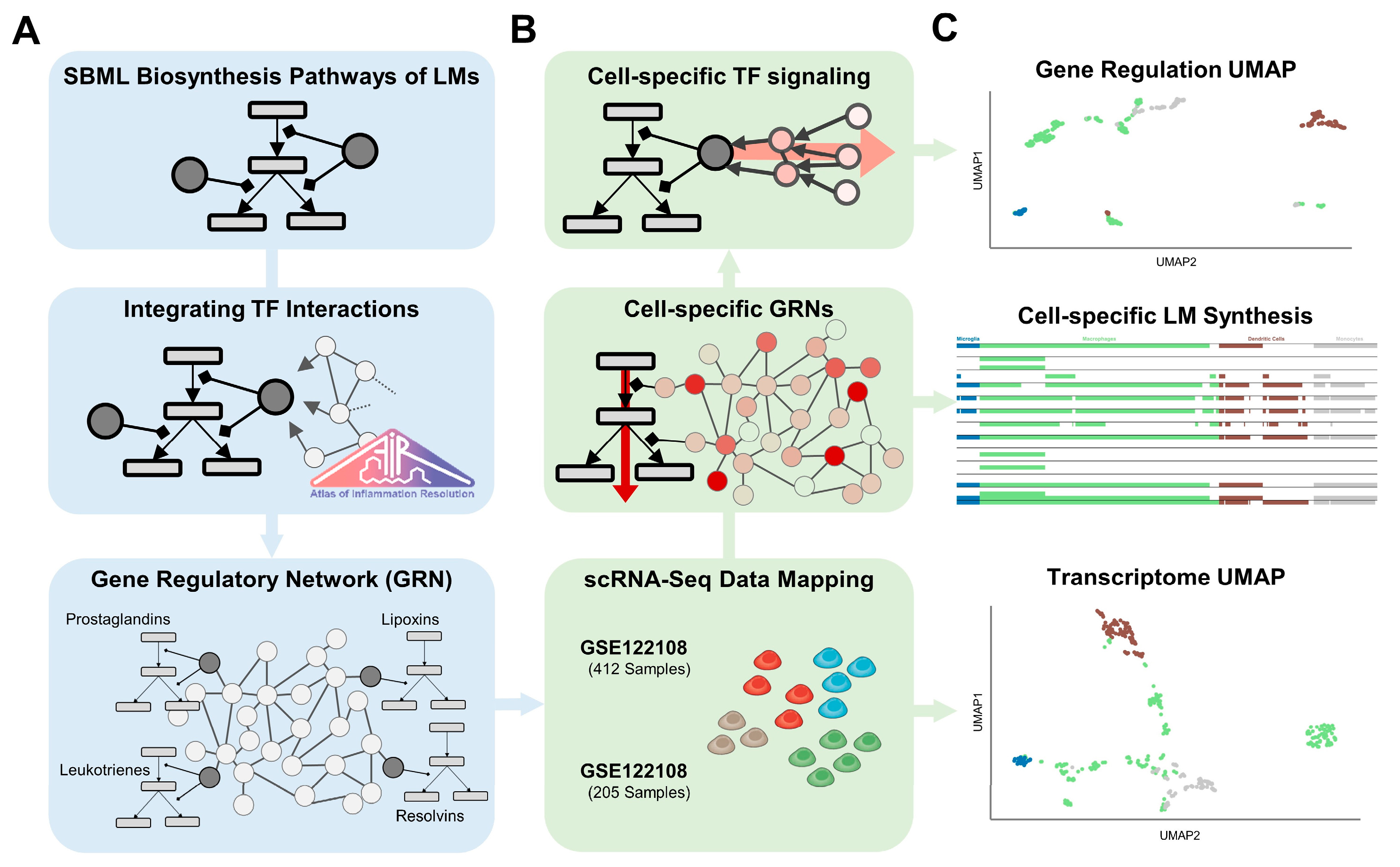
4. Materials and Methods
4.1. Network Curation
4.2. Data Processing and Integration
4.3. Topological Analysis
4.4. Topological Weighting
4.5. Feature Extraction
4.6. Cell Type Clustering
4.7. Statistical Evaluation of Features
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory Responses and Inflammation-Associated Diseases in Organs. Oncotarget 2018, 9, 7204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, J.M.; Reeves, G.; Billman, G.E.; Sturmberg, J.P. Inflammation-Nature’s Way to Efficiently Respond to All Types of Challenges: Implications for Understanding and Managing “the Epidemic” of Chronic Diseases. Front. Med. 2018, 5, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C. Points of Control in Inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef]
- Shimizu, T. Lipid Mediators in Health and Disease: Enzymes and Receptors as Therapeutic Targets for the Regulation of Immunity and Inflammation. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 123–150. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.; Kim, M.; Hwang, S.W. Molecular Mechanisms Underlying the Actions of Arachidonic Acid-Derived Prostaglandins on Peripheral Nociception. J. Neuroinflammation 2020, 17, 1–27. [Google Scholar] [CrossRef]
- Ricciotti, E.; Fitzgerald, G.A. Prostaglandins and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism Pathways of Arachidonic Acids: Mechanisms and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2021, 6, 1–30. [Google Scholar] [CrossRef]
- Dennis, E.A.; Norris, P.C. Eicosanoid Storm in Infection and Inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Chiurchiù, V.; Leuti, A.; Maccarrone, M. Bioactive Lipids and Chronic Inflammation: Managing the Fire Within. Front. Immunol. 2018, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 2016, 7, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega-Gómez, A.; Perretti, M.; Soehnlein, O. Resolution of Inflammation: An Integrated View. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Vago, J.P.; Perretti, M.; Teixeira, M.M. Mediators of the Resolution of the Inflammatory Response. Trends Immunol. 2019, 40, 212–227. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-Resolving Lipid Mediators Are Leads for Resolution Physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Chiang, N.; Dalli, J.; Levy, B.D. Lipid Mediators in the Resolution of Inflammation. Cold Spring Harb. Perspect. Biol. 2015, 7, a016311. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norris, P.C.; Serhan, C.N. Metabololipidomic Profiling of Functional Immunoresolvent Clusters and Eicosanoids in Mammalian Tissues. Biochem. Biophys. Res. Commun. 2018, 504, 553. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Komshian, S.; Sansbury, B.E.; Wu, B.; Mottola, G.; Chen, M.; Spite, M.; Conte, M.S. Biosynthesis of Proresolving Lipid Mediators by Vascular Cells and Tissues. FASEB J. 2017, 31, 3393. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Lu, Y.; Tian, H.; Alapure, B.V.; Wang, Q.; Bunnell, B.A.; Laborde, J.M. Maresin-like Lipid Mediators Are Produced by Leukocytes and Platelets and Rescue Reparative Function of Diabetes-Impaired Macrophages. Chem. Biol. 2014, 21, 1318–1329. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H. What Is a Cell Type and How to Define It? Cell 2022, 185, 2739–2755. [Google Scholar] [CrossRef]
- Ye, Z.; Sarkar, C.A. Towards a Quantitative Understanding of Cell Identity. Trends Cell Biol. 2018, 28, 1030. [Google Scholar] [CrossRef]
- Kiselev, V.Y.; Andrews, T.S.; Hemberg, M. Challenges in Unsupervised Clustering of Single-Cell RNA-Seq Data. Nat. Rev. Genet. 2018 205 2019, 20, 273–282. [Google Scholar] [CrossRef]
- Liu, J.; Fan, Z.; Zhao, W.; Zhou, X. Machine Intelligence in Single-Cell Data Analysis: Advances and New Challenges. Front. Genet. 2021, 12, 807. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, W.; Han, H.; Liu, J.; Liu, B.; Wang, Y. ScGSLC: An Unsupervised Graph Similarity Learning Framework for Single-Cell RNA-Seq Data Clustering. Comput. Biol. Chem. 2021, 90, 107415. [Google Scholar] [CrossRef]
- Serhan, C.N.; Gupta, S.K.; Perretti, M.; Godson, C.; Brennan, E.; Li, Y.; Soehnlein, O.; Shimizu, T.; Werz, O.; Chiurchiù, V.; et al. The Atlas of Inflammation Resolution (AIR). Mol. Aspects Med. 2020, 74, 100894. [Google Scholar] [CrossRef] [PubMed]
- Hoch, M.; Smita, S.; Cesnulevicius, K.; Lescheid, D.; Schultz, M.; Wolkenhauer, O.; Gupta, S. Network- and Enrichment-Based Inference of Phenotypes and Targets from Large-Scale Disease Maps. NPJ Syst. Biol. Appl. 2022, 8, 13. [Google Scholar] [CrossRef]
- Li, H.; Wang, F.; Guo, X.; Jiang, Y. Decreased MEF2A Expression Regulated by Its Enhancer Methylation Inhibits Autophagy and May Play an Important Role in the Progression of Alzheimer’s Disease. Front. Neurosci. 2021, 15, 669. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kaur, S.; Kapil, L.; Singh, C.; Singh, A. HDAC11: A Novel Inflammatory Biomarker in Huntington’s Disease. EXCLI J. 2022, 21, 647. [Google Scholar] [CrossRef]
- Sun, L.; Telles, E.; Karl, M.; Cheng, F.; Luetteke, N.; Sotomayor, E.M.; Miller, R.H.; Seto, E. Loss of HDAC11 Ameliorates Clinical Symptoms in a Multiple Sclerosis Mouse Model. Life Sci. Alliance 2018, 1. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, L.; Xu, Y.; Tang, X.; Wang, X. Substantia Nigra Smad3 Signaling Deficiency: Relevance to Aging and Parkinson’s Disease and Roles of Microglia, Proinflammatory Factors, and MAPK. J. Neuroinflammation 2020, 17, 342. [Google Scholar] [CrossRef]
- Deczkowska, A.; Matcovitch-Natan, O.; Tsitsou-Kampeli, A.; Ben-Hamo, S.; Dvir-Szternfeld, R.; Spinrad, A.; Singer, O.; David, E.; Winter, D.R.; Smith, L.K.; et al. Mef2C Restrains Microglial Inflammatory Response and Is Lost in Brain Ageing in an IFN-I-Dependent Manner. Nat. Commun. 2017, 8, 717. [Google Scholar] [CrossRef] [Green Version]
- Gong, M.; Shi, R.; Liu, Y.; Ke, J.; Liu, X.; Du, H.Z.; Liu, C.M. Abnormal Microglial Polarization Induced by Arid1a Deletion Leads to Neuronal Differentiation Deficits. Cell Prolif. 2022, 55, e13314. [Google Scholar] [CrossRef]
- Su, L.; Zhang, M.; Ji, F.; Zhao, J.; Wang, Y.; Wang, W.; Zhang, S.; Ma, H.; Wang, Y.; Jiao, J. Microglia Homeostasis Mediated by Epigenetic ARID1A Regulates Neural Progenitor Cells Response and Leads to Autism-like Behaviors. Mol. Psychiatry 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell Transcriptomics Reveals Distinct Inflammation-induced Microglia Signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef]
- Xiang, S.; Huang, Z.; Wang, T.; Han, Z.; Yu, C.Y.; Ni, D.; Huang, K.; Zhang, J. Condition-Specific Gene Co-Expression Network Mining Identifies Key Pathways and Regulators in the Brain Tissue of Alzheimer’s Disease Patients. BMC Med. Genom. 2018, 11, 39–51. [Google Scholar] [CrossRef]
- Jantaratnotai, N.; Ling, A.; Cheng, J.; Schwab, C.; Mcgeer, P.L.; Mclarnon, J.G. Upregulation and Expression Patterns of the Angiogenic Transcription Factor Ets-1 in Alzheimer’s Disease Brain. J. Alzheimers. Dis. 2013, 37, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Waetzig, V.; Czeloth, K.; Hidding, U.; Mielke, K.; Kanzow, M.; Brecht, S.; Goetz, M.; Lucius, R.; Herdegen, T.; Hanisch, U.R. C-Jun N-Terminal Kinases (JNKs) Mediate pro-Inflammatory Actions of Microglia. Glia 2005, 50, 235–246. [Google Scholar] [CrossRef]
- Diaz-Aparicio, I.; Paris, I.; Sierra-Torre, V.; Plaza-Zabala, A.; Rodríguez-Iglesias, N.; Márquez-Ropero, M.; Beccari, S.; Huguet, P.; Abiega, O.; Alberdi, E.; et al. Microglia Actively Remodel Adult Hippocampal Neurogenesis through the Phagocytosis Secretome. J. Neurosci. 2020, 40, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruchot, J.; Lein, F.; Lewen, I.; Reiche, L.; Weyers, V.; Petzsch, P.; Göttle, P.; Köhrer, K.; Hartung, H.-P.; Küry, P.; et al. Siponimod Modulates the Reaction of Microglial Cells to Pro-Inflammatory Stimulation. Int. J. Mol. Sci. 2022, 23, 13278. [Google Scholar] [CrossRef]
- Aaker, J.D.; Elbaz, B.; Wu, Y.; Looney, T.J.; Zhang, L.; Lahn, B.T.; Popko, B. Transcriptional Fingerprint of Hypomyelination in Zfp191null and Shiverer (Mbpshi) Mice. ASN Neuro 2016, 8, 1759091416670749. [Google Scholar] [CrossRef] [Green Version]
- Shippy, D.C.; Watters, J.J.; Ulland, T.K. Transcriptional Response of Murine Microglia in Alzheimer’s Disease and Inflammation. BMC Genomics 2022, 23, 1–12. [Google Scholar] [CrossRef]
- Prasad, S.; Singh, K. Interaction of USF1/USF2 and α-Pal/Nrf1 to Fmr-1 Promoter Increases in Mouse Brain during Aging. Biochem. Biophys. Res. Commun. 2008, 376, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, S.Y.; El Jeon, S.; Choi, J.H.; Lee, C.J.; Jang, T.Y.; Yun, H.J.; Lee, Y.; Kim, P.; Cho, S.H.; et al. DCLK1 Promotes Colorectal Cancer Stemness and Aggressiveness via the XRCC5/COX2 Axis. Theranostics 2022, 12, 5258–5271. [Google Scholar] [CrossRef]
- Joo, M.; Kwon, M.; Cho, Y.J.; Hu, N.; Pedchenko, T.V.; Sadikot, R.T.; Blackwell, T.S.; Christman, J.W. Lipopolysaccharide-Dependent Interaction between PU.1 and c-Jun Determines Production of Lipocalin-Type Prostaglandin D Synthase and Prostaglandin D2 in Macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L771–L779. [Google Scholar] [CrossRef] [Green Version]
- Lappas, M. Identification of SMAD3 as a Novel Mediator of Inflammation in Human Myometrium In Vitro. Mediat. Inflamm. 2018, 2018, 3140420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Zafar, M.I.; Jüttner, S.; Höcker, M.; Wiedenmann, B. Expression and Molecular Regulation of the Cox2 Gene in Gastroenteropancreatic Neuroendocrine Tumors and Antiproliferation of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs). Med. Sci. Monit. 2018, 24, 8125–8140. [Google Scholar] [CrossRef]
- Cilenti, F.; Barbiera, G.; Caronni, N.; Iodice, D.; Montaldo, E.; Barresi, S.; Lusito, E.; Cuzzola, V.; Vittoria, F.M.; Mezzanzanica, L.; et al. A PGE2-MEF2A Axis Enables Context-Dependent Control of Inflammatory Gene Expression. Immunity 2021, 54, 1665–1682. [Google Scholar] [CrossRef]
- Hausburg, F.; Jung, J.J.; Hoch, M.; Wolfien, M.; Rimmbach, C.; David, R. (Re-)Programming of Subtype Specific Cardiomyocytes. Adv. Drug Deliv. Rev. 2017, 120, 142–167. (In Revision) [Google Scholar] [CrossRef]
- Wahlestedt, M.; Ladopoulos, V.; Hidalgo, I.; Sanchez Castillo, M.; Hannah, R.; Säwén, P.; Wan, H.; Dudenhöffer-Pfeifer, M.; Magnusson, M.; Norddahl, G.L.; et al. Critical Modulation of Hematopoietic Lineage Fate by Hepatic Leukemia Factor. Cell Rep. 2017, 21, 2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Ke, M.; Yue, S.; Xiao, W.; Yan, Y.; Deng, X.; Ying, Q.L.; Li, J.; Ke, B. Blockade of Notch Signaling Promotes Acetaminophen-Induced Liver Injury. Immunol. Res. 2017, 65, 739–749. [Google Scholar] [CrossRef]
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-Inducible Factor 2α Regulates Macrophage Function in Mouse Models of Acute and Tumor Inflammation. J. Clin. Invest. 2010, 120, 2699. [Google Scholar] [CrossRef] [Green Version]
- Veremeyko, T.; Yung, A.W.Y.; Anthony, D.C.; Strekalova, T.; Ponomarev, E.D. Early Growth Response Gene-2 Is Essential for M1 and M2 Macrophage Activation and Plasticity by Modulation of the Transcription Factor CEBPβ. Front. Immunol. 2018, 9, 2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, Y.; Spann, N.J.; Link, V.M.; Muse, E.D.; Strid, T.; Edillor, C.; Kolar, M.J.; Matsuzaka, T.; Hayakawa, S.; Tao, J.; et al. SREBP1 Contributes to Resolution of Pro-Inflammatory TLR4 Signaling by Reprogramming Fatty Acid Metabolism. Cell Metab. 2017, 25, 412–427. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.T.; Chang, H.W.; Wu, M.J.; Lai, Y.T.; Wu, W.C.; Yu, W.C.Y.; Chang, V.H.S. Klf10 Deficiency in Mice Exacerbates Pulmonary Inflammation by Increasing Expression of the Proinflammatory Molecule NPRA. Int. J. Biochem. Cell Biol. 2016, 79, 231–238. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Q.; Li, D.; Li, J.; Aki, D.; Liu, Y.C. The E3 Ligase VHL Controls Alveolar Macrophage Function via Metabolic–Epigenetic Regulation. J. Exp. Med. 2018, 215, 3180. [Google Scholar] [CrossRef]
- Li, S.; Zhao, L.; Li, X.; Shang, G.; Gao, L.; Song, Z.; Li, T. Mir-204 Regulates LPS-Induced A549 Cell Damage by Targeting FOXK2. J. Healthc. Eng. 2021, 2021, 7404671. [Google Scholar] [CrossRef]
- Nejati Moharrami, N.; Tande, E.B.; Ryan, L.; Espevik, T.; Boyartchuk, V. RORα Controls Inflammatory State of Human Macrophages. PLoS ONE 2018, 13, e0207374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Gan, C.P.; Zhang, S.; Zhou, X.K.; Li, X.F.; Wei, Y.Q.; Yang, J.L.; Wu, M. FIP200 Is Involved in Murine Pseudomonas Infection by Regulating HMGB1 Intracellular Translocation. Cell. Physiol. Biochem. 2014, 33, 1733–1744. [Google Scholar] [CrossRef]
- Cochrane, C.R.; Vaghjiani, V.; Szczepny, A.; Jayasekara, W.S.N.; Gonzalez-Rajal, A.; Kikuchi, K.; McCaughan, G.W.; Burgess, A.; Gough, D.J.; Watkins, D.N.; et al. Trp53 and Rb1 Regulate Autophagy and Ligand-Dependent Hedgehog Signaling. J. Clin. Invest. 2020, 130, 4006–4018. [Google Scholar] [CrossRef]
- Moresi, V.; Carrer, M.; Grueter, C.E.; Rifki, O.F.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone Deacetylases 1 and 2 Regulate Autophagy Flux and Skeletal Muscle Homeostasis in Mice. Proc. Natl. Acad. Sci. USA 2012, 109, 1649–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCaffrey, T.A.; Fu, C.; Du, B.; Eksinar, S.; Kent, K.C.; Harry, B.; Kreiger, K.; Rosengart, T.; Cybulsky, M.I.; Silverman, E.S.; et al. High-Level Expression of Egr-1 and Egr-1–Inducible Genes in Mouse and Human Atherosclerosis. J. Clin. Investig. 2000, 105, 653. [Google Scholar] [CrossRef] [Green Version]
- Patino, W.D.; Kang, J.G.; Matoba, S.; Mian, O.Y.; Gochuico, B.R.; Hwang, P.M. Atherosclerotic Plaque Macrophage Transcriptional Regulators Are Expressed in Blood and Modulated by Tristetraprolin. Circ. Res. 2006, 98, 1282–1289. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; An, F.S.; Zhang, W.; Gong, L.; Wei, S.J.; Qin, W.D.; Wang, X.P.; Zhao, Y.X.; Zhang, Y.; Zhang, C.; et al. Inhibition of C-Jun N-Terminal Kinase Attenuates Low Shear Stress–Induced Atherogenesis in Apolipoprotein E–Deficient Mice. Mol. Med. 2011, 17, 990. [Google Scholar] [CrossRef]
- Harja, E.; Bucciarelli, L.G.; Lu, Y.; Stern, D.M.; Zou, Y.S.; Schmidt, A.M.; Yan, S.F. Early Growth Response-1 Promotes Atherogenesis: Mice Deficient in Early Growth Response-1 and Apolipoprotein E Display Decreased Atherosclerosis and Vascular Inflammation. Circ. Res. 2004, 94, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Stula, M.; Orzechowski, H.D.; Gschwend, S.; Vetter, R.; Von Harsdorf, R.; Dietz, R.; Paul, M. Influence of Sustained Mechanical Stress on Egr-1 MRNA Expression in Cultured Human Endothelial Cells. Mol. Cell. Biochem. 2000, 210, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.C. Lipidomic Profiling of Bioactive Lipids by Mass Spectrometry during Microbial Infections. Semin. Immunol. 2013, 25, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Perez-Hernandez, J.; Chiurchiù, V.; Perruche, S.; You, S. Regulation of T-Cell Immune Responses by Pro-Resolving Lipid Mediators. Front. Immunol. 2021, 12, 4767. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Puccetti, A.; Fiore, P.F.; Pelosi, A.; Tinazzi, E.; Patuzzo, G.; Argentino, G.; Moretta, F.; Lunardi, C.; Dolcino, M. Gene Expression Profiling in Behcet’s Disease Indicates an Autoimmune Component in the Pathogenesis of the Disease and Opens New Avenues for Targeted Therapy. J. Immunol. Res. 2018, 2018, 4246965. [Google Scholar] [CrossRef]
- Ma, J.; Wang, J.; Ghoraie, L.S.; Men, X.; Liu, L.; Dai, P. Network-Based Method for Drug Target Discovery at the Isoform Level. Sci. Rep. 2019, 9, 13868. [Google Scholar] [CrossRef] [Green Version]
- Gal-Oz, S.T.; Maier, B.; Yoshida, H.; Seddu, K.; Elbaz, N.; Czysz, C.; Zuk, O.; Stranger, B.E.; Ner-Gaon, H.; Shay, T. ImmGen Report: Sexual Dimorphism in the Immune System Transcriptome. Nat. Commun. 2019, 10, 4295. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Lareau, C.A.; Ramirez, R.N.; Rose, S.A.; Maier, B.; Wroblewska, A.; Desland, F.; Chudnovskiy, A.; Mortha, A.; Dominguez, C.; et al. The Cis-Regulatory Atlas of the Mouse Immune System. Cell 2019, 176, 897–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baras, A.S.; Mitchell, C.J.; Myers, J.R.; Gupta, S.; Weng, L.-C.; Ashton, J.M.; Cornish, T.C.; Pandey, A.; Halushka, M.K. MiRge—A Multiplexed Method of Processing Small RNA-Seq Data to Determine MicroRNA Entropy. PLoS ONE 2015, 10, e0143066. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Cho, K.H. Topological Estimation of Signal Flow in Complex Signaling Networks. Sci. Rep. 2018, 8, 5262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waskom, M.L. Seaborn: Statistical Data Visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoch, M.; Rauthe, J.; Cesnulevicius, K.; Schultz, M.; Lescheid, D.; Wolkenhauer, O.; Chiurchiù, V.; Gupta, S. Cell-Type-Specific Gene Regulatory Networks of Pro-Inflammatory and Pro-Resolving Lipid Mediator Biosynthesis in the Immune System. Int. J. Mol. Sci. 2023, 24, 4342. https://doi.org/10.3390/ijms24054342
Hoch M, Rauthe J, Cesnulevicius K, Schultz M, Lescheid D, Wolkenhauer O, Chiurchiù V, Gupta S. Cell-Type-Specific Gene Regulatory Networks of Pro-Inflammatory and Pro-Resolving Lipid Mediator Biosynthesis in the Immune System. International Journal of Molecular Sciences. 2023; 24(5):4342. https://doi.org/10.3390/ijms24054342
Chicago/Turabian StyleHoch, Matti, Jannik Rauthe, Konstantin Cesnulevicius, Myron Schultz, David Lescheid, Olaf Wolkenhauer, Valerio Chiurchiù, and Shailendra Gupta. 2023. "Cell-Type-Specific Gene Regulatory Networks of Pro-Inflammatory and Pro-Resolving Lipid Mediator Biosynthesis in the Immune System" International Journal of Molecular Sciences 24, no. 5: 4342. https://doi.org/10.3390/ijms24054342