
Delayed TBI-Induced Neuronal Death in the Ipsilateral Hippocampus and Behavioral Deficits in Rats: Influence of Corticosterone-Dependent Survivorship Bias?
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Time Course of Corticosterone during 3 Months after TBI and Mortality
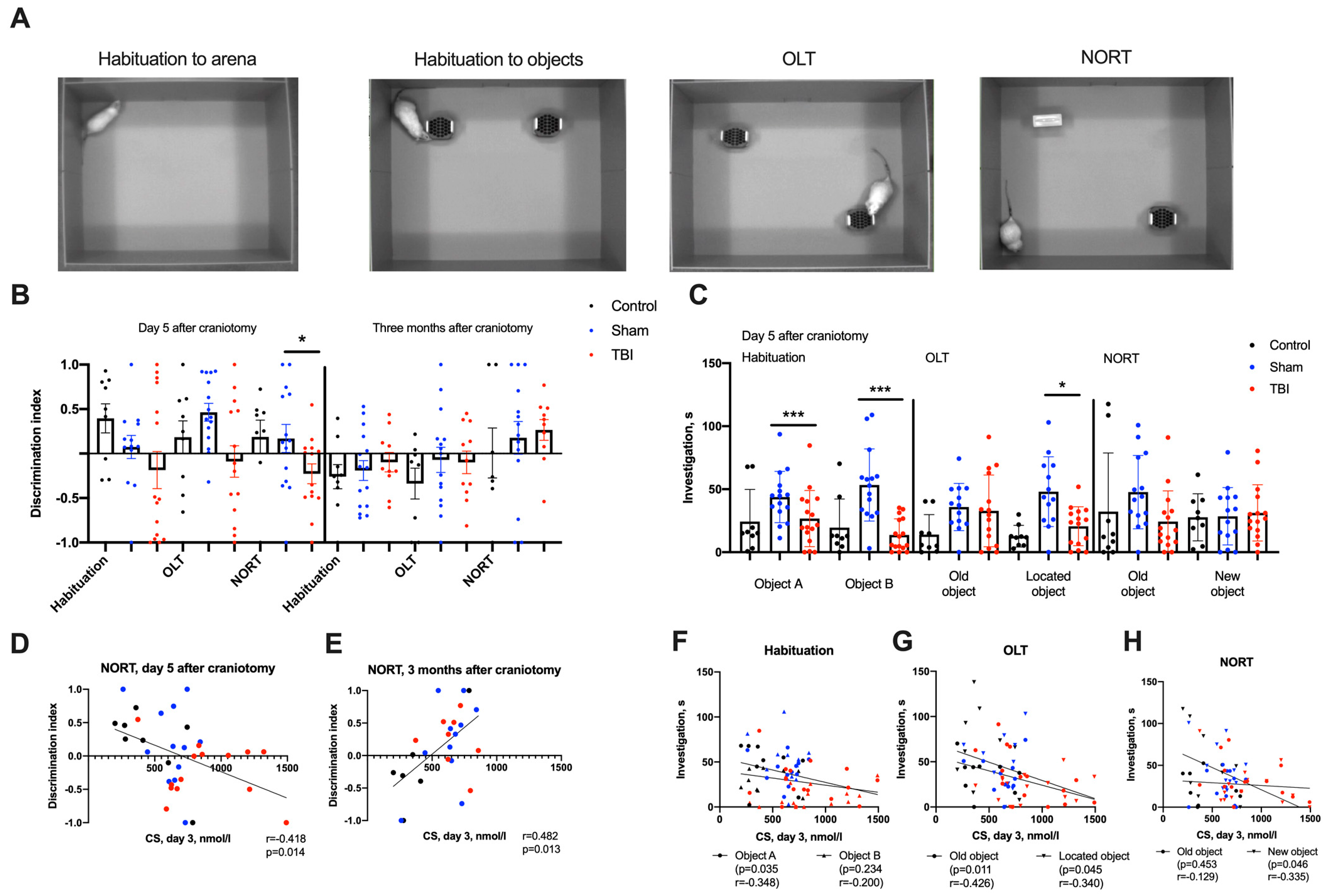
2.2. Behavioral Changes in Acute and Remote Periods of TBI
2.3. Neurodegeneration and Neuroinflammation in the Hippocampus 3 Months after TBI
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Lateral Fluid Percussion Injury Modelling
4.3. Behavior Testing
4.4. Histology, Immunohistochemistry and Morphometry
4.5. Assessment of CS
4.6. Statistical Methods
5. Conclusions
Limitations
- We did not use tissue samples collected from spontaneously dead animals to confirm bilateral hippocampal damage in rats with CS elevation > CS > 860 nmol/L because of the impracticability of standardizing the delay of tissue collection to avoid unbiased assessment.
- We used the same cohort of animals with the same behavioral tests in the acute period of TBI and 3 months after. The lack of possible differences in the second trial of cognitive tests could be possibly explained by experience bias. To avoid bias, we used three groups of animals (control, Sham and TBI), long inter-trial periods for repetitive testing (3 months) and the re-learning paradigm in the Barnes maze test.
- We used a single immunohistochemical marker for each type of glial cell (Iba1 for microglial cells and GFAP for astroglial cells). It was not enough to prove cell types in all visible marker-positive cells, and additional makers might improve the accuracy of the assessment. Immunohistochemistry also cannot be used to assess changes in gene and protein expression in cells without precise immunochemical analysis (Western blot).
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CS | corticosterone |
| DG | dentate gyrus, hippocampal field |
| EPM | elevated plus mase |
| gcl | granular cells layer |
| GCs | glucocorticoids |
| HPA | hypothalamo-pituitary axis |
| LPS | lipopolysaccharide |
| LFPI | lateral fluid percussion brain injury |
| NORT | new object recognition test |
| OF | open field test |
| OLT | object location test |
| pcl | pyramidal cells layer |
| TBI | traumatic brain injury |
References
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Gulyaeva, N.V. Biochemical Mechanisms and Translational Relevance of Hippocampal Vulnerability to Distant Focal Brain Injury: The Price of Stress Response. Biochemistry 2019, 84, 1306–1328. [Google Scholar] [CrossRef] [PubMed]
- Bigler, E.D.; Blatter, D.D.; Anderson, C.V.; Johnson, S.C.; Gale, S.D.; Hopkins, R.O.; Burnett, B. Hippocampal volume in normal aging and traumatic brain injury. Am. J. Neuroradiol. 1997, 18. [Google Scholar]
- Gupta, P.K.; Sayed, N.; Ding, K.; Agostini, M.A.; Van Ness, P.C.; Yablon, S.; Madden, C.; Mickey, B.; D’Ambrosio, R.; Diaz-Arrastia, R. Subtypes of Post-Traumatic Epilepsy: Clinical, Electrophysiological, and Imaging Features. J. Neurotrauma 2014, 31, 1439–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terpstra, A.R.; Girard, T.A.; Colella, B.; Green, R.E.A. Higher Anxiety Symptoms Predict Progressive Hippocampal Atrophy in the Chronic Stages of Moderate to Severe Traumatic Brain Injury. Neurorehabil. Neural Repair 2017, 31, 1063–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, H.J.; Lifshitz, J.; Marklund, N.; Grady, M.S.; Graham, D.I.; Hovda, D.A.; McIntosh, T.K. Lateral Fluid Percussion Brain Injury: A 15-Year Review and Evaluation. J. Neurotrauma 2005, 22, 42–75. [Google Scholar] [CrossRef]
- Hicks, R.; Soares, H.; Smith, D.; McIntosh, T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996, 91, 236–246. [Google Scholar] [CrossRef]
- Cortez, S.C.; McIntosh, T.K.; Noble, L.J. Experimental fluid percussion brain injury: Vascular disruption and neuronal and glial alterations. Brain Res. 1989, 482, 271–282. [Google Scholar] [CrossRef]
- Lescot, T.; Fulla-Oller, L.; Po, C.; Chen, X.R.; Puybasset, L.; Gillet, B.; Plotkine, M.; Meric, P.; Marchand-Leroux, C. Temporal and Regional Changes after Focal Traumatic Brain Injury. J. Neurotrauma 2010, 27, 85–94. [Google Scholar] [CrossRef]
- Komoltsev, I.G.; Volkova, A.A.; Levshina, I.P.; Novikova, M.R.; Manolova, A.O.; Stepanichev, M.; Gulyaeva, N. The Number of IgG-Positive Neurons in the Rat Hippocampus Increases after Dosed Traumatic Brain Injury. Neurochem. J. 2018, 12, 256–261. [Google Scholar] [CrossRef]
- Tran, L.D.; Lifshitz, J.; Witgen, B.M.; Schwarzbach, E.; Cohen, A.S.; Grady, M.S. Response of the Contralateral Hippocampus to Lateral Fluid Percussion Brain Injury. J. Neurotrauma 2006, 23, 1330–1342. [Google Scholar] [CrossRef]
- Aungst, S.L.; Kabadi, S.V.; Thompson, S.M.; Stoica, B.A.; Faden, A.I. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J. Cereb. Blood Flow Metab. 2014, 34, 1223–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lifshitz, J.; Witgen, B.M.; Grady, M.S. Acute cognitive impairment after lateral fluid percussion brain injury recovers by 1 month: Evaluation by conditioned fear response. Behav. Brain Res. 2007, 177, 347–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spain, A.; Daumas, S.; Lifshitz, J.; Rhodes, J.; Andrews, P.J.; Horsburgh, K.; Fowler, J.H. Mild fluid percussion injury in mice produces evolving selective axonal pathology and cognitive deficits relevant to human brain injury. J. Neurotrauma 2010, 27, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Komoltsev, I.G.; Tret’yakova, L.V.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Butuzov, A.V.; Novikova, M.R.; Kvichansky, A.A.; Moiseeva, Y.V.; Onufriev, M.V.; et al. Neuroinflammatory Cytokine Response, Neuronal Death, and Microglial Proliferation in the Hippocampus of Rats during the Early Period after Lateral Fluid Percussion-Induced Traumatic Injury of the Neocortex. Mol. Neurobiol. 2021, 59, 1151–1167. [Google Scholar] [CrossRef] [PubMed]
- Komoltsev, I.G.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Onufriev, M.V.; Moiseeva, J.V.; Novikova, M.R.; Gulyaeva, N.V. Neuroinflammation and Neuronal Loss in the Hippocampus Are Associated with Immediate Posttraumatic Seizures and Corticosterone Elevation in Rats. Int. J. Mol. Sci. 2021, 22, 5883. [Google Scholar] [CrossRef] [PubMed]
- Komoltsev, I.G.; Gulyaeva, N.V. Brain Trauma, Glucocorticoids and Neuroinflammation: Dangerous Liaisons for the Hippocampus. Biomedicines 2022, 10, 1139. [Google Scholar] [CrossRef]
- Bensalah, M.; Donaldson, M.; Aribi, Y.; Iabassen, M.; Cherfi, L.; Nebbal, M.; Medjaher, M.; Haffaf, E.; Abdennebi, B.; Guenane, K.; et al. Cortisol evaluation during the acute phase of traumatic brain injury—A prospective study. Clin. Endocrinol. 2018, 88, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Chandra, A.; Yadav, A.; Ojha, B.K.; Srivastava, C.; Verma, R.; Ali, W. Dynamic change in cortisol levels associated with severity, progression, and survival of patients with traumatic brain injury. Clin. Neurol. Neurosurg. 2022, 222, 107419. [Google Scholar] [CrossRef]
- Chiu, C.-C.; Liao, Y.-E.; Yang, L.-Y.; Wang, J.-Y.; Tweedie, D.; Karnati, H.K.; Greig, N.H.; Wang, J.-Y. Neuroinflammation in animal models of traumatic brain injury. J. Neurosci. Methods 2016, 272, 38–49. [Google Scholar] [CrossRef] [Green Version]
- Grady, M.S.; Charleston, J.S.; Maris, D.; Witgen, B.M.; Lifshitz, J. Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: Analysis by stereological estimation. J. Neurotrauma 2003, 20, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.; Houle, S.; Cotter, C.; Zimomra, Z.; Martens, K.M.; Vonder Haar, C.; Kokiko-Cochran, O.N. Lateral Fluid Percussion Injury Causes Sex-Specific Deficits in Anterograde but Not Retrograde Memory. Front. Behav. Neurosci. 2022, 16, 806598. [Google Scholar] [CrossRef] [PubMed]
- Fedor, M.; Berman, R.F.; Muizelaar, J.P.; Lyeth, B.G. Hippocampal θ dysfunction after lateral fluid percussion injury. J. Neurotrauma 2010, 27, 1605–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, D.R.; Law, L.M.; Young, C.; Fuentes, A.; Truran, S.; Karamanova, N.; Bell, L.C.; Turner, G.; Emerson, H.; Mastroeni, D.; et al. Chronic Cognitive and Cerebrovascular Function after Mild Traumatic Brain Injury in Rats. J. Neurotrauma 2022, 39, 1429–1441. [Google Scholar] [CrossRef]
- Tapp, Z.M.; Godbout, J.P.; Kokiko-Cochran, O.N. A Tilted Axis: Maladaptive Inflammation and HPA Axis Dysfunction Contribute to Consequences of TBI. Front. Neurol. 2019, 10, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef]
- Sorrells, S.F.; Caso, J.R.; Munhoz, C.D.; Sapolsky, R.M. The Stressed CNS: When Glucocorticoids Aggravate Inflammation. Neuron 2009, 64, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolshakov, A.P.; Tret’yakova, L.V.; Kvichansky, A.A.; Gulyaeva, N.V. Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation. Biochemistry 2021, 86, 156–167. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Functional Neurochemistry of the Ventral and Dorsal Hippocampus: Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem. Res. 2019, 44, 1306–1322. [Google Scholar] [CrossRef]
- Tretyakova, L.V.; Kvichansky, A.A.; Bolshakov, A.P.; Gulyaeva, N.V. Dexamethasone Modulates Lipopolysaccharide-Induced Expression of Proinflammatory Cytokines in Rat Hippocampus. Neurochem. J. 2021, 15, 302–307. [Google Scholar] [CrossRef]
- Skupio, U.; Tertil, M.; Sikora, M.; Golda, S.; Wawrzczak-Bargiela, A.; Przewlocki, R. Behavioral and molecular alterations in mice resulting from chronic treatment with dexamethasone: Relevance to depression. Neuroscience 2015, 286, 141–150. [Google Scholar] [CrossRef]
- Frank, M.G.; Hershman, S.A.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Chronic exposure to exogenous glucocorticoids primes microglia to pro-inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology 2014, 40, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Fenn, A.M.; Gensel, J.C.; Huang, Y.; Popovich, P.G.; Lifshitz, J.; Godbout, J.P. Immune Activation Promotes Depression 1 Month After Diffuse Brain Injury: A Role for Primed Microglia. Biol. Psychiatry 2014, 76, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Kloet, E.R.; Meijer, O.C.; de Nicola, A.F.; de Rijk, R.H.; Joëls, M. Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front. Neuroendocrinol. 2018, 49, 124–145. [Google Scholar] [CrossRef] [PubMed]
- Sorrells, S.F.; Munhoz, C.D.; Manley, N.C.; Yen, S.; Sapolsky, R.M. Glucocorticoids Increase Excitotoxic Injury and Inflammation in the Hippocampus of Adult Male Rats. Neuroendocrinology 2014, 100, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Rowe, R.K.; Ortiz, J.B.; Thomas, T.C. Mild and Moderate Traumatic Brain Injury and Repeated Stress Affect Corticosterone in the Rat. Neurotrauma Rep. 2020, 1, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Elston, D.M. Survivorship bias. J. Am. Acad. Dermatol. 2021. [Google Scholar] [CrossRef]
- Ho, A.M.; Dion, P.W.; Yeung, J.H.; Joynt, G.M.; Lee, A.; Ng, C.S.; Chang, A.; So, F.L.; Cheung, C.W. Simulation of survivorship bias in observational studies on plasma to red blood cell ratios in massive transfusion for trauma. Br. J. Surg. 2012, 99 (Suppl. 1), 132–139. [Google Scholar] [CrossRef]
- Ng, C.S.; Ho, A.M.; Underwood, M.J.; Graham, T.R. Survivorship bias after military thoracic injuries. World J. Surg. 2011, 35, 2826–2827, author reply 2828. [Google Scholar] [CrossRef]
- Scheen, A.J. Possible survivorship bias rather than reverse causality in EMPA-REG OUTCOME. Diabetes Res. Clin. Pract. 2017, 127, 290. [Google Scholar] [CrossRef]
- Toi, Y.; Sugawara, S. Survivorship Bias in Analyses of Immune Checkpoint Inhibitor Trials-In Reply. JAMA Oncol. 2019, 5, 1226–1227. [Google Scholar] [CrossRef] [PubMed]
- Czeisler, M.É.; Wiley, J.F.; Czeisler, C.A.; Rajaratnam, S.M.W.; Howard, M.E. Uncovering survivorship bias in longitudinal mental health surveys during the COVID-19 pandemic. Epidemiol. Psychiatr. Sci. 2021, 30, e45. [Google Scholar] [CrossRef] [PubMed]
- Aydin, E.; Torlak, N.; Haberman, B.; Lim, F.Y.; Peiro, J.L. The Survivorship Bias in Congenital Diaphragmatic Hernia. Children 2022, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, M.C.; Hochman, S.M.; Milne, W.K.; Swaminathan, A.K. Immediate Versus Delayed Cardiac Catheterization in Post-Arrest Patients without ST Elevation Myocardial Infarction: Is Survivorship Bias an Important Influence?: March 2022 Annals of Emergency Medicine Journal Club. Ann. Emerg. Med. 2022, 79, 315–331. [Google Scholar]
- Bermúdez-Guzmán, L.; Jimenez-Huezo., G.; Arguedas, A.; Leal, A. Mutational survivorship bias: The case of PNKP. PLoS ONE 2020, 15, e0237682. [Google Scholar] [CrossRef]
- Walf, A.A.; Frye, C.A. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat. Protoc. 2007, 2, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Denninger, J.K.; Smith, B.M.; Kirby, E.D. Novel Object Recognition and Object Location Behavioral Testing in Mice on a Budget. J. Vis. Exp. 2018, 2018. [Google Scholar] [CrossRef]
- Barnes, C.A.; Nadel, L.; Honig, W.K. Spatial memory deficit in senescent rats. Can. J. Psychol./Rev. Can. Psychol. 1980, 34, 29–39. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komoltsev, I.; Shalneva, D.; Kostyunina, O.; Volkova, A.; Frankevich, S.; Shirobokova, N.; Belikova, A.; Balan, S.; Chizhova, O.; Salyp, O.; et al. Delayed TBI-Induced Neuronal Death in the Ipsilateral Hippocampus and Behavioral Deficits in Rats: Influence of Corticosterone-Dependent Survivorship Bias? Int. J. Mol. Sci. 2023, 24, 4542. https://doi.org/10.3390/ijms24054542
Komoltsev I, Shalneva D, Kostyunina O, Volkova A, Frankevich S, Shirobokova N, Belikova A, Balan S, Chizhova O, Salyp O, et al. Delayed TBI-Induced Neuronal Death in the Ipsilateral Hippocampus and Behavioral Deficits in Rats: Influence of Corticosterone-Dependent Survivorship Bias? International Journal of Molecular Sciences. 2023; 24(5):4542. https://doi.org/10.3390/ijms24054542
Chicago/Turabian StyleKomoltsev, Ilia, Daria Shalneva, Olga Kostyunina, Aleksandra Volkova, Stepan Frankevich, Natalia Shirobokova, Anastasia Belikova, Sofia Balan, Olesya Chizhova, Olga Salyp, and et al. 2023. "Delayed TBI-Induced Neuronal Death in the Ipsilateral Hippocampus and Behavioral Deficits in Rats: Influence of Corticosterone-Dependent Survivorship Bias?" International Journal of Molecular Sciences 24, no. 5: 4542. https://doi.org/10.3390/ijms24054542