

Molecular Basis beyond Interrelated Bone Resorption/Regeneration in Periodontal Diseases: A Concise Review

 , , , , ,
, , , , ,  and
and 
Abstract
:1. Introduction
2. Bone Cells
3. The Effect of the Periodontal Inflammatory Milieu on Stem Cells
4. Cytokines Effect on Osteoblastogenesis in Periodontal Inflammation
4.1. IL-1
4.2. IL-6
4.3. IL-10
4.4. IL-17
4.5. TNF-α
4.6. IL-1β and TNF-α
4.7. IFN-γ
4.8. TGF-β
4.9. Other Cytokines and Osteoblastogenesis
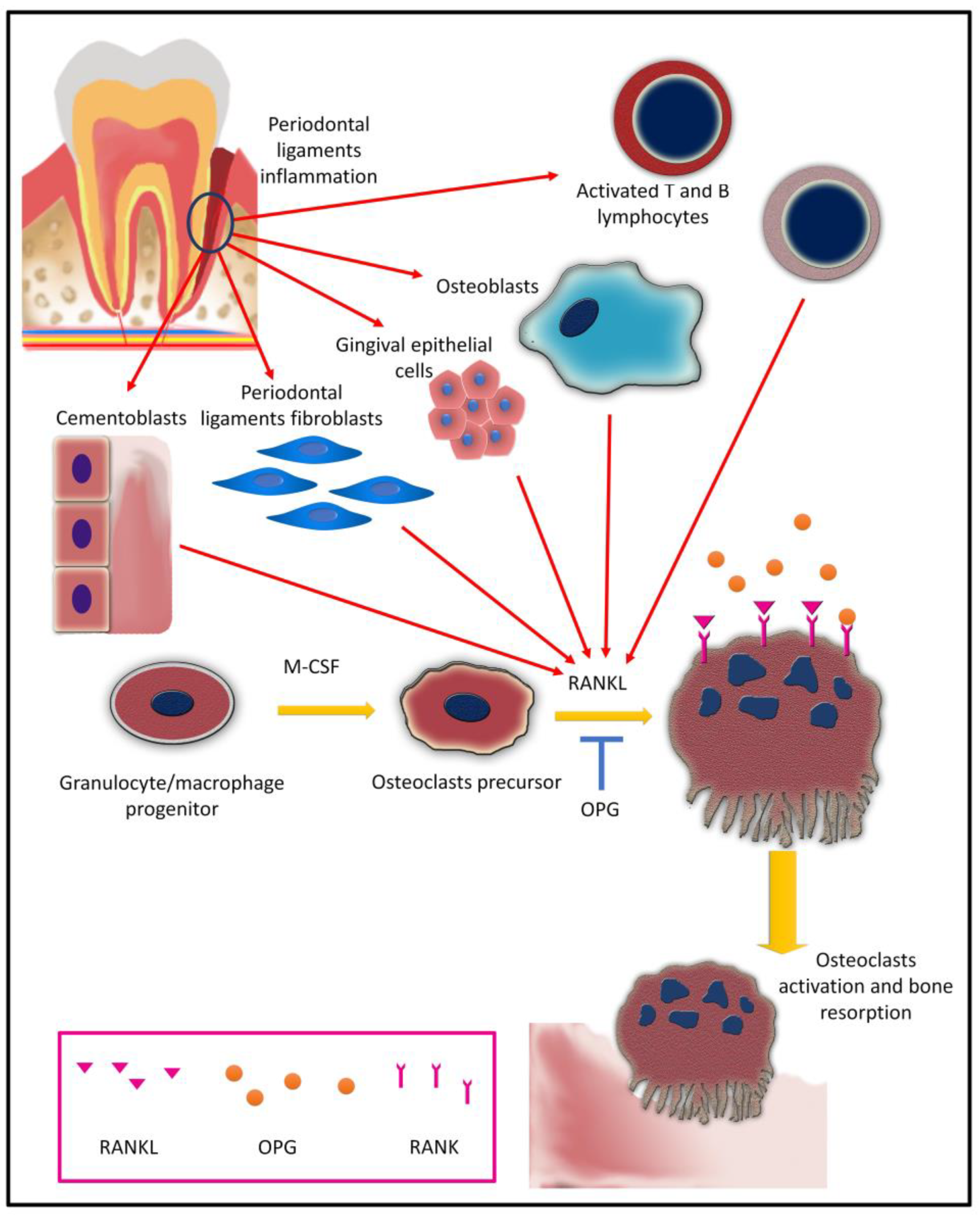
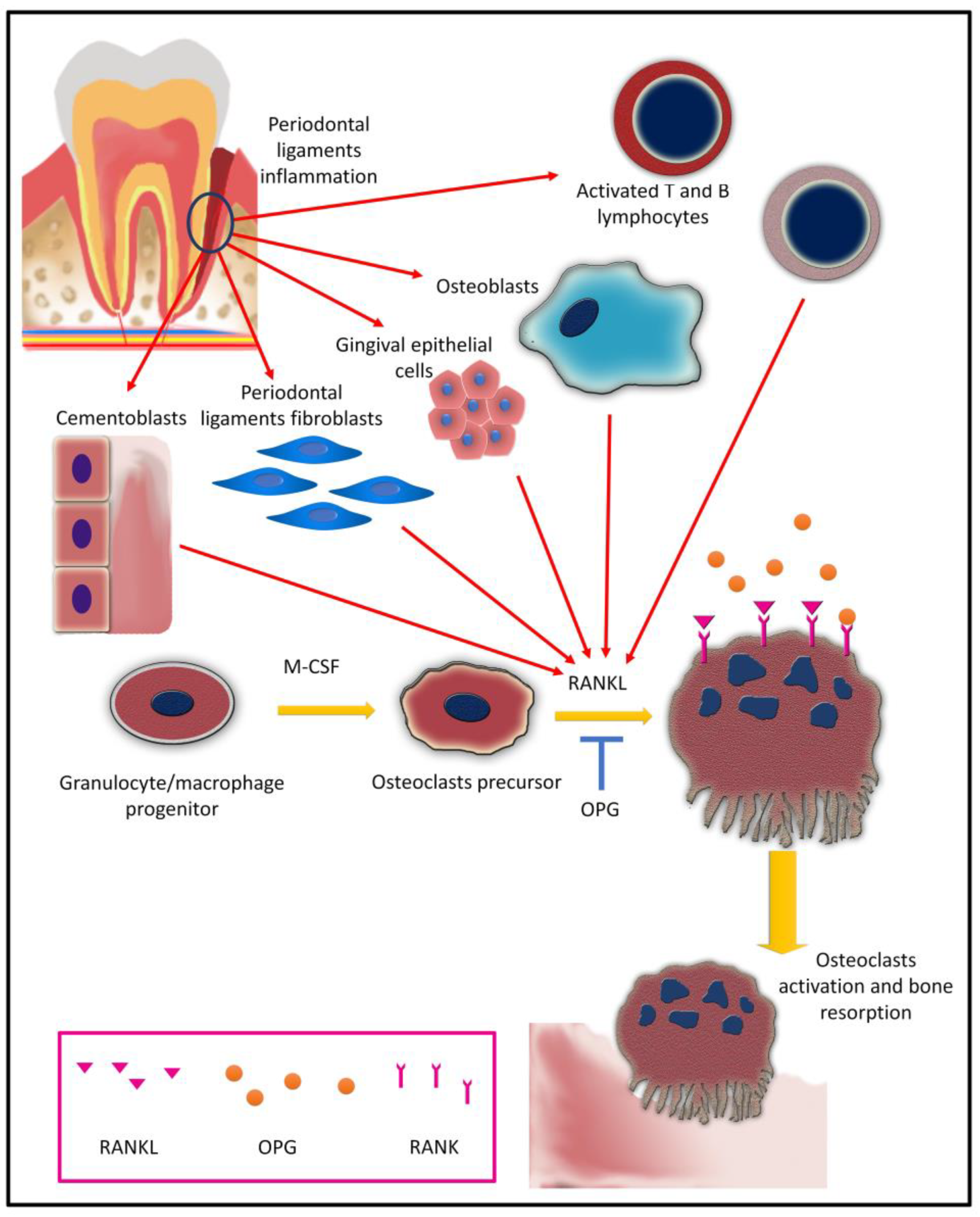
5. Osteoclasts and Osteoclastogenesis in Periodontal Inflammation
5.1. Osteoclast Differentiation (Osteoclastogenesis)
5.2. Cytokines and Osteoclastogenesis in Periodontitis
5.2.1. IL-1 Super Family and Osteoclastogenesis
5.2.2. IL-6 and Osteoclastogenesis
5.2.3. IL-17A and Osteoclastogenesis
5.2.4. IL-22 and Osteoclastogenesis
5.2.5. Other Interleukins and Osteoclastogenesis
5.2.6. TNF and Osteoclastogenesis
5.3. Micro RNA and Osteoclastogenesis in Periodontitis
5.4. Bacterial Factors and Osteoclastogenesis in Periodontitis
5.4.1. Periodontal Bacteria
5.4.2. Lipopolysaccharides
5.4.3. Peptidoglycan
6. Osteocytes and Periodontal Inflammation
6.1. Crosstalk among Osteocytes, Osteoclasts, and Osteoblasts
6.2. Osteocytes in Periodontitis
6.3. Osteocyte Senescence in Periodontitis
6.4. Apoptotic Osteocyte in Periodontitis
7. Clinical Implications
7.1. The Potentiality of Using the GCF and the Salivary Biomarkers as Diagnostic Tools for Periodontal Diseases
7.2. Role of Immunomodulation as a Therapeutic Strategy in Periodontitis
8. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ALP | Alkaline phosphatase |
| ALT | Alanine aminotransferase |
| AST | Aspartate aminotransferase |
| BMP | Bone morphogenetic proteins |
| c-Fms | Colony-stimulating factor-1 receptor |
| CRP | C-reactive protein |
| COL-I | Type I collagen |
| CXCL8 | CXC-chemokine ligand 8 |
| DKK1 | Dickkopf-related protein 1 |
| DMSCs | Dental mesenchymal stem cells |
| DPSCs | Dental pulp stem cells |
| ECM | Extracellular matrix |
| ERK | Extracellular signal-regulated kinase |
| GCF | Gingival crevicular fluid |
| GM-CSF | Granulocyte–macrophage colony-stimulating factor |
| GMSCs | Gingival mesenchymal stem cells |
| Grb 2 | Growth factor receptor bound protein 2 |
| ICTP | Carboxyterminal telopeptide pyridinoline cross-links of type I collagen |
| IDO | Indoleamine 2,3-dioxygenase |
| IFN-γ | Interferon-gamma |
| IL | Interleukin |
| IL-1ra | IL-1 receptor antagonist |
| IL-6R | IL-6 receptor |
| JAK | Janus kinase |
| JNK | c-Jun N-terminal kinase |
| LDH | Lactate dehydrogenase |
| LPS | Lipopolysaccharide |
| LRP5/6 | Lipoprotein receptor-related protein-5/6 |
| MAPKs | Mitogen-activated protein kinases |
| MCP-1 | Monocyte chemoattractant protein-1 |
| M-CSF | Macrophage colony-stimulating factor |
| MIP-1 α | Macrophage inflammatory protein-1 alpha |
| MMPs | Matrix metalloproteinases |
| NFATc1 | Nuclear factor of activated T cells 1 |
| NF-κB | Nuclear factor-kappa B |
| NOD | Nucleotide-binding oligomerization domain |
| OC | Osteocalcin |
| ON | Osteonectin |
| OPG | Osteoprotegrin |
| OPN | Osteopontin |
| OSCAR | Osteoclasts-associated receptor |
| OSX | Osterix |
| P. gingivalis-LPS | Porphyromonas gingivalis lipopolysaccharides |
| PDLSCs | Periodontal ligament stem cells |
| PGE2 | Prostaglandin E2 |
| PGN | Peptidoglycan |
| PI3K | Phosphoinositide 3-kinase |
| PTX3 | Pentraxin3 |
| RANKL | Receptor activator of nuclear factor kappa-B ligand |
| ROS | Reactive oxygen species |
| RUNX2 | Runt-related transcription factor 2 |
| SCAP | Stem cells from the apical papilla |
| SHP2 | Src homology 2 |
| sIL-6R | Soluble interleukin-6 receptor |
| STAT | Signal transducer and activator of transcription |
| TGF-β | Transforming growth factor-β |
| TH | T helper |
| TLRs | Toll-like receptors |
| TNF | Tumor necrosis factor |
| TRAFs | TNF receptor-associated factors |
| TRAP | Tartrate-resistant acid phosphatase |
| TREM-1 | Triggering receptor expressed on myeloid cells-1 |
References
- Gulati, M.; Anand, V.; Jain, N.; Anand, B.; Bahuguna, R.; Govila, V.; Rastogi, P. Essentials of periodontal medicine in preventive medicine. Int. J. Prev. Med. 2013, 4, 988–994. [Google Scholar] [PubMed]
- Eke, P.I.; Dye, B.A.; Wei, L.; Slade, G.D.; Thornton-Evans, G.O.; Borgnakke, W.S.; Taylor, G.W.; Page, R.C.; Beck, J.D.; Genco, R.J. Update on Prevalence of Periodontitis in Adults in the United States: NHANES 2009 to 2012. J. Periodontol. 2015, 86, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Buduneli, N. Biomarkers in Periodontal Health and Disease; Springer International Publishing: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Hajishengallis, G. Immunomicrobial pathogenesis of periodontitis: Keystones, pathobionts, and host response. Trends Immunol. 2014, 35, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurdziński, K.T.; Potempa, J.; Grabiec, A.M. Epigenetic regulation of inflammation in periodontitis: Cellular mechanisms and therapeutic potential. Clin. Epigenetics 2020, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, D.; Zhang, Y.; Li, M. Inflammation, mesenchymal stem cells and bone regeneration. Histochem. Cell Biol. 2018, 149, 393–404. [Google Scholar] [CrossRef]
- Alfakry, H.; Malle, E.; Koyani, C.N.; Pussinen, P.J.; Sorsa, T. Neutrophil proteolytic activation cascades: A possible mechanistic link between chronic periodontitis and coronary heart disease. Innate Immun. 2016, 22, 85–99. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Chakravarti, A.; Raquil, M.-A.; Tessier, P.; Poubelle, P.E. Surface RANKL of Toll-like receptor 4-stimulated human neutrophils activates osteoclastic bone resorption. Blood 2009, 114, 1633–1644. [Google Scholar] [CrossRef]
- Berglundh, T.; Donati, M.; Zitzmann, N. B cells in periodontitis—friends or enemies? Periodontol. 2000 2007, 45, 51–66. [Google Scholar] [CrossRef]
- Sun, X.; Gao, J.; Meng, X.; Lu, X.; Zhang, L.; Chen, R. Polarized Macrophages in Periodontitis: Characteristics, Function, and Molecular Signaling. Front. Immunol. 2021, 12, 4953. [Google Scholar] [CrossRef]
- Pan, W.; Wang, Q.; Chen, Q. The cytokine network involved in the host immune response to periodontitis. Int. J. Oral Sci. 2019, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florencio-Silva, R.; Sasso, G.R.d.S.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of bone tissue: Structure, function, and factors that influence bone cells. BioMed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, N.; Rosenberg, O.; Soudry, M. Osteoblasts in bone physiology—Mini review. Rambam Maimonides Med. J. 2012, 3, e0013. [Google Scholar] [CrossRef] [Green Version]
- Fierro, F.A.; Nolta, J.A.; Adamopoulos, I.E. Concise review: Stem cells in osteoimmunology. Stem Cells 2017, 35, 1461–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmagid, S.M.; Sondag, G.R.; Moussa, F.M.; Belcher, J.Y.; Yu, B.; Stinnett, H.; Novak, K.; Mbimba, T.; Khol, M.; Hankenson, K.D. Mutation in osteoactivin promotes receptor activator of NFκB ligand (RANKL)-mediated osteoclast differentiation and survival but inhibits osteoclast function. J. Biol. Chem. 2015, 290, 20128–20146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsimbri, P. The biology of normal bone remodelling. Eur. J. Cancer Care 2017, 26, e12740. [Google Scholar] [CrossRef] [PubMed]
- Cremasco, V.; Decker, C.E.; Stumpo, D.; Blackshear, P.J.; Nakayama, K.I.; Nakayama, K.; Lupu, T.S.; Graham, D.B.; Novack, D.V.; Faccio, R. Protein kinase C–delta deficiency perturbs bone homeostasis by selective uncoupling of cathepsin K secretion and ruffled border formation in osteoclasts. J. Bone Miner. Res. 2012, 27, 2452–2463. [Google Scholar] [CrossRef] [Green Version]
- Robling, A.G.; Bonewald, L.F. The osteocyte: New insights. Annu. Rev. Physiol. 2020, 82, 485–506. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Chen, J.; Liu, S.; Jin, Y. Stem cell-based bone and dental regeneration: A view of microenvironmental modulation. Int. J. Oral Sci. 2019, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Monjaraz, B.; Santiago-Osorio, E.; Monroy-García, A.; Ledesma-Martínez, E.; Mendoza-Núñez, V.M. Mesenchymal Stem Cells of Dental Origin for Inducing Tissue Regeneration in Periodontitis: A Mini-Review. Int. J. Mol. Sci. 2018, 19, 944. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhou, L.; Zhou, C.; Zhang, S.; Jing, J.; Xie, L.; Sun, N.; Duan, X.; Jing, W.; Liang, X.; et al. GDF11 decreases bone mass by stimulating osteoclastogenesis and inhibiting osteoblast differentiation. Nat. Commun. 2016, 7, 12794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalisserry, E.P.; Nam, S.Y.; Park, S.H.; Anil, S. Therapeutic potential of dental stem cells. J. Tissue Eng. 2017, 8, 2041731417702531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.C.; Wang, M.Y.; Zhang, S.W.; Wu, Y.S.; Zhou, C.C.; Zheng, R.X.; Shao, B.; Wang, Y.; Xie, L.; Liu, W.Q.; et al. Ubiquitin-specific protease USP34 controls osteogenic differentiation and bone formation by regulating BMP2 signaling. EMBO J. 2018, 37, e99398. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xie, J.; Wang, C.; Zhong, D.; Xie, L.; Fang, H. Immunomodulatory Properties of Stem Cells in Periodontitis: Current Status and Future Prospective. Stem Cells Int. 2020, 2020, 9836518. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Jin, L.; Ma, P.; Fan, Z.; Wang, S. Allogeneic stem cells from deciduous teeth in treatment for periodontitis in miniature swine. J. Periodontol. 2014, 85, 845–851. [Google Scholar] [CrossRef]
- Tang, H.N.; Xia, Y.; Yu, Y.; Wu, R.X.; Gao, L.N.; Chen, F.M. Stem cells derived from “inflamed” and healthy periodontal ligament tissues and their sheet functionalities: A patient-matched comparison. J. Clin. Periodontol. 2016, 43, 72–84. [Google Scholar] [CrossRef]
- Liu, N.; Shi, S.; Deng, M.; Tang, L.; Zhang, G.; Liu, N.; Ding, B.; Liu, W.; Liu, Y.; Shi, H.; et al. High levels of β-catenin signaling reduce osteogenic differentiation of stem cells in inflammatory microenvironments through inhibition of the noncanonical Wnt pathway. J. Bone Miner. Res. 2011, 26, 2082–2095. [Google Scholar] [CrossRef]
- Liu, D.; Xu, J.; Liu, O.; Fan, Z.; Liu, Y.; Wang, F.; Ding, G.; Wei, F.; Zhang, C.; Wang, S. Mesenchymal stem cells derived from inflamed periodontal ligaments exhibit impaired immunomodulation. J. Clin. Periodontol. 2012, 39, 1174–1182. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Tan, J.; Wang, T.; Wang, Q. The immunomodulatory properties of periodontal ligament stem cells isolated from inflamed periodontal granulation. Cells Tissues Organs 2014, 199, 256–265. [Google Scholar] [CrossRef]
- Qiu, J.; Wang, X.; Zhou, H.; Zhang, C.; Wang, Y.; Huang, J.; Liu, M.; Yang, P.; Song, A. Enhancement of periodontal tissue regeneration by conditioned media from gingiva-derived or periodontal ligament-derived mesenchymal stem cells: A comparative study in rats. Stem Cell Res. Ther. 2020, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- Alongi, D.J.; Yamaza, T.; Song, Y.; Fouad, A.F.; Romberg, E.E.; Shi, S.; Tuan, R.S.; Huang, G.T. Stem/progenitor cells from inflamed human dental pulp retain tissue regeneration potential. Regen. Med. 2010, 5, 617–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attar, A.; Eslaminejad, M.B.; Tavangar, M.S.; Karamzadeh, R.; Dehghani-Nazhvani, A.; Ghahramani, Y.; Malekmohammadi, F.; Hosseini, S.M. Dental pulp polyps contain stem cells comparable to the normal dental pulps. J. Clin. Exp. Dent. 2014, 6, e53–e59. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Nan, X.; Zhong, T.Y.; Li, T.; Li, A. Treatment of Periodontal Bone Defects with Stem Cells from Inflammatory Dental Pulp Tissues in Miniature Swine. Tissue Eng. Regen. Med. 2019, 16, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, L.; Mauceri, R.; Coppola, A.; Pitrone, M.; Pizzo, G.; Campisi, G.; Pizzolanti, G.; Giordano, C. Mesenchymal stem cells derived from inflamed dental pulpal and gingival tissue: A potential application for bone formation. Stem Cell Res. Ther. 2017, 8, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Jiang, Y.; Huang, X.; Liu, Q.; Zhang, Y.; Luo, W.; Zhang, F.; Zhou, P.; Lin, J.; Zhang, H. Pro-Inflammatory Cytokine TNF-α Attenuates BMP9-Induced Osteo/Odontoblastic Differentiation of the Stem Cells of Dental Apical Papilla (SCAPs). Cell. Physiol. Biochem. 2017, 41, 1725–1735. [Google Scholar] [CrossRef] [Green Version]
- Whiting, D.; Chung, W.O.; Johnson, J.D.; Paranjpe, A. Characterization of the Cellular Responses of Dental Mesenchymal Stem Cells to the Immune System. J. Endod. 2018, 44, 1126–1131. [Google Scholar] [CrossRef]
- Stadler, A.F.; Angst, P.D.; Arce, R.M.; Gomes, S.C.; Oppermann, R.V.; Susin, C. Gingival crevicular fluid levels of cytokines/chemokines in chronic periodontitis: A meta-analysis. J. Clin. Periodontol. 2016, 43, 727–745. [Google Scholar] [CrossRef]
- Zheng, X.Y.; Mao, C.Y.; Qiao, H.; Zhang, X.; Yu, L.; Wang, T.Y.; Lu, E.Y. Plumbagin suppresses chronic periodontitis in rats via down-regulation of TNF-α, IL-1β and IL-6 expression. Acta Pharmacol. Sin. 2017, 38, 1150–1160. [Google Scholar] [CrossRef] [Green Version]
- Sonomoto, K.; Yamaoka, K.; Oshita, K.; Fukuyo, S.; Zhang, X.; Nakano, K.; Okada, Y.; Tanaka, Y. Interleukin-1β induces differentiation of human mesenchymal stem cells into osteoblasts via the Wnt-5a/receptor tyrosine kinase-like orphan receptor 2 pathway. Arthritis Rheum. 2012, 64, 3355–3363. [Google Scholar] [CrossRef]
- Hoogduijn, M.J.; Popp, F.; Verbeek, R.; Masoodi, M.; Nicolaou, A.; Baan, C.; Dahlke, M.H. The immunomodulatory properties of mesenchymal stem cells and their use for immunotherapy. Int. Immunopharmacol. 2010, 10, 1496–1500. [Google Scholar] [CrossRef]
- Redlich, K.; Smolen, J.S. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 2012, 11, 234–250. [Google Scholar] [CrossRef]
- Zwerina, J.; Redlich, K.; Polzer, K.; Joosten, L.; Krönke, G.; Distler, J.; Hess, A.; Pundt, N.; Pap, T.; Hoffmann, O.; et al. TNF-induced structural joint damage is mediated by IL-1. Proc. Natl. Acad. Sci. USA 2007, 104, 11742–11747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Cai, B.; Zhu, W.; Shi, J.; Wang, Y.; Si, M. IL-1 Receptor Antagonist Protects the Osteogenesis Capability of Gingival-Derived Stem/Progenitor Cells under Inflammatory Microenvironment Induced by Porphyromonas gingivalis Lipopolysaccharides. Stem Cells Int. 2021, 2021, 6638575. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.T.; Lee, S.M.; Kou, X.; Karabucak, B. The Role of Interleukin 6 in Osteogenic and Neurogenic Differentiation Potentials of Dental Pulp Stem Cells. J. Endod. 2019, 45, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K.; Komaki, M.; Mimori, K.; Leon, E.; Izumi, Y.; Ishikawa, I. IL-6 induces osteoblastic differentiation of periodontal ligament cells. J. Dent. Res. 2008, 87, 937–942. [Google Scholar] [CrossRef]
- Taguchi, Y.; Yamamoto, M.; Yamate, T.; Lin, S.C.; Mocharla, H.; DeTogni, P.; Nakayama, N.; Boyce, B.F.; Abe, E.; Manolagas, S.C. Interleukin-6-type cytokines stimulate mesenchymal progenitor differentiation toward the osteoblastic lineage. Proc. Assoc. Am. Physicians 1998, 110, 559–574. [Google Scholar]
- Xie, Z.; Tang, S.; Ye, G.; Wang, P.; Li, J.; Liu, W.; Li, M.; Wang, S.; Wu, X.; Cen, S.; et al. Interleukin-6/interleukin-6 receptor complex promotes osteogenic differentiation of bone marrow-derived mesenchymal stem cells. Stem Cell Res. Ther. 2018, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Bakker, A.D.; Kulkarni, R.N.; Klein-Nulend, J.; Lems, W.F. IL-6 alters osteocyte signaling toward osteoblasts but not osteoclasts. J. Dent. Res. 2014, 93, 394–399. [Google Scholar] [CrossRef] [Green Version]
- Kaneshiro, S.; Ebina, K.; Shi, K.; Higuchi, C.; Hirao, M.; Okamoto, M.; Koizumi, K.; Morimoto, T.; Yoshikawa, H.; Hashimoto, J. IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/Akt2 pathways in vitro. J. Bone Miner. Metab. 2014, 32, 378–392. [Google Scholar] [CrossRef]
- Hughes, F.J.; Howells, G.L. Interleukin-6 inhibits bone formation in vitro. Bone Miner. 1993, 21, 21–28. [Google Scholar] [CrossRef]
- De Benedetti, F.; Rucci, N.; Del Fattore, A.; Peruzzi, B.; Paro, R.; Longo, M.; Vivarelli, M.; Muratori, F.; Berni, S.; Ballanti, P.; et al. Impaired skeletal development in interleukin-6-transgenic mice: A model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 2006, 54, 3551–3563. [Google Scholar] [CrossRef] [PubMed]
- Dresner-Pollak, R.; Gelb, N.; Rachmilewitz, D.; Karmeli, F.; Weinreb, M. Interleukin 10-deficient mice develop osteopenia, decreased bone formation, and mechanical fragility of long bones. Gastroenterology 2004, 127, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Hou, L.; Belani, A.; Wang, C.Y.; Uchiyama, T.; Müller, R.; Stashenko, P. IL-10, but not IL-4, suppresses infection-stimulated bone resorption in vivo. J. Immunol. 2000, 165, 3626–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; You, H.; Qin, N.; Zuo, W. Interleukin-10 Modulates the Metabolism and Osteogenesis of Human Dental Pulp Stem Cells. Cell. Reprogramming 2021, 23, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Zhang, C.; Jin, L.; Yang, Y. IL-17 alters the mesenchymal stem cell niche towards osteogenesis in cooperation with osteocytes. J. Cell. Physiol. 2020, 235, 4466–4480. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Tan, J.; Lei, L.; Sun, W.; Wu, Y.; Ding, P.; Chen, L. The positive effects of secreting cytokines IL-17 and IFN-γ on the early-stage differentiation and negative effects on the calcification of primary osteoblasts in vitro. Int. Immunopharmacol. 2018, 57, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Croes, M.; Öner, F.C.; van Neerven, D.; Sabir, E.; Kruyt, M.C.; Blokhuis, T.J.; Dhert, W.J.A.; Alblas, J. Proinflammatory T cells and IL-17 stimulate osteoblast differentiation. Bone 2016, 84, 262–270. [Google Scholar] [CrossRef]
- Nam, D.; Mau, E.; Wang, Y.; Wright, D.; Silkstone, D.; Whetstone, H.; Whyne, C.; Alman, B. T-lymphocytes enable osteoblast maturation via IL-17F during the early phase of fracture repair. PLoS ONE 2012, 7, e40044. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Seo, S.J.; Kim, J.Y.; Kim, Y.G.; Lee, Y. IL-17 promotes osteoblast differentiation, bone regeneration, and remodeling in mice. Biochem. Biophys. Res. Commun. 2020, 524, 1044–1050. [Google Scholar] [CrossRef]
- Zhang, J.R.; Pang, D.D.; Tong, Q.; Liu, X.; Su, D.F.; Dai, S.M. Different Modulatory Effects of IL-17, IL-22, and IL-23 on Osteoblast Differentiation. Mediat. Inflamm. 2017, 2017, 5950395. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Park, J.W.; Lee, J.M.; Suh, J.Y.; Lee, J.K.; Chang, B.S.; Um, H.S.; Kim, J.Y.; Lee, Y. IL-17 inhibits osteoblast differentiation and bone regeneration in rat. Arch. Oral Biol. 2014, 59, 897–905. [Google Scholar] [CrossRef]
- Đorđević, I.O.; Kukolj, T.; Krstić, J.; Trivanović, D.; Obradović, H.; Santibañez, J.F.; Mojsilović, S.; Ilić, V.; Bugarski, D.; Jauković, A. The inhibition of periodontal ligament stem cells osteogenic differentiation by IL-17 is mediated via MAPKs. Int. J. Biochem. Cell Biol. 2016, 71, 92–101. [Google Scholar] [CrossRef]
- Osta, B.; Benedetti, G.; Miossec, P. Classical and Paradoxical Effects of TNF-α on Bone Homeostasis. Front. Immunol. 2014, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Feng, G.; Xing, J.; Shen, B.; Li, L.; Tan, W.; Xu, Y.; Liu, S.; Liu, H.; Jiang, J.; et al. TNF-α triggers osteogenic differentiation of human dental pulp stem cells via the NF-κB signalling pathway. Cell Biol. Int. 2013, 37, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Gerstenfeld, L.C.; Cho, T.J.; Kon, T.; Aizawa, T.; Cruceta, J.; Graves, B.D.; Einhorn, T.A. Impaired intramembranous bone formation during bone repair in the absence of tumor necrosis factor-alpha signaling. Cells Tissues Organs 2001, 169, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Kon, T.; Cho, T.J.; Aizawa, T.; Yamazaki, M.; Nooh, N.; Graves, D.; Gerstenfeld, L.C.; Einhorn, T.A. Expression of osteoprotegerin, receptor activator of NF-kappaB ligand (osteoprotegerin ligand) and related proinflammatory cytokines during fracture healing. J. Bone Miner. Res. 2001, 16, 1004–1014. [Google Scholar] [CrossRef]
- Gerstenfeld, L.C.; Cullinane, D.M.; Barnes, G.L.; Graves, D.T.; Einhorn, T.A. Fracture healing as a post-natal developmental process: Molecular, spatial, and temporal aspects of its regulation. J. Cell. Biochem. 2003, 88, 873–884. [Google Scholar] [CrossRef]
- Mountziaris, P.M.; Mikos, A.G. Modulation of the inflammatory response for enhanced bone tissue regeneration. Tissue Eng. Part B Rev. 2008, 14, 179–186. [Google Scholar] [CrossRef]
- Rundle, C.H.; Wang, H.; Yu, H.; Chadwick, R.B.; Davis, E.I.; Wergedal, J.E.; Lau, K.H.; Mohan, S.; Ryaby, J.T.; Baylink, D.J. Microarray analysis of gene expression during the inflammation and endochondral bone formation stages of rat femur fracture repair. Bone 2006, 38, 521–529. [Google Scholar] [CrossRef]
- Gerstenfeld, L.C.; Cho, T.J.; Kon, T.; Aizawa, T.; Tsay, A.; Fitch, J.; Barnes, G.L.; Graves, D.T.; Einhorn, T.A. Impaired fracture healing in the absence of TNF-alpha signaling: The role of TNF-alpha in endochondral cartilage resorption. J. Bone Miner. Res. 2003, 18, 1584–1592. [Google Scholar] [CrossRef]
- Huang, H.; Zhao, N.; Xu, X.; Xu, Y.; Li, S.; Zhang, J.; Yang, P. Dose-specific effects of tumor necrosis factor alpha on osteogenic differentiation of mesenchymal stem cells. Cell Prolif. 2011, 44, 420–427. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, Y.; Li, C.; Jiang, Q.; Zhu, L. Effect of TNF-α on the proliferation and osteogenesis of human periodontal mesenchymal stem cells. Exp. Ther. Med. 2021, 21, 434. [Google Scholar] [CrossRef] [PubMed]
- Mountziaris, P.M.; Spicer, P.P.; Kasper, F.K.; Mikos, A.G. Harnessing and modulating inflammation in strategies for bone regeneration. Tissue Eng. Part B Rev. 2011, 17, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diarra, D.; Stolina, M.; Polzer, K.; Zwerina, J.; Ominsky, M.S.; Dwyer, D.; Korb, A.; Smolen, J.; Hoffmann, M.; Scheinecker, C.; et al. Dickkopf-1 is a master regulator of joint remodeling. Nat. Med. 2007, 13, 156–163. [Google Scholar] [CrossRef]
- Kato, H.; Taguchi, Y.; Tominaga, K.; Umeda, M.; Tanaka, A. Porphyromonas gingivalis LPS inhibits osteoblastic differentiation and promotes pro-inflammatory cytokine production in human periodontal ligament stem cells. Arch. Oral Biol. 2014, 59, 167–175. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Kikuiri, T.; Akiyama, K.; Chen, C.; Xu, X.; Yang, R.; Chen, W.; Wang, S.; Shi, S. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-γ and TNF-α. Nat. Med. 2011, 17, 1594–1601. [Google Scholar] [CrossRef]
- Kukolj, T.; Trivanović, D.; Djordjević, I.O.; Mojsilović, S.; Krstić, J.; Obradović, H.; Janković, S.; Santibanez, J.F.; Jauković, A.; Bugarski, D. Lipopolysaccharide can modify differentiation and immunomodulatory potential of periodontal ligament stem cells via ERK1,2 signaling. J. Cell. Physiol. 2018, 233, 447–462. [Google Scholar] [CrossRef]
- Croes, M.; Oner, F.C.; Kruyt, M.C.; Blokhuis, T.J.; Bastian, O.; Dhert, W.J.; Alblas, J. Proinflammatory Mediators Enhance the Osteogenesis of Human Mesenchymal Stem Cells after Lineage Commitment. PLoS ONE 2015, 10, e0132781. [Google Scholar] [CrossRef]
- Lin, T.; Pajarinen, J.; Nabeshima, A.; Lu, L.; Nathan, K.; Jämsen, E.; Yao, Z.; Goodman, S.B. Preconditioning of murine mesenchymal stem cells synergistically enhanced immunomodulation and osteogenesis. Stem Cell Res. Ther. 2017, 8, 277. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Gao, L.N.; An, Y.; Hu, C.H.; Jin, F.; Zhou, J.; Jin, Y.; Chen, F.M. Comparison of mesenchymal stem cells derived from gingival tissue and periodontal ligament in different incubation conditions. Biomaterials 2013, 34, 7033–7047. [Google Scholar] [CrossRef] [PubMed]
- Schindeler, A.; McDonald, M.M.; Bokko, P.; Little, D.G. Bone remodeling during fracture repair: The cellular picture. Semin. Cell Dev. Biol. 2008, 19, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.L.; Tang, K.C.; Patel, A.P.; Bonilla, L.M.; Pierobon, N.; Ponzio, N.M.; Rameshwar, P. Antigen-presenting property of mesenchymal stem cells occurs during a narrow window at low levels of interferon-gamma. Blood 2006, 107, 4817–4824. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.K.; Lau, A.S.; Li, J.C.; Law, H.K.; Lau, Y.L.; Chan, G.C. MHC expression kinetics and immunogenicity of mesenchymal stromal cells after short-term IFN-gamma challenge. Exp. Hematol. 2008, 36, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Lawitschka, A.; Ball, L.; Peters, C. Nonpharmacologic treatment of chronic graft-versus-host disease in children and adolescents. Biol. Blood Marrow Transplant. 2012, 18, S74–S81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duque, G.; Huang, D.C.; Macoritto, M.; Rivas, D.; Yang, X.F.; Ste-Marie, L.G.; Kremer, R. Autocrine regulation of interferon gamma in mesenchymal stem cells plays a role in early osteoblastogenesis. Stem Cells 2009, 27, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Duque, G.; Huang, D.C.; Dion, N.; Macoritto, M.; Rivas, D.; Li, W.; Yang, X.F.; Li, J.; Lian, J.; Marino, F.T.; et al. Interferon-γ plays a role in bone formation in vivo and rescues osteoporosis in ovariectomized mice. J. Bone Miner. Res. 2011, 26, 1472–1483. [Google Scholar] [CrossRef]
- Dighe, A.S.; Yang, S.; Madhu, V.; Balian, G.; Cui, Q. Interferon gamma and T cells inhibit osteogenesis induced by allogeneic mesenchymal stromal cells. J. Orthop. Res. 2013, 31, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Grassi, F.; Ryan, M.R.; Terauchi, M.; Page, K.; Yang, X.; Weitzmann, M.N.; Pacifici, R. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J. Clin. Investig. 2007, 117, 122–132. [Google Scholar] [CrossRef]
- Chen, G.; Deng, C.; Li, Y.P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [Green Version]
- Corcione, A.; Benvenuto, F.; Ferretti, E.; Giunti, D.; Cappiello, V.; Cazzanti, F.; Risso, M.; Gualandi, F.; Mancardi, G.L.; Pistoia, V.; et al. Human mesenchymal stem cells modulate B-cell functions. Blood 2006, 107, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Liu, J.; Gan, Y.; Dai, K.; Zhao, J.; Huang, M.; Huang, Y.; Zhuang, Y.; Zhang, X. High-Dose TGF-β1 Impairs Mesenchymal Stem Cell-Mediated Bone Regeneration via Bmp2 Inhibition. J. Bone Miner. Res. 2020, 35, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Dudakov, J.A.; Hanash, A.M.; Jenq, R.R.; Young, L.F.; Ghosh, A.; Singer, N.V.; West, M.L.; Smith, O.M.; Holland, A.M.; Tsai, J.J.; et al. Interleukin-22 drives endogenous thymic regeneration in mice. Science 2012, 336, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, B.; Liu, S.; Liu, G.; Yan, W.; Wang, Y.; Li, Z.; Fan, C. Macrophages derived from THP-1 promote the osteogenic differentiation of mesenchymal stem cells through the IL-23/IL-23R/β-catenin pathway. Exp. Cell Res. 2015, 339, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Mansoori, M.N.; Kakaji, M.; Shukla, M.; Gupta, S.K.; Singh, D. Interleukin 27 (IL-27) Alleviates Bone Loss in Estrogen-deficient Conditions by Induction of Early Growth Response-2 Gene. J. Biol. Chem. 2017, 292, 4686–4699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Shavit, Z. The osteoclast: A multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J. Cell. Biochem. 2007, 102, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L. Osteoclasts: What do they do and how do they do it? Am. J. Pathol. 2007, 170, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Kondo, M.; Wagers, A.J.; Manz, M.G.; Prohaska, S.S.; Scherer, D.C.; Beilhack, G.F.; Shizuru, J.A.; Weissman, I.L. Biology of hematopoietic stem cells and progenitors: Implications for clinical application. Annu. Rev. Immunol. 2003, 21, 759–806. [Google Scholar] [CrossRef]
- Metcalf, D. Hematopoietic cytokines. Blood 2008, 111, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw. 2018, 18, e8. [Google Scholar] [CrossRef]
- Suda, T.; Takahashi, N.; Udagawa, N.; Jimi, E.; Gillespie, M.T.; Martin, T.J. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr. Rev. 1999, 20, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Nanci, A. Ten Cate’s Oral Histology-e-Book: Development, Structure, and Function; Elsevier Health Sciences: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Fujihara, R.; Usui, M.; Yamamoto, G.; Nishii, K.; Tsukamoto, Y.; Okamatsu, Y.; Sato, T.; Asou, Y.; Nakashima, K.; Yamamoto, M. Tumor necrosis factor-α enhances RANKL expression in gingival epithelial cells via protein kinase A signaling. J. Periodontal Res. 2014, 49, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Usui, M.; Sato, T.; Yamamoto, G.; Okamatsu, Y.; Hanatani, T.; Moritani, Y.; Sano, K.; Yamamoto, M.; Nakashima, K. Gingival epithelial cells support osteoclastogenesis by producing receptor activator of nuclear factor kappa B ligand via protein kinase A signaling. J. Periodontal Res. 2016, 51, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, H.; Chiba, M.; Shimizu, Y.; Mitani, H. Dual regulation of osteoclast differentiation by periodontal ligament cells through RANKL stimulation and OPG inhibition. J. Dent. Res. 2001, 80, 887–891. [Google Scholar] [CrossRef]
- Kawai, T.; Matsuyama, T.; Hosokawa, Y.; Makihira, S.; Seki, M.; Karimbux, N.Y.; Goncalves, R.B.; Valverde, P.; Dibart, S.; Li, Y.P.; et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am. J. Pathol. 2006, 169, 987–998. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Mehta, C.K.; Hsu, T.Y.; Alsulaimani, F.F. Bacteria induce osteoclastogenesis via an osteoblast-independent pathway. Infect. Immun. 2002, 70, 3143–3148. [Google Scholar] [CrossRef] [Green Version]
- Boabaid, F.; Berry, J.E.; Koh, A.J.; Somerman, M.J.; McCcauley, L.K. The role of parathyroid hormone-related protein in the regulation of osteoclastogenesis by cementoblasts. J. Periodontol. 2004, 75, 1247–1254. [Google Scholar] [CrossRef]
- Huynh, N.C.; Everts, V.; Pavasant, P.; Ampornaramveth, R.S. Interleukin-1β induces human cementoblasts to support osteoclastogenesis. Int. J. Oral Sci. 2017, 9, e5. [Google Scholar] [CrossRef]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Lüthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Udagawa, N.; Takahashi, N.; Katagiri, T.; Tamura, T.; Wada, S.; Findlay, D.M.; Martin, T.J.; Hirota, H.; Taga, T.; Kishimoto, T.; et al. Interleukin (IL)-6 induction of osteoclast differentiation depends on IL-6 receptors expressed on osteoblastic cells but not on osteoclast progenitors. J. Exp. Med. 1995, 182, 1461–1468. [Google Scholar] [CrossRef]
- Kudo, O.; Sabokbar, A.; Pocock, A.; Itonaga, I.; Fujikawa, Y.; Athanasou, N.A. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone 2003, 32, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S.L. IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan Tat, S.; Padrines, M.; Théoleyre, S.; Heymann, D.; Fortun, Y. IL-6, RANKL, TNF-alpha/IL-1: Interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev. 2004, 15, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Assuma, R.; Oates, T.; Cochran, D.; Amar, S.; Graves, D.T. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J. Immunol. 1998, 160, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Delima, A.J.; Oates, T.; Assuma, R.; Schwartz, Z.; Cochran, D.; Amar, S.; Graves, D.T. Soluble antagonists to interleukin-1 (IL-1) and tumor necrosis factor (TNF) inhibits loss of tissue attachment in experimental periodontitis. J. Clin. Periodontol. 2001, 28, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Delima, A.J.; Assuma, R.; Amar, S.; Oates, T.; Cochran, D. Interleukin-1 and tumor necrosis factor antagonists inhibit the progression of inflammatory cell infiltration toward alveolar bone in experimental periodontitis. J. Periodontol. 1998, 69, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Bloemen, V.; Schoenmaker, T.; de Vries, T.J.; Everts, V. IL-1β favors osteoclastogenesis via supporting human periodontal ligament fibroblasts. J. Cell. Biochem. 2011, 112, 1890–1897. [Google Scholar] [CrossRef]
- Salla, J.T.; Taddei, S.R.; Queiroz-Junior, C.M.; Andrade Junior, I.; Teixeira, M.M.; Silva, T.A. The effect of IL-1 receptor antagonist on orthodontic tooth movement in mice. Arch. Oral Biol. 2012, 57, 519–524. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, I.; Jimi, E. Regulation of osteoclast differentiation and function by interleukin-1. Vitam. Horm. 2006, 74, 357–370. [Google Scholar] [CrossRef]
- Kim, J.H.; Jin, H.M.; Kim, K.; Song, I.; Youn, B.U.; Matsuo, K.; Kim, N. The mechanism of osteoclast differentiation induced by IL-1. J. Immunol. 2009, 183, 1862–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, H.; Jimi, E.; Okamoto, F.; Motokawa, W.; Okabe, K. IL-1-induced receptor activator of NF-kappa B ligand in human periodontal ligament cells involves ERK-dependent PGE2 production. Bone 2005, 36, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Jules, J.; Zhang, P.; Ashley, J.W.; Wei, S.; Shi, Z.; Liu, J.; Michalek, S.M.; Feng, X. Molecular basis of requirement of receptor activator of nuclear factor κB signaling for interleukin 1-mediated osteoclastogenesis. J. Biol. Chem. 2012, 287, 15728–15738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mun, S.H.; Ko, N.Y.; Kim, H.S.; Kim, J.W.; Kim, D.K.; Kim, A.R.; Lee, S.H.; Kim, Y.G.; Lee, C.K.; Lee, S.H.; et al. Interleukin-33 stimulates formation of functional osteoclasts from human CD14(+) monocytes. Cell. Mol. Life Sci. 2010, 67, 3883–3892. [Google Scholar] [CrossRef] [Green Version]
- Mine, Y.; Makihira, S.; Yamaguchi, Y.; Tanaka, H.; Nikawa, H. Involvement of ERK and p38 MAPK pathways on Interleukin-33-induced RANKL expression in osteoblastic cells. Cell Biol. Int. 2014, 38, 655–662. [Google Scholar] [CrossRef]
- Duka, M.; Eraković, M.; Dolićanin, Z.; Stefanović, D.; Čolić, M. Production of Soluble Receptor Activator of Nuclear Factor Kappa-Β Ligand and Osteoprotegerin by Apical Periodontitis Cells in Culture and Their Modulation by Cytokines. Mediat. Inflamm. 2019, 2019, 8325380. [Google Scholar] [CrossRef] [Green Version]
- Lapérine, O.; Cloitre, A.; Caillon, J.; Huck, O.; Bugueno, I.M.; Pilet, P.; Sourice, S.; Le Tilly, E.; Palmer, G.; Davideau, J.L.; et al. Interleukin-33 and RANK-L Interplay in the Alveolar Bone Loss Associated to Periodontitis. PLoS ONE 2016, 11, e0168080. [Google Scholar] [CrossRef] [Green Version]
- Keller, J.; Catala-Lehnen, P.; Wintges, K.; Schulze, J.; Bickert, T.; Ito, W.; Horst, A.K.; Amling, M.; Schinke, T. Transgenic over-expression of interleukin-33 in osteoblasts results in decreased osteoclastogenesis. Biochem. Biophys. Res. Commun 2012, 417, 217–222. [Google Scholar] [CrossRef]
- Schulze, J.; Bickert, T.; Beil, F.T.; Zaiss, M.M.; Albers, J.; Wintges, K.; Streichert, T.; Klaetschke, K.; Keller, J.; Hissnauer, T.N.; et al. Interleukin-33 is expressed in differentiated osteoblasts and blocks osteoclast formation from bone marrow precursor cells. J. Bone Miner. Res. 2011, 26, 704–717. [Google Scholar] [CrossRef]
- Lima, I.L.; Macari, S.; Madeira, M.F.; Rodrigues, L.F.; Colavite, P.M.; Garlet, G.P.; Soriani, F.M.; Teixeira, M.M.; Fukada, S.Y.; Silva, T.A. Osteoprotective Effects of IL-33/ST2 Link to Osteoclast Apoptosis. Am. J. Pathol. 2015, 185, 3338–3348. [Google Scholar] [CrossRef] [Green Version]
- Gegen, T.; Zhu, Y.; Sun, Q.; Hou, B. Role of interleukin-33 in the clinical pathogenesis of chronic apical periodontitis. J. Int. Med. Res. 2019, 47, 3332–3343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloitre, A.; Halgand, B.; Sourice, S.; Caillon, J.; Huck, O.; Bugueno, I.M.; Batool, F.; Guicheux, J.; Geoffroy, V.; Lesclous, P. IL-36γ is a pivotal inflammatory player in periodontitis-associated bone loss. Sci. Rep. 2019, 9, 19257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Mata, J.; Uy, H.L.; Guise, T.A.; Story, B.; Boyce, B.F.; Mundy, G.R.; Roodman, G.D. Interleukin-6 enhances hypercalcemia and bone resorption mediated by parathyroid hormone-related protein in vivo. J. Clin. Investig. 1995, 95, 2846–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, T.; Udagawa, N.; Takahashi, N.; Miyaura, C.; Tanaka, S.; Yamada, Y.; Koishihara, Y.; Ohsugi, Y.; Kumaki, K.; Taga, T.; et al. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 11924–11928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axmann, R.; Böhm, C.; Krönke, G.; Zwerina, J.; Smolen, J.; Schett, G. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. 2009, 60, 2747–2756. [Google Scholar] [CrossRef]
- Apolinário Vieira, G.H.; Aparecida Rivas, A.C.; Figueired.do Costa, K.; Ferreira Oliveira, L.F.; Tanaka Suzuki, K.; Reis Messora, M.; Sprone Ricoldi, M.; Gonçalves de Almeida, A.L.; Taba, M., Jr. Specific inhibition of IL-6 receptor attenuates inflammatory bone loss in experimental periodontitis. J. Periodontol. 2021, 92, 1460–1469. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, X.; Huang, D.; Ji, Y.; Kang, F. IL-6 Enhances Osteocyte-Mediated Osteoclastogenesis by Promoting JAK2 and RANKL Activity In Vitro. Cell Physiol. Biochem. 2017, 41, 1360–1369. [Google Scholar] [CrossRef]
- Johnson, R.W.; McGregor, N.E.; Brennan, H.J.; Crimeen-Irwin, B.; Poulton, I.J.; Martin, T.J.; Sims, N.A. Glycoprotein130 (Gp130)/interleukin-6 (IL-6) signalling in osteoclasts promotes bone formation in periosteal and trabecular bone. Bone 2015, 81, 343–351. [Google Scholar] [CrossRef]
- Feng, W.; Liu, H.; Luo, T.; Liu, D.; Du, J.; Sun, J.; Wang, W.; Han, X.; Yang, K.; Guo, J.; et al. Combination of IL-6 and sIL-6R differentially regulate varying levels of RANKL-induced osteoclastogenesis through NF-κB, ERK and JNK signaling pathways. Sci. Rep. 2017, 7, 41411. [Google Scholar] [CrossRef] [Green Version]
- Yoshitake, F.; Itoh, S.; Narita, H.; Ishihara, K.; Ebisu, S. Interleukin-6 directly inhibits osteoclast differentiation by suppressing receptor activator of NF-kappaB signaling pathways. J. Biol. Chem. 2008, 283, 11535–11540. [Google Scholar] [CrossRef] [Green Version]
- Zhong, J.; Wang, Z.; Yuan, W.; Shen, Y.; Chen, L. Interleukin-17 promotes osteoclastogenesis and periodontal damage via autophagy in vitro and in vivo. Int. Immunopharmacol. 2022, 107, 108631. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Tan, J.; Wang, Z.; Ding, P.; Tang, Q.; Xia, M.; Wei, Y.; Chen, L. Interleukin-17A facilitates osteoclast differentiation and bone resorption via activation of autophagy in mouse bone marrow macrophages. Mol. Med. Rep. 2019, 19, 4743–4752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, C.M.F.; Maltos, K.L.M.; Shehabeldin, M.S.; Thomas, L.L.; Zhuang, Z.; Yoshizawa, S.; Verdelis, K.; Gaffen, S.L.; Garlet, G.P.; Little, S.R.; et al. Local Sustained Delivery of Anti-IL-17A Antibodies Limits Inflammatory Bone Loss in Murine Experimental Periodontitis. J. Immunol. 2021, 206, 2386–2392. [Google Scholar] [CrossRef] [PubMed]
- Bostanci, N.; Abe, T.; Belibasakis, G.N.; Hajishengallis, G. TREM-1 Is Upregulated in Experimental Periodontitis, and Its Blockade Inhibits IL-17A and RANKL Expression and Suppresses Bone loss. J. Clin. Med. 2019, 8, 1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Zúñiga, J.; Melgar-Rodríguez, S.; Rojas, L.; Alvarez, C.; Monasterio, G.; Carvajal, P.; Vernal, R. Increased levels of the T-helper 22-associated cytokine (interleukin-22) and transcription factor (aryl hydrocarbon receptor) in patients with periodontitis are associated with osteoclast resorptive activity and severity of the disease. J. Periodontal Res. 2017, 52, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Monasterio, G.; Budini, V.; Fernández, B.; Castillo, F.; Rojas, C.; Alvarez, C.; Cafferata, E.A.; Vicencio, E.; Cortés, B.I.; Cortez, C.; et al. IL-22-expressing CD4(+) AhR(+) T lymphocytes are associated with RANKL-mediated alveolar bone resorption during experimental periodontitis. J. Periodontal Res. 2019, 54, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, Q.; Yu, S.; Xu, C.; Li, X.; Zhang, C.; Gao, L. Effect of interleukin-22 on osteogenic differentiation and the osteoclastogenic response of human periodontal ligament fibroblasts in vitro. J. Periodontol. 2020, 91, 1085–1097. [Google Scholar] [CrossRef]
- Song, L.; Dong, G.; Guo, L.; Graves, D.T. The function of dendritic cells in modulating the host response. Mol. Oral Microbiol. 2018, 33, 13–21. [Google Scholar] [CrossRef]
- Hofbauer, L.; Lacey, D.; Dunstan, C.; Spelsberg, T.; Riggs, B.; Khosla, S. Interleukin-1β and tumor necrosis factor-α, but not interleukin-6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells. Bone 1999, 25, 255–259. [Google Scholar] [CrossRef]
- Boström, E.A.; Lundberg, P. The newly discovered cytokine IL-34 is expressed in gingival fibroblasts, shows enhanced expression by pro-inflammatory cytokines, and stimulates osteoclast differentiation. PLoS ONE 2013, 8, e81665. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Kitaura, H.; Sands, M.S.; Aya, K.; Zhou, P.; Hirayama, T.; Uthgenannt, B.; Wei, S.; Takeshita, S.; Novack, D.V.; Silva, M.J.; et al. Marrow stromal cells and osteoclast precursors differentially contribute to TNF-alpha-induced osteoclastogenesis in vivo. J. Immunol. 2004, 173, 4838–4846. [Google Scholar] [CrossRef] [Green Version]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-α Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Front. Immunol. 2019, 10, 2925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragab, A.A.; Nalepka, J.L.; Bi, Y.; Greenfield, E.M. Cytokines synergistically induce osteoclast differentiation: Support by immortalized or normal calvarial cells. Am. J. Physiol. Cell Physiol. 2002, 283, C679–C687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacey, D.C.; Simmons, P.J.; Graves, S.E.; Hamilton, J.A. Proinflammatory cytokines inhibit osteogenic differentiation from stem cells: Implications for bone repair during inflammation. Osteoarthr. Cartil. 2009, 17, 735–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, E.; Zhan, F.; Walker, R.; Rasmussen, E.; Ma, Y.; Barlogie, B.; Shaughnessy, J.D., Jr. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N. Engl. J. Med. 2003, 349, 2483–2494. [Google Scholar] [CrossRef]
- Wang, H.; Young, S.R.; Gerard-O’Riley, R.; Hum, J.M.; Yang, Z.; Bidwell, J.P.; Pavalko, F.M. Blockade of TNFR1 signaling: A role of oscillatory fluid shear stress in osteoblasts. J. Cell. Physiol. 2011, 226, 1044–1051. [Google Scholar] [CrossRef]
- Ma, T.; Miyanishi, K.; Suen, A.; Epstein, N.J.; Tomita, T.; Smith, R.L.; Goodman, S.B. Human interleukin-1-induced murine osteoclastogenesis is dependent on RANKL, but independent of TNF-alpha. Cytokine 2004, 26, 138–144. [Google Scholar] [CrossRef]
- Li, P.; Schwarz, E.M.; O’Keefe, R.J.; Ma, L.; Boyce, B.F.; Xing, L. RANK signaling is not required for TNFalpha-mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J. Bone Miner. Res. 2004, 19, 207–213. [Google Scholar] [CrossRef]
- Jules, J.; Shi, Z.; Liu, J.; Xu, D.; Wang, S.; Feng, X. Receptor activator of NF-{kappa}B (RANK) cytoplasmic IVVY535-538 motif plays an essential role in tumor necrosis factor-{alpha} (TNF)-mediated osteoclastogenesis. J. Biol. Chem. 2010, 285, 37427–37435. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Kaji, K.; Katogi, R.; Takeshita, S.; Kudo, A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J. Biol. Chem. 2000, 275, 4858–4864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Sarosi, I.; Yan, X.Q.; Morony, S.; Capparelli, C.; Tan, H.L.; McCabe, S.; Elliott, R.; Scully, S.; Van, G.; et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 1566–1571. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Yin, Y.; Liu, J.; Li, Z.; Lu, X. MiR-124 Attenuates Osteoclastogenic Differentiation of Bone Marrow Monocytes Via Targeting Rab27a. Cell. Physiol. Biochem. 2017, 43, 1663–1672. [Google Scholar] [CrossRef]
- Guo, J.; Zeng, X.; Miao, J.; Liu, C.; Wei, F.; Liu, D.; Zheng, Z.; Ting, K.; Wang, C.; Liu, Y. MiRNA-218 regulates osteoclast differentiation and inflammation response in periodontitis rats through Mmp9. Cell. Microbiol. 2019, 21, e12979. [Google Scholar] [CrossRef]
- Mizoguchi, F.; Murakami, Y.; Saito, T.; Miyasaka, N.; Kohsaka, H. miR-31 controls osteoclast formation and bone resorption by targeting RhoA. Arthritis Res. Ther. 2013, 15, R102. [Google Scholar] [CrossRef] [Green Version]
- Kagiya, T.; Nakamura, S. Expression profiling of microRNAs in RAW264.7 cells treated with a combination of tumor necrosis factor alpha and RANKL during osteoclast differentiation. J. Periodontal Res. 2013, 48, 373–385. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D. Periodontal microbial ecology. Periodontol. 2000 2005, 38, 135–187. [Google Scholar] [CrossRef]
- Darveau, R.; Hajishengallis, G.; Curtis, M. Porphyromonas gingivalis as a potential community activist for disease. J. Dent. Res. 2012, 91, 816–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, P.; Dixon, M.; Evans, R.; Roopenian, D. Heterogeneity of Porphyromonas gingivalis strains in the induction of alveolar bone loss in mice. Oral Microbiol. Immunol. 2000, 15, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Wray, D.; Grahame, L. Periodontal bone loss in mice induced by different periodontopathic organisms. Arch. Oral Biol. 1992, 37, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Genco, C.A.; Van Dyke, T.; Amar, S. Animal models for Porphyromonas gingivalis-mediated periodontal disease. Trends Microbiol. 1998, 6, 444–449. [Google Scholar] [CrossRef]
- Zhang, W.; Ju, J.; Rigney, T.; Tribble, G. Porphyromonas gingivalis infection increases osteoclastic bone resorption and osteoblastic bone formation in a periodontitis mouse model. BMC Oral Health 2014, 14, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.K.; Moon, S.Y.; Cha, J.H.; Kim, K.W.; Yoo, Y.J. Prostaglandin E2 is a main mediator in receptor activator of nuclear factor-κB ligand-dependent osteoclastogenesis induced by Porphyromonas gingivalis, Treponema denticola, and Treponema socranskii. J. Periodontol. 2005, 76, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Okahashi, N.; Inaba, H.; Nakagawa, I.; Yamamura, T.; Kuboniwa, M.; Nakayama, K.; Hamada, S.; Amano, A. Porphyromonas gingivalis induces receptor activator of NF-κB ligand expression in osteoblasts through the activator protein 1 pathway. Infect. Immun. 2004, 72, 1706–1714. [Google Scholar] [CrossRef] [Green Version]
- Polak, D.; Wilensky, A.; Shapira, L.; Halabi, A.; Goldstein, D.; Weiss, E.I.; Houri-Haddad, Y. Mouse model of experimental periodontitis induced by Porphyromonas gingivalis/Fusobacterium nucleatum infection: Bone loss and host response. J. Clin. Periodontol. 2009, 36, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Kesavalu, L.; Sathishkumar, S.; Bakthavatchalu, V.; Matthews, C.; Dawson, D.; Steffen, M.; Ebersole, J.L. Rat model of polymicrobial infection, immunity, and alveolar bone resorption in periodontal disease. Infect. Immun. 2007, 75, 1704–1712. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Kang, M.; Zhang, M.J.; Reza Sailani, M.; Kuraji, R.; Martinez, A.; Ye, C.; Kamarajan, P.; Le, C.; Zhan, L. Polymicrobial periodontal disease triggers a wide radius of effect and unique virome. Npj Biofilms Microbiomes 2020, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; Mazmanian, S.K. Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat. Immunol. 2013, 14, 668–675. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Wu, W.; Liu, Z.; Cong, Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J. Gastroenterol. 2017, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Yagi, T. Bioactive mechanism of Porphyromonas gingivalis lipid A. Periodontol. 2000 2010, 54, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.P.; Meghji, S.; Wilson, M.; Reddi, K.; White, P.; Henderson, B. Bacterially induced bone destruction: Mechanisms and misconceptions. Infect. Immun. 1996, 64, 2371–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujihara, M.; Muroi, M.; Tanamoto, K.; Suzuki, T.; Azuma, H.; Ikeda, H. Molecular mechanisms of macrophage activation deactivation by lipopolysaccharide: Roles of the receptor complex. Pharmacol. Ther. 2003, 100, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Zúñiga, J.; Monasterio, G.; Alvarez, C.; Melgar-Rodríguez, S.; Benítez, A.; Ciuchi, P.; García, M.; Arias, J.; Sanz, M.; Vernal, R. Variability of the dendritic cell response triggered by different serotypes of Aggregatibacter actinomycetemcomitans or Porphyromonas gingivalis is toll-like receptor 2 (TLR2) or TLR4 dependent. J. Periodontol. 2015, 86, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akatsu, T.; Takahashi, N.; Debari, K.; Morita, I.; Murota, S.; Nagata, N.; Takatani, O.; Suda, T. Prostaglandins promote osteoclastlike cell formation by a mechanism involving cyclic adenosine 3′, 5′-monophosphate in mouse bone marrow cell cultures. J. Bone Miner. Res. 1989, 4, 29–35. [Google Scholar] [CrossRef]
- Watanabe, K.; Iizuka, T.; Adeleke, A.; Pham, L.; Shlimon, A.; Yasin, M.; Horvath, P.; Unterman, T. Involvement of toll-like receptor 4 in alveolar bone loss and glucose homeostasis in experimental periodontitis. J. Periodontal Res. 2011, 46, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Taubman, M.A.; Valverde, P.; Han, X.; Kawai, T. Immune response: The key to bone resorption in periodontal disease. J. Periodontol. 2005, 76, 2033–2041. [Google Scholar] [CrossRef]
- Islam, S.; Hassan, F.; Tumurkhuu, G.; Dagvadorj, J.; Koide, N.; Naiki, Y.; Mori, I.; Yoshida, T.; Yokochi, T. Bacterial lipopolysaccharide induces osteoclast formation in RAW 264.7 macrophage cells. Biochem. Biophys. Res. Commun. 2007, 360, 346–351. [Google Scholar] [CrossRef]
- Kikuchi, T.; Matsuguchi, T.; Tsuboi, N.; Mitani, A.; Tanaka, S.; Matsuoka, M.; Yamamoto, G.; Hishikawa, T.; Noguchi, T.; Yoshikai, Y. Gene expression of osteoclast differentiation factor is induced by lipopolysaccharide in mouse osteoblasts via Toll-like receptors. J. Immunol. 2001, 166, 3574–3579. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Sun, F.; Li, X.; Zhou, Y.; Yin, S.; Zhou, X. Porphyromonas endodontalis lipopolysaccharides induce RANKL by mouse osteoblast in a way different from that of Escherichia coli lipopolysaccharide. J. Endod. 2011, 37, 1653–1658. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Kim, N.; Rho, J.; Choi, Y. Stimulation by toll-like receptors inhibits osteoclast differentiation. J. Immunol. 2002, 169, 1516–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, W.; Bar-Shavit, Z. Dual modulation of osteoclast differentiation by lipopolysaccharide. J. Bone Miner. Res. 2002, 17, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Umezu, A.; Kaneko, N.; Toyama, Y.; Watanabe, Y.; Itoh, H. Appearance of osteoclasts by injections of lipopolysaccharides in rat periodontal tissue. J. Periodontal Res. 1989, 24, 378–383. [Google Scholar] [CrossRef]
- Rogers, J.E.; Li, F.; Coatney, D.D.; Rossa, C., Jr.; Bronson, P.; Krieder, J.M.; Giannobile, W.V.; Kirkwood, K.L. Actinobacillus actinomycetemcomitans lipopolysaccharide-mediated experimental bone loss model for aggressive periodontitis. J. Periodontol. 2007, 78, 550–558. [Google Scholar] [CrossRef] [Green Version]
- Nishida, E.; Hara, Y.; Kaneko, T.; Ikeda, Y.; Ukai, T.; Kato, I. Bone resorption and local interleukin-1α and interleukin-1β synthesis induced by Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis lipopolysaccharide. J. Periodontal. Res. 2001, 36, 1–8. [Google Scholar] [CrossRef]
- Hou, G.-Q.; Guo, C.; Song, G.-H.; Fang, N.; Fan, W.-J.; Chen, X.-D.; Yuan, L.; Wang, Z.-Q. Lipopolysaccharide (LPS) promotes osteoclast differentiation and activation by enhancing the MAPK pathway and COX-2 expression in RAW264. 7 cells. Int. J. Mol. Med. 2013, 32, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Bristol-Rothstein, L.A.; Schwab, J.H. Bone-resorbing activity is expressed by rat macrophages in response to arthropathic streptococcal cell wall polymers. Inflammation 1992, 16, 485–496. [Google Scholar] [CrossRef]
- Hotokezaka, H.; Sakai, E.; Ohara, N.; Hotokezaka, Y.; Gonzales, C.; Matsuo, K.i.; Fujimura, Y.; Yoshida, N.; Nakayama, K. Molecular analysis of RANKL-independent cell fusion of osteoclast-like cells induced by TNF-α, lipopolysaccharide, or peptidoglycan. J. Cell. Biochem. 2007, 101, 122–134. [Google Scholar] [CrossRef]
- Ozaki, Y.; Ukai, T.; Yamaguchi, M.; Yokoyama, M.; Haro, E.R.A.; Yoshimoto, M.; Kaneko, T.; Yoshinaga, M.; Nakamura, H.; Shiraishi, C. Locally administered T cells from mice immunized with lipopolysaccharide (LPS) accelerate LPS-induced bone resorption. Bone 2009, 44, 1169–1176. [Google Scholar] [CrossRef]
- Bar-Shavit, Z. Taking a toll on the bones: Regulation of bone metabolism by innate immune regulators. Autoimmunity 2008, 41, 195–203. [Google Scholar] [CrossRef]
- Kishimoto, T.; Kaneko, T.; Ukai, T.; Yokoyama, M.; Ayon Haro, R.; Yoshinaga, Y.; Yoshimura, A.; Hara, Y. Peptidoglycan and lipopolysaccharide synergistically enhance bone resorption and osteoclastogenesis. J. Periodontal Res. 2012, 47, 446–454. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Toll-like receptors; their physiological role and signal transduction system. Int. Immunopharmacol. 2001, 1, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Chamaillard, M.; Hashimoto, M.; Horie, Y.; Masumoto, J.; Qiu, S.; Saab, L.; Ogura, Y.; Kawasaki, A.; Fukase, K.; Kusumoto, S. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 2003, 4, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Girardin, S.E.; Boneca, I.G.; Viala, J.; Chamaillard, M.; Labigne, A.; Thomas, G.; Philpott, D.J.; Sansonetti, P. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 2003, 278, 8869–8872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girardin, S.E.; Boneca, I.G.; Carneiro, L.A.; Antignac, A.; Jéhanno, M.; Viala, J.; Tedin, K.; Taha, M.-K.; Labigne, A.; Zäthringer, U. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 2003, 300, 1584–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inohara, N.; Nunez, G. NODs: Intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol. 2003, 3, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Inohara, N.; Benito, A.; Chen, F.F.; Yamaoka, S.; Núñez, G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J. Biol. Chem. 2001, 276, 4812–4818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masumoto, J.; Yang, K.; Varambally, S.; Hasegawa, M.; Tomlins, S.A.; Qiu, S.; Fujimoto, Y.; Kawasaki, A.; Foster, S.J.; Horie, Y. Nod1 acts as an intracellular receptor to stimulate chemokine production and neutrophil recruitment in vivo. J. Exp. Med. 2006, 203, 203–213. [Google Scholar] [CrossRef]
- Prates, T.; Taira, T.; Holanda, M.; Bignardi, L.; Salvador, S.; Zamboni, D.S.; Cunha, F.d.Q.; Fukada, S.Y. NOD2 contributes to Porphyromonas gingivalis–induced bone resorption. J. Dent. Res. 2014, 93, 1155–1162. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Piemontese, M.; Onal, M.; Campbell, J.; Goellner, J.J.; Dusevich, V.; Bonewald, L.; Manolagas, S.C.; O’Brien, C.A. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS ONE 2015, 10, e0138189. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.J.; Sims, N.A. RANKL/OPG; Critical role in bone physiology. Rev. Endocr. Metab. Disord. 2015, 16, 131–139. [Google Scholar] [CrossRef]
- Monroe, D.G.; McGee-Lawrence, M.E.; Oursler, M.J.; Westendorf, J.J. Update on Wnt signaling in bone cell biology and bone disease. Genes Chromosomes Cancer 2012, 492, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, T.P.; Später, D.; Taketo, M.M.; Birchmeier, W.; Hartmann, C. Canonical Wnt/β-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev. Cell 2005, 8, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, E.; Kido, J.-i.; Takagi, R.; Inagaki, Y.; Naruishi, K.; Nagata, T.; Yumoto, H. Advanced glycation end-product 2 and Porphyromonas gingivalis lipopolysaccharide increase sclerostin expression in mouse osteocyte-like cells. Bone 2019, 122, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Ma, Y.; Li, X.; Wu, X.; Liu, W.; Li, X.; Shen, J.; Wang, H. Lipopolysaccharide increases IL-6 secretion via activation of the ERK1/2 signaling pathway to up-regulate RANKL gene expression in MLO-Y4 cells. Cell Biol. Int. 2017, 41, 84–92. [Google Scholar] [CrossRef]
- Pacios, S.; Xiao, W.; Mattos, M.; Lim, J.; Tarapore, R.S.; Alsadun, S.; Yu, B.; Wang, C.-Y.; Graves, D.T. Osteoblast lineage cells play an essential role in periodontal bone loss through activation of nuclear factor-kappa B. Sci. Rep. 2015, 5, 16694. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Xu, J.; Figliomeni, L.; Huang, L.; Pavlos, N.; Rogers, M.; Tan, A.; Price, P.; Zheng, M. Expression of RANKL and OPG mRNA in periodontal disease: Possible involvement in bone destruction. Int. J. Mol. Med. 2003, 11, 17–21. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Honma, M.; Ikebuchi, Y.; Kariya, Y.; Hayashi, M.; Hayashi, N.; Aoki, S.; Suzuki, H. RANKL subcellular trafficking and regulatory mechanisms in osteocytes. J. Bone Miner. Res. 2013, 28, 1936–1949. [Google Scholar] [CrossRef]
- Esfahrood, Z.R.; Yadegari, Z.; Veysari, S.K.; Kadkhodazadeh, M. Gingival crevicular fluid levels of sclerostin in chronic periodontitis and healthy subjects. J. Korean Assoc. Oral Maxillofac. Surg. 2018, 44, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Baek, K.; Hwang, H.R.; Park, H.J.; Kwon, A.; Qadir, A.S.; Ko, S.H.; Woo, K.M.; Ryoo, H.M.; Kim, G.S.; Baek, J.H. TNF-α upregulates sclerostin expression in obese mice fed a high-fat diet. J. Cell. Physiol. 2014, 229, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Heiland, G.R.; Zwerina, K.; Baum, W.; Kireva, T.; Distler, J.H.; Grisanti, M.; Asuncion, F.; Li, X.; Ominsky, M.; Richards, W. Neutralisation of Dkk-1 protects from systemic bone loss during inflammation and reduces sclerostin expression. Ann. Rheum. Dis. 2010, 69, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Kurimoto, P.; Zhang, J.; Niu, Q.; Stolina, M.; Dechow, P.; Feng, J.; Hesterman, J.; Silva, M.; Ominsky, M. Sclerostin and DKK1 inhibition preserves and augments alveolar bone volume and architecture in rats with alveolar bone loss. J. Dent. Res. 2018, 97, 1031–1038. [Google Scholar] [CrossRef]
- Witcher, P.C.; Miner, S.E.; Horan, D.J.; Bullock, W.A.; Lim, K.-E.; Kang, K.S.; Adaniya, A.L.; Ross, R.D.; Loots, G.G.; Robling, A.G. Sclerostin neutralization unleashes the osteoanabolic effects of Dkk1 inhibition. JCI Insight 2018, 3, e98673. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Huang, D.; Zhou, W.; Yan, L.; Yue, J.; Lu, W.; Song, D.; Zhou, X.; Ye, L.; Zhang, L. Dickkopf-1 may regulate bone coupling by attenuating wnt/β-catenin signaling in chronic apical periodontitis. Arch. Oral Biol. 2018, 86, 94–100. [Google Scholar] [CrossRef]
- Wijenayaka, A.R.; Kogawa, M.; Lim, H.P.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS ONE 2011, 6, e25900. [Google Scholar] [CrossRef] [Green Version]
- Goes, P.; Dutra, C.; Lösser, L.; Hofbauer, L.C.; Rauner, M.; Thiele, S. Loss of Dkk-1 in osteocytes mitigates alveolar bone loss in mice with periodontitis. Front. Immunol. 2019, 2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmatian, H.; Bakker, A.D.; Klein-Nulend, J.; van Lenthe, G.H. Aging, osteocytes, and mechanotransduction. Curr. Osteoporos. Rep. 2017, 15, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Tchkonia, T.; Zhu, Y.; Van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Aquino-Martinez, R.; Rowsey, J.L.; Fraser, D.G.; Eckhardt, B.A.; Khosla, S.; Farr, J.N.; Monroe, D.G. LPS-induced premature osteocyte senescence: Implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone 2020, 132, 115220. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Jun, H.-K.; Jung, Y.-J.; Choi, B.-K. Treponema denticola, Porphyromonas gingivalis, and Tannerella forsythia induce cell death and release of endogenous danger signals. Arch. Oral Biol. 2017, 73, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-W.; He, H.-B. Apoptosis of periodontium cells in streptozototocin-and ligature-induced experimental diabetic periodontitis in rats. Acta Odontol. Scand. 2013, 71, 1206–1215. [Google Scholar] [CrossRef]
- Al-Dujaili, S.A.; Lau, E.; Al-Dujaili, H.; Tsang, K.; Guenther, A.; You, L. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J. Cell. Biochem. 2011, 112, 2412–2423. [Google Scholar] [CrossRef]
- Bellido, T. Osteocyte-driven bone remodeling. Calcif. Tissue Int. 2014, 94, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. Functions of the osteocyte network in the regulation of bone mass. Cell Tissue Res. 2013, 352, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algate, K.; Haynes, D.; Bartold, P.; Crotti, T.; Cantley, M. The effects of tumour necrosis factor-α on bone cells involved in periodontal alveolar bone loss; osteoclasts, osteoblasts and osteocytes. J. Periodontal Res. 2016, 51, 549–566. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Zhang, F.; Wu, J.; Xu, N.; Liang, M. Gingipains disrupt F-actin and cause osteoblast apoptosis via integrin β1. J. Periodontal Res. 2018, 53, 762–776. [Google Scholar] [CrossRef] [PubMed]
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef]
- Tomás, I.; Arias-Bujanda, N.; Alonso-Sampedro, M.; Casares-de-Cal, M.A.; Sánchez-Sellero, C.; Suárez-Quintanilla, D.; Balsa-Castro, C. Cytokine-based Predictive Models to Estimate the Probability of Chronic Periodontitis: Development of Diagnostic Nomograms. Sci. Rep. 2017, 7, 11580. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, K.R.; Kim, H.N. The Potential Impact of Salivary IL-1 on the Diagnosis of Periodontal Disease: A Pilot Study. Healthcare 2021, 9, 729. [Google Scholar] [CrossRef]
- Gürlek, Ö.; Gümüş, P.; Nile, C.J.; Lappin, D.F.; Buduneli, N. Biomarkers and Bacteria Around Implants and Natural Teeth in the Same Individuals. J. Periodontol. 2017, 88, 752–761. [Google Scholar] [CrossRef]
- Nair, V.; Grover, V.; Arora, S.; Das, G.; Ahmad, I.; Ohri, A.; Sainudeen, S.; Saluja, P.; Saha, A. Comparative Evaluation of Gingival Crevicular Fluid Interleukin-17, 18 and 21 in Different Stages of Periodontal Health and Disease. Medicina 2022, 58, 1042. [Google Scholar] [CrossRef]
- Madureira, D.F.; Lucas De Abreu Lima, I.; Costa, G.C.; Lages, E.M.B.; Martins, C.C.; Aparecida Da Silva, T. Tumor Necrosis Factor-alpha in Gingival Crevicular Fluid as a Diagnostic Marker for Periodontal Diseases: A Systematic Review. J. Evid. Based Dent. Pract. 2018, 18, 315–331. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, H.N. Changes in Inflammatory Cytokines in Saliva after Non-Surgical Periodontal Therapy: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2020, 18, 194. [Google Scholar] [CrossRef]
- Izadi Borujeni, S.; Mayer, M.; Eickholz, P. Activated matrix metalloproteinase-8 in saliva as diagnostic test for periodontal disease? A case-control study. Med. Microbiol. Immunol. 2015, 204, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Cafiero, C.; Spagnuolo, G.; Marenzi, G.; Martuscelli, R.; Colamaio, M.; Leuci, S. Predictive periodontitis: The most promising salivary biomarkers for early diagnosis of periodontitis. J. Clin. Med. 2021, 10, 1488. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G. Periodontitis: From microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Pang, X.; Li, Z.; Chen, Z.; Wang, Y. Immunomodulation in the Treatment of Periodontitis: Progress and Perspectives. Front. Immunol. 2021, 12, 781378. [Google Scholar] [CrossRef] [PubMed]
- Orihuela-Campos, R.C.; Tamaki, N.; Mukai, R.; Fukui, M.; Miki, K.; Terao, J.; Ito, H.O. Biological impacts of resveratrol, quercetin, and N-acetylcysteine on oxidative stress in human gingival fibroblasts. J. Clin. Biochem. Nutr. 2015, 56, 220–227. [Google Scholar] [CrossRef]
- Garzón, H.; Suárez, L.J.; Muñoz, S.; Cardona, J.; Fontalvo, M.; Alfonso-Rodríguez, C.A. Biomaterials Used for Periodontal Disease Treatment: Focusing on Immunomodulatory Properties. Int. J. Biomater. 2022, 2022, 7693793. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Cell | Characteristics | Express | Biological Activity |
|---|---|---|---|
| Osteoblast | Cuboidal in shape, they are found on a bone surface, they contact each other by adherens and gap junctions | -Alkaline phosphatase -Osteocalcin -Osteopontin -Bone sialoprotien -Collα1 -Osterix -RUNX2 | -They synthesize the extracellular bone matrix and promote its mineralization by matrix vesicles. |
| Osteocyte | Stellate shape, the cell body lies in lacunae and cell processes run in the canaliculi in all directions connected with gap junction | -Insulin-like growth factor-I -c-fos -RUNX2 -Bone sialoprotien | -They are involved in bone turnover. -Widely spread interconnected cells allow the diffusion of substances through the bone. -They act as mechanoreceptors of bone. |
| Osteoclast | Large multinucleated phagocytic cells | -Tartrate-resistant acid phosphatase -Carbonic anhydrase II -Vacuolar proton ATPase -Vitronectin receptor -Calcitonin receptor | -They are involved in bone resorption (bone remodeling) during growth or changing mechanical stresses. -They participate in the long-term maintenance of blood calcium homeostasis. |
| Cytokines | Osteoblastogenesis | Osteoclastogenesis | ||
|---|---|---|---|---|
| Low doses enhance osteoblastogenesis via | High doses decrease osteoblastogenesis via | Enhance osteoclastogenesis via | Inhibit osteoclastogenesis via | |
| IL-1 | -Noncanonical Wnt-5a/Ror2 pathway. -BMP/Smad signaling pathway. | -Activating NF-κB and MAPK signaling pathway. -Suppressing BMP/signaling. -Upregulating the Wnt signaling pathway antagonists DKK1 and sclerostin. | -Potentiating RANKL via p38 MAPK pathway. -Downregulation of OPG mRNA expression via ERK pathway. -Upregulating transcriptional factors, including JNK, P38, and ERK. -Inducing osteoclast-specific genes, such as OSCAR and TRAP -Upregulating IL-1 and IL-1RI expression via c-Fos and NFATc1. | |
| IL-6 | -Upregulation of RUNX2 and ALP activity through insulin-like growth factor. -Activation of transcription (STAT3) pathway. | -Downregulation of RUNX2, osterix (OSX), and OC via activation of Src homology 2 (SHP2)/MAPK/extracellular signal-regulated kinase (ERK), Janus kinase (JAK)/STAT3, and SHP2/phosphoinositide 3-kinase (PI3K)/Akt2 signaling pathways. | -Upregulating RANKL expression on osteoblasts -GP130 signaling pathway. -With high level of RANKL upregulated osteoclast differentiation through upregulation of NF-κB, ERK, and JNK phosphorylation. | -Suppression of NF-κB pathways. -With low level of RANKL, IL-6, and sIL-6R downregulated osteoclast differentiation. |
| IL-17 | -Upregulating ALP, OC, RUNX2, and RANKL expression. | -Inhibiting BMP-2-induced Osteoblastogenesis with reduced expression of ALP, OC, RUNX2, and OSX expression. -Activation of ERK1,2 and c-Jun N-terminal kinase (JNK) MAPK. | -RANKL upregulation through triggering receptor expressed on myeloid cells-1 (TREM-1). | |
| TNF-α | -NF-κB through increasing expression of BMP-2, OSX, RUNX2, OC, and Wnt signaling pathway. | -Increasing production of DKK-1 and Wnt signaling pathway antagonist. | -Upregulating RANK expression on osteoclasts precursor and sensitizing precursor cells to RANKL via activation of NF-kB and SAPK/JNK. -Inhibiting WNT signaling pathway. | |
| IL-1β and TNF-α | -No change. | -Boosting the canonical Wnt/-catenin pathway and blocking the noncanonical Wnt/Ca2+ pathway. | ||
| IFN-γ | -Enhancing stem cells’ antigen-presenting activities, minimizing their lysis. -Inducing bone marrow MSCs secrete functional indoleamine 2,3-dioxygenase and IL-10. | -T cell activation. | ||
| TGF-β | -Canonical Smad-dependent and non-canonical Smad-independent pathways (e.g., p38, MAPK). -Forming a complex with Smad4, which interacts with RUNX2, resulting in numerous osteogenic genes to be activated -Activating Smad3 and enhancing BMP-2 production in bone marrow MSCs. | -Modifying Smad3 binding sites cause BMP-2 suppression on its promoter -Elevating tomoregulin-1levels in mice, causing BMP-2 suppression. | ||
| IL-10 | -Activating oxidative phosphorylation. | |||
| IL-23 | -Canonical Wnt-catenin pathway. | |||
| IL-27 | -Inhibiting osteoblast death. | |||
| IL-33 | -Potentiating RANKL via ERK and p38 MAPK. -Activating spleen associated tyrosine kinase, phospholipase Cc2, Grb2-associated-binding protein 2, MAPK, TAK-1, NF-kB. IL-33, TNF-α receptor-associated factor 6 (TRAF6), nuclear factor of activated T cells cytoplasmic 1, c-Fos, c-Src, cathepsin K, and calcitonin receptor. | -Impeding osteoclast differentiation factors, such as NFATc1. -Upregulation of osteoclasts apoptosis via upregulation of proapoptotic molecules, such as BAX, Fas, and FasL. -Lower levels are corelated with OPG expression in chronic periodontitis. | ||
| IL-36 γ | -Correlated with RANKL to OPG ratio. | |||
| IL-22 | -Upregulation of RANKL expression via the MAPK signaling pathway. | |||
| IL-8 | -Stimulating inflammatory cells to secrete IFN-γ, IL-17, TNF-α, IL-1β, and RANKL. | |||
| IL-11 | -Upregulating RANKL expression by osteoblasts. | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadek, K.M.; El Moshy, S.; Radwan, I.A.; Rady, D.; Abbass, M.M.S.; El-Rashidy, A.A.; Dörfer, C.E.; Fawzy El-Sayed, K.M. Molecular Basis beyond Interrelated Bone Resorption/Regeneration in Periodontal Diseases: A Concise Review. Int. J. Mol. Sci. 2023, 24, 4599. https://doi.org/10.3390/ijms24054599
Sadek KM, El Moshy S, Radwan IA, Rady D, Abbass MMS, El-Rashidy AA, Dörfer CE, Fawzy El-Sayed KM. Molecular Basis beyond Interrelated Bone Resorption/Regeneration in Periodontal Diseases: A Concise Review. International Journal of Molecular Sciences. 2023; 24(5):4599. https://doi.org/10.3390/ijms24054599
Chicago/Turabian StyleSadek, Khadiga M., Sara El Moshy, Israa Ahmed Radwan, Dina Rady, Marwa M. S. Abbass, Aiah A. El-Rashidy, Christof E. Dörfer, and Karim M. Fawzy El-Sayed. 2023. "Molecular Basis beyond Interrelated Bone Resorption/Regeneration in Periodontal Diseases: A Concise Review" International Journal of Molecular Sciences 24, no. 5: 4599. https://doi.org/10.3390/ijms24054599