
Unraveling the Bombus terrestris Hemolymph, an Indicator of the Immune Response to Microbial Infections, through Complementary Mass Spectrometry Approaches

 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
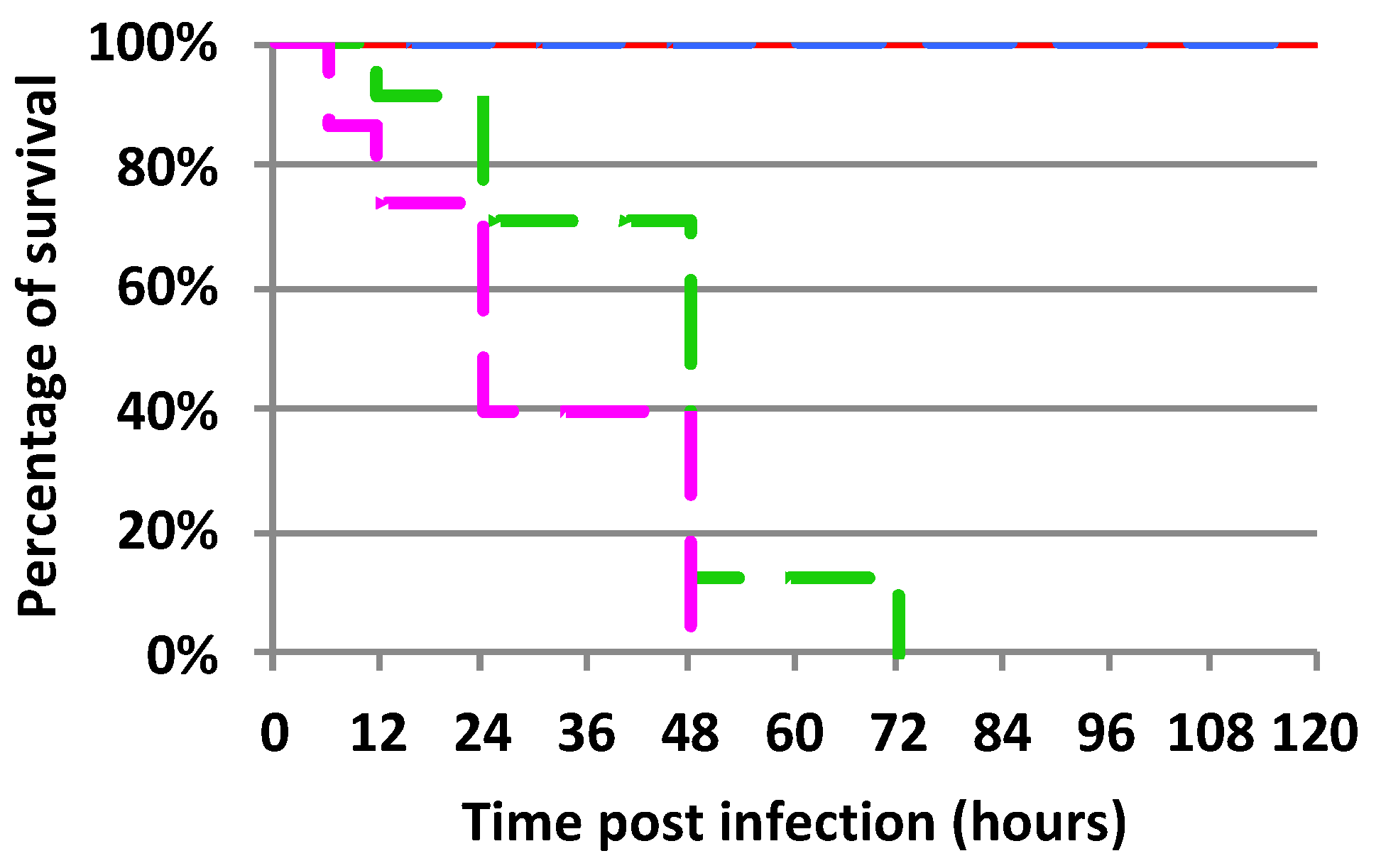
2.1. Mortality Rate of Experimentally Infected Bumble Bees Versus Control
2.2. Molecular Hemolymph Signatures by MALDI BeeTyping® to Follow the Impact of Bacterial Infections in B. terrestris
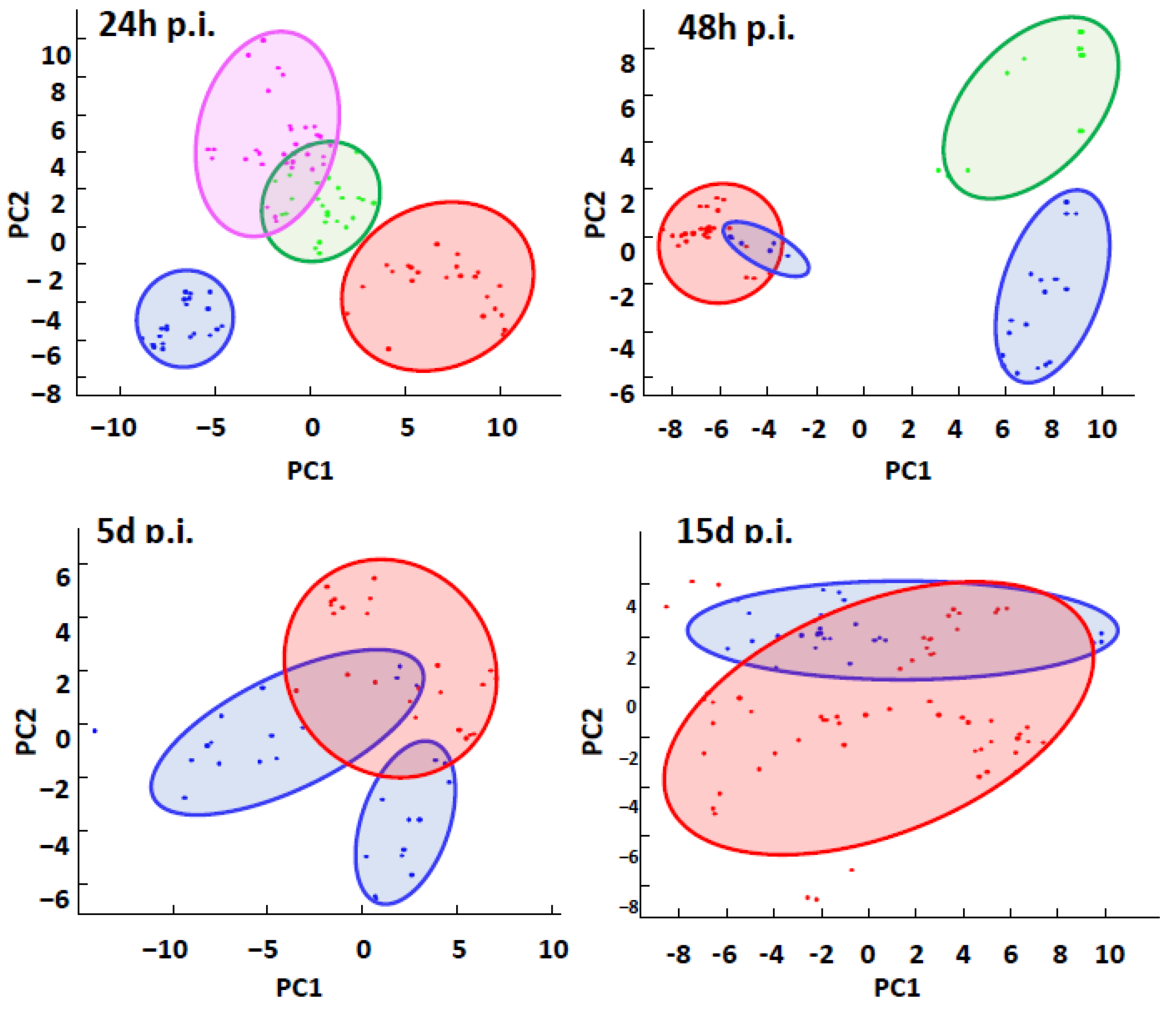
2.2.1. Global Analysis of MFPs by MALDI BeeTyping® Discriminates between Different Bacterial Infections
2.2.2. The Immune Response to Microbial Infections
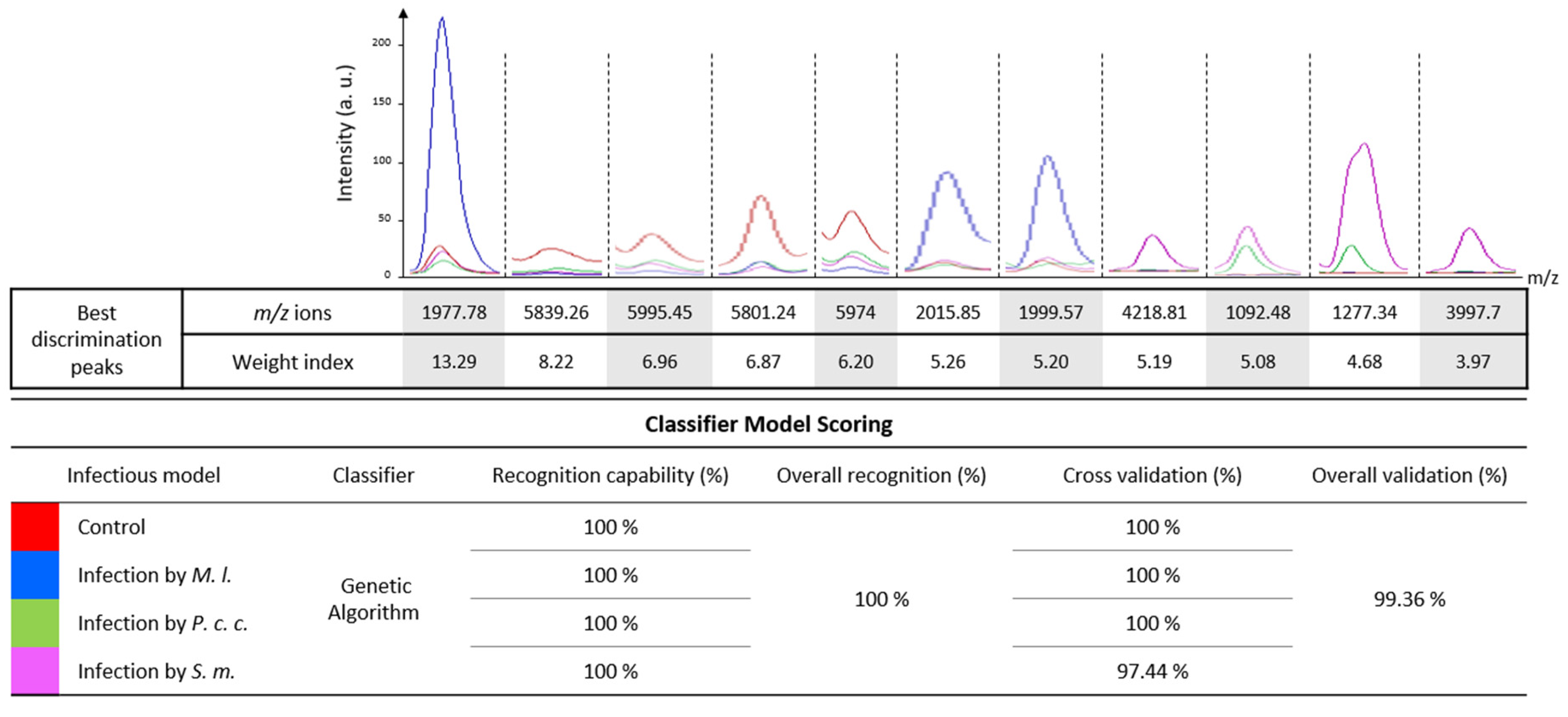
2.3. Development of a Computational Model to Discriminate between the Stressor and Its Impact
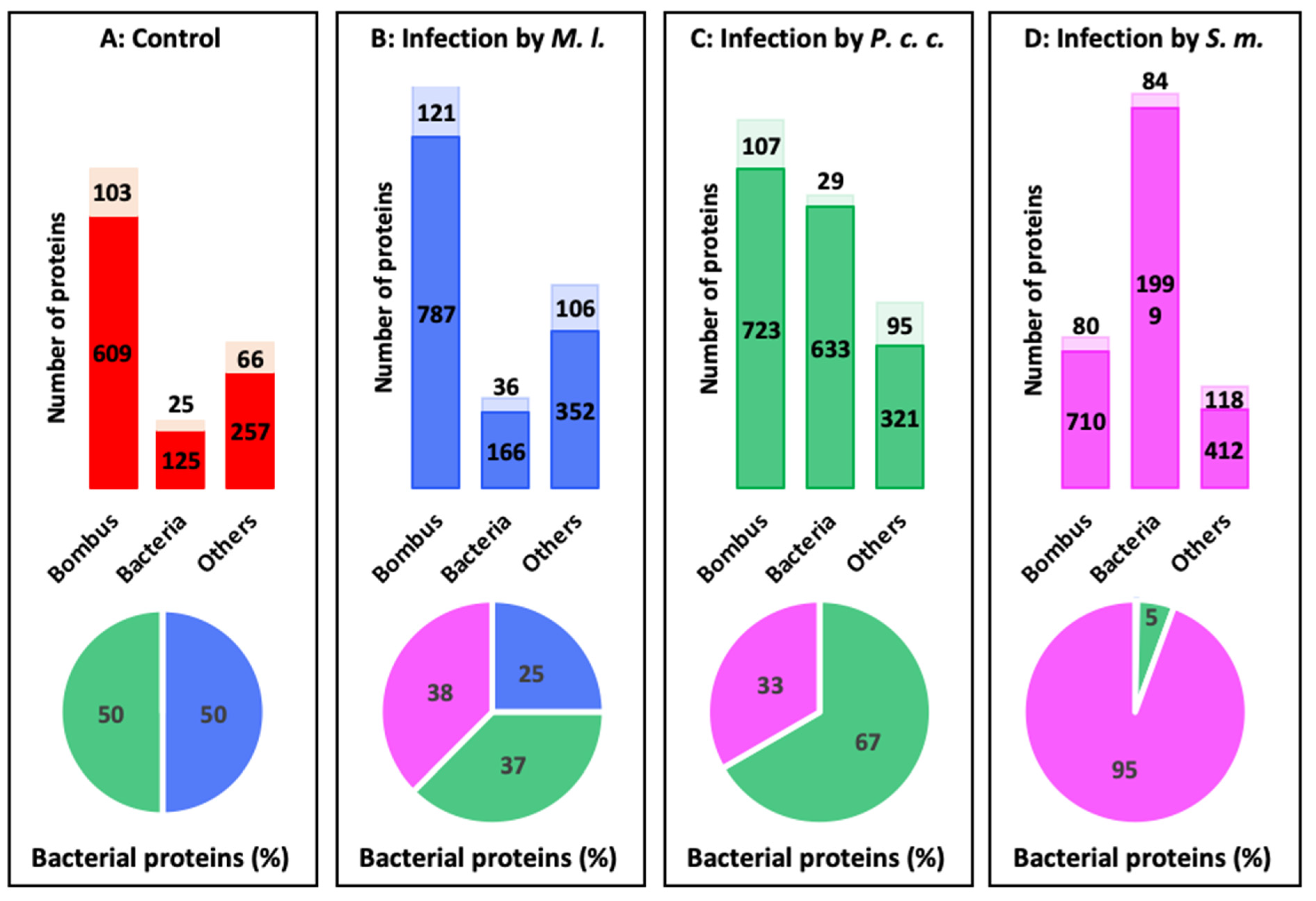
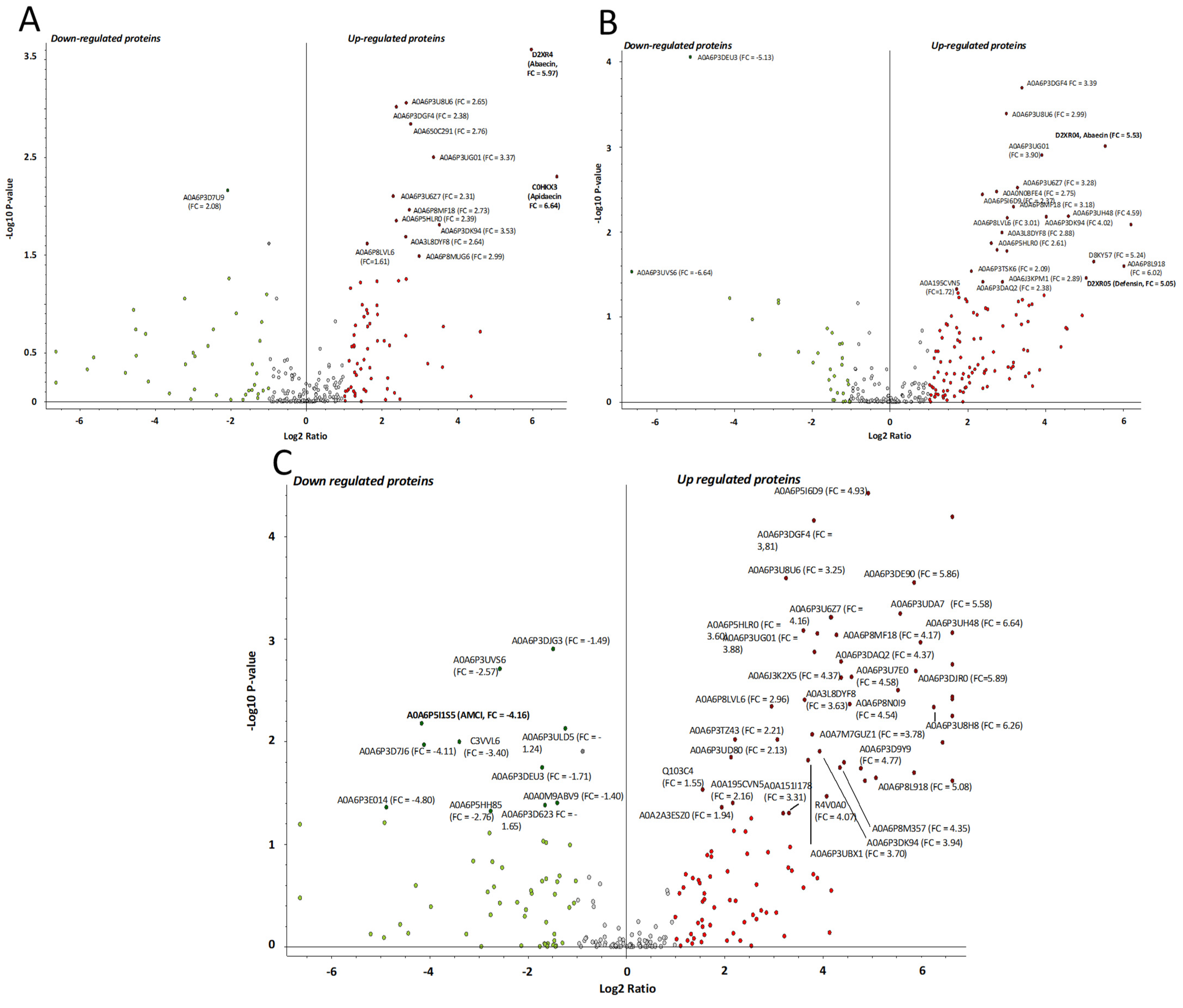
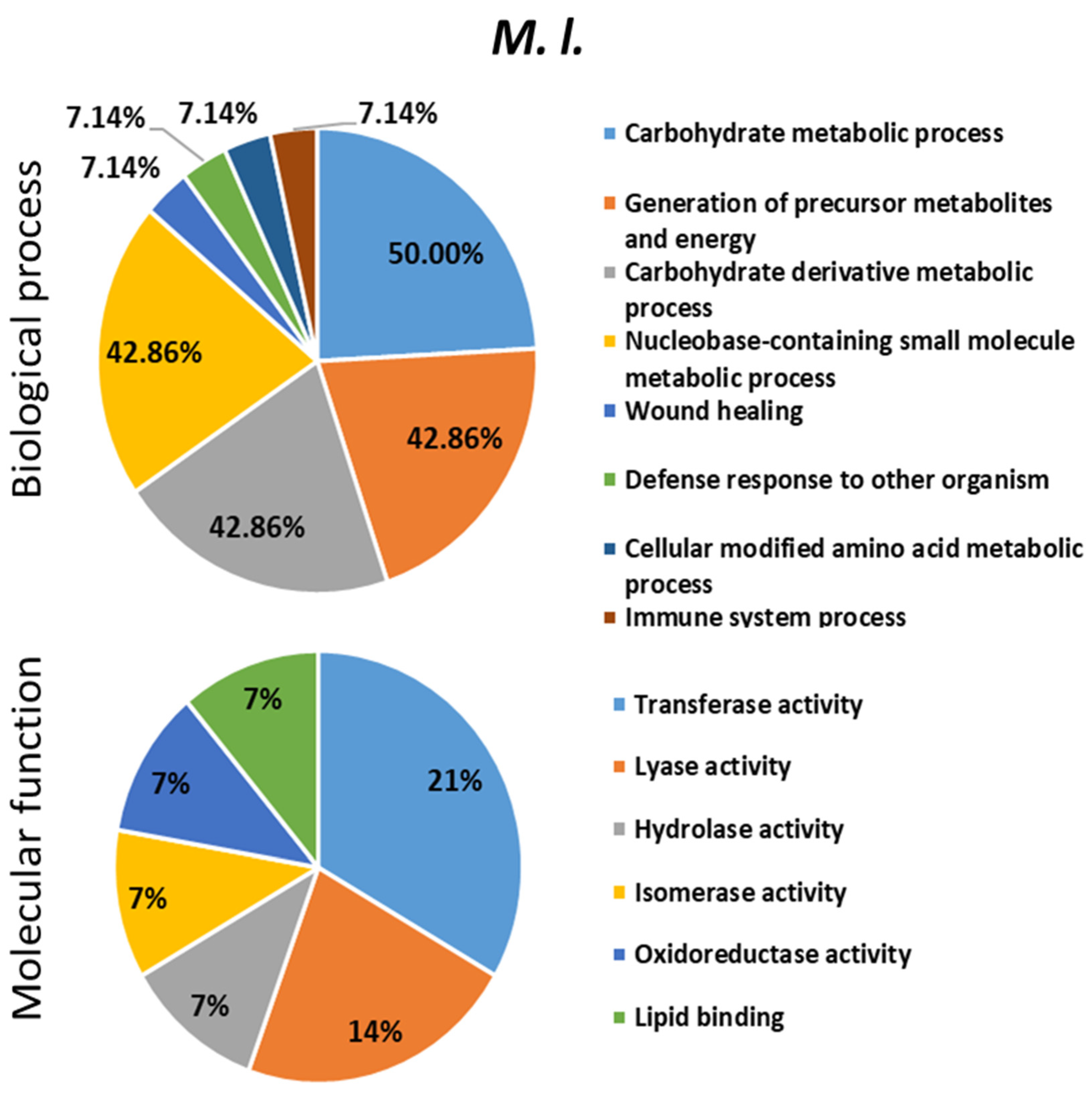
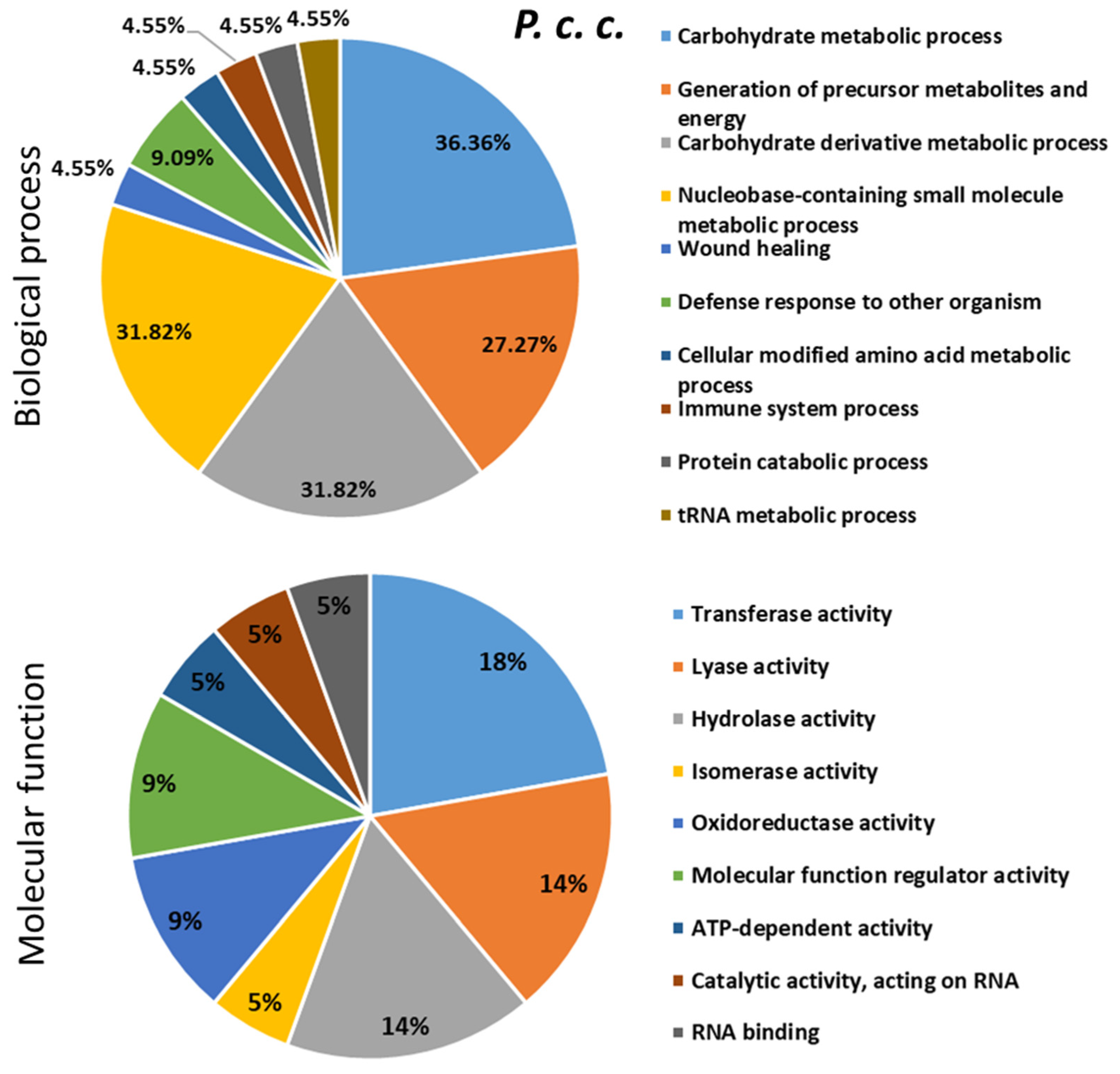
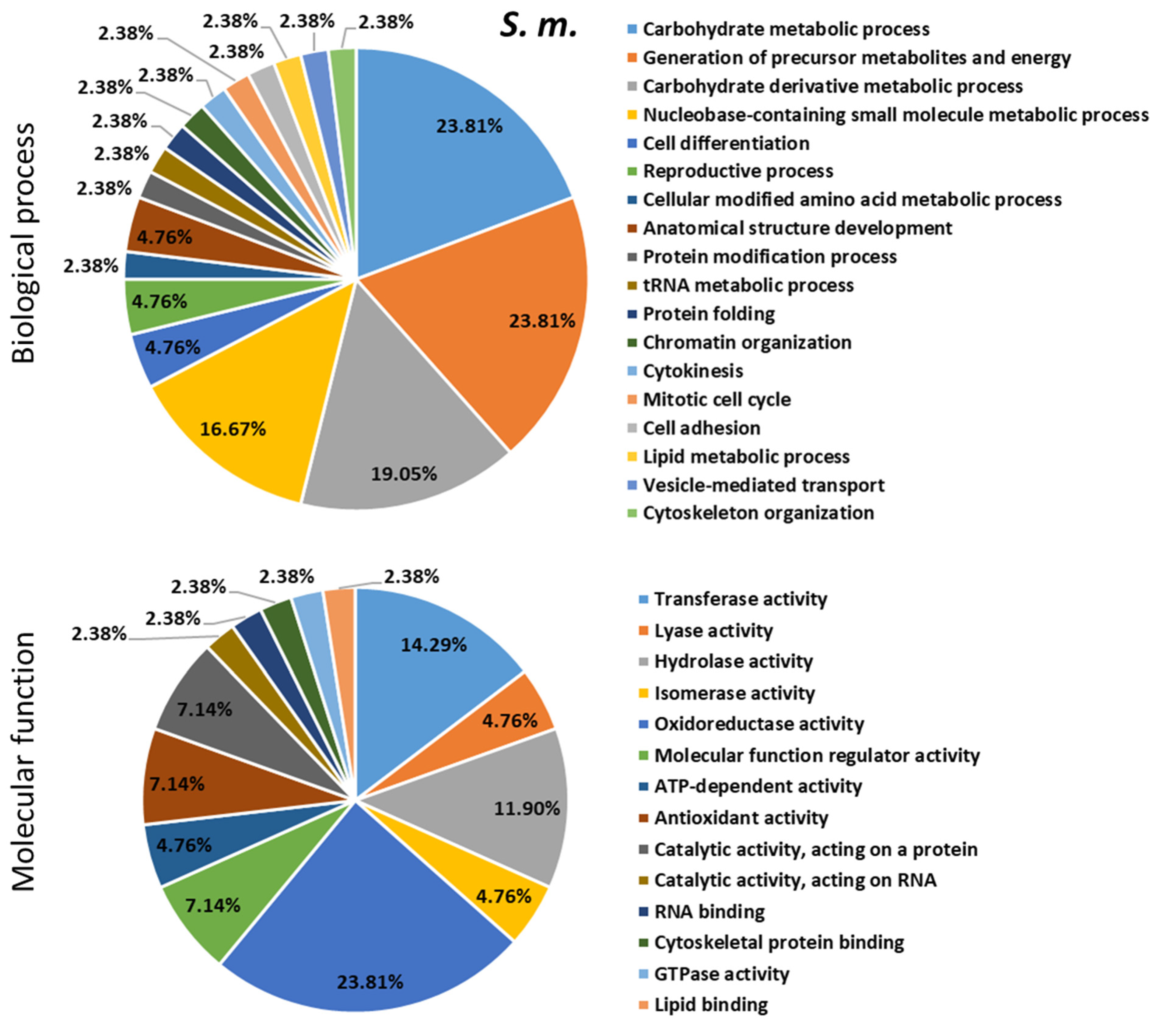
2.4. Proteomics Analysis of the Hemolymph of B. terrestris in the Context of Experimental Bacterial Infections
2.5. Treatment-Dependent Protein Intensity
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Bacterial Strains used for the Experimental Infections
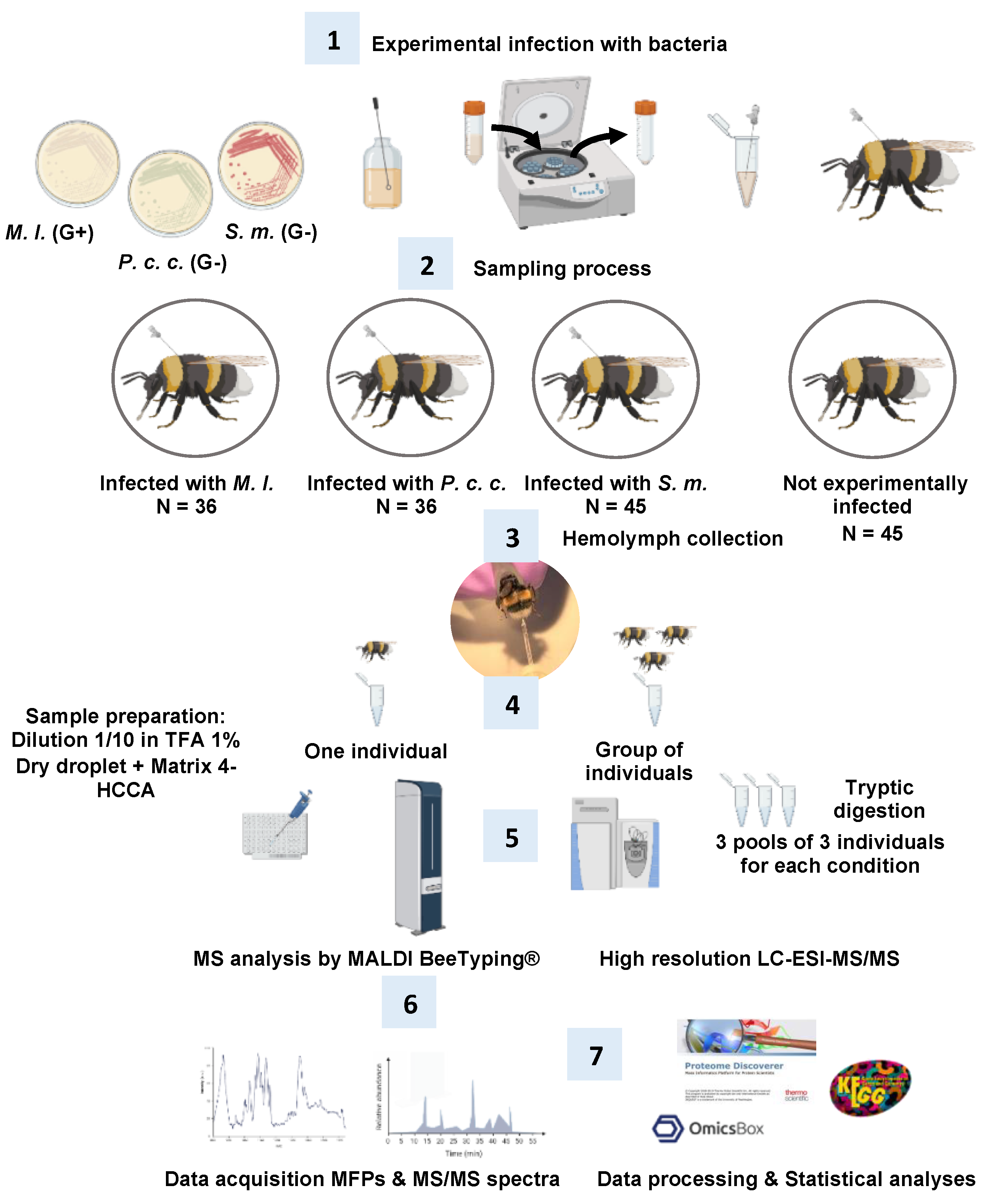
3.3. Bumble Bees, Infection Experiments, and Hemolymph Collection (Figure 10, Steps 2 and 3)
3.3.1. Bumble Bee Colonies
3.3.2. Bacterial Infections
3.3.3. Hemolymph Collection
3.3.4. Survival Rate Assessment
3.4. MALDI Molecular Mass Fingerprints or MALDI BeeTyping® (Figure 10, Steps 4–6)
3.4.1. Sample Preparation
3.4.2. Sample Analysis and Data Acquisition
3.4.3. MALDI Data Processing
3.5. Bottom-Up Proteomics: Off-Gel Digestion and LC-ESI-MS/MS (Figure 10, Steps 5–7)
3.5.1. Sample Preparation
3.5.2. Nano-LC-MS/MS Analysis
3.5.3. Database Searching and Protein Identification
3.6. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klein, A.M.; Vaissière, B.E.; Cane, J.H.; Steffan-Dewenter, I.; Cunningham, S.A.; Kremen, C.; Tscharntke, T. Importance of Pollinators in Changing Landscapes for World Crops. Proc. R. Soc. B Biol. Sci. 2007, 274, 303–313. [Google Scholar] [CrossRef]
- IPBES. Global Assessment Report on Biodiversity and Ecosystem Services of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services; Zenodo: Genève, Switzerland, 2019. [Google Scholar]
- Sánchez-Bayo, F.; Wyckhuys, K.A.G. Worldwide Decline of the Entomofauna: A Review of Its Drivers. Biol. Conserv. 2019, 232, 8–27. [Google Scholar] [CrossRef]
- Carvalho, F.P. Pesticides, Environment, and Food Safety. Food Energy Secur. 2017, 6, 48–60. [Google Scholar] [CrossRef]
- Dafni, A.; Kevan, P.; Gross, C.L.; Goka, K. Bombus terrestris, Pollinator, Invasive and Pest: An Assessment of Problems Associated with Its Widespread Introductions for Commercial Purposes. Appl. Entomol. Zool. 2010, 45, 101–113. [Google Scholar] [CrossRef]
- Velthuis, H.H.W.; van Doorn, A. A Century of Advances in Bumblebee Domestication and the Economic and Environmental Aspects of Its Commercialization for Pollination. Apidologie 2006, 37, 421–451. [Google Scholar] [CrossRef]
- Goulson, D.; Lye, G.C.; Darvill, B. Decline and Conservation of Bumble Bees. Annu. Rev. Entomol. 2008, 53, 191–208. [Google Scholar] [CrossRef]
- Goulson, D.; Nicholls, E.; Botias, C.; Rotheray, E.L. Bee Declines Driven by Combined Stress from Parasites, Pesticides, and Lack of Flowers. Science 2015, 347, 1255957. [Google Scholar] [CrossRef] [PubMed]
- Welchman, D.P.; Aksoy, S.; Jiggins, F.; Lemaitre, B. Insect Immunity: From Pattern Recognition to Symbiont-Mediated Host Defense. Cell Host Microbe 2009, 6, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Ali Mohammadie Kojour, M.; Han, Y.S.; Jo, Y.H. An Overview of Insect Innate Immunity. Entomol. Res. 2020, 50, 282–291. [Google Scholar] [CrossRef]
- Hoffmann, J.; Akira, S. Innate Immunity. Curr. Opin. Immunol. 2013, 25, 1–3. [Google Scholar] [CrossRef]
- Harder, J.; Schröder, J.-M. (Eds.) Antimicrobial Peptides; Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-24197-5. [Google Scholar]
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial Peptides: Application Informed by Evolution. Science 2020, 368, eaau5480. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.A. The Immune Response of Drosophila. Nature 2003, 426, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, G.M.; Robinson, G.E.; Gibbs, R.A.; Worley, K.C.; Evans, J.D.; Maleszka, R.; Robertson, H.M.; Weaver, D.B.; Beye, M.; Bork, P.; et al. Insights into Social Insects from the Genome of the Honeybee Apis mellifera. Nature 2006, 443, 931–949. [Google Scholar] [CrossRef]
- Eleftherianos, I.; Zhang, W.; Heryanto, C.; Mohamed, A.; Contreras, G.; Tettamanti, G.; Wink, M.; Bassal, T. Diversity of Insect Antimicrobial Peptides and Proteins—A Functional Perspective: A Review. Int. J. Biol. Macromol. 2021, 191, 277–287. [Google Scholar] [CrossRef]
- Choi, Y.S.; Choo, Y.M.; Lee, K.S.; Yoon, H.J.; Kim, I.; Je, Y.H.; Sohn, H.D.; Jin, B.R. Cloning and Expression Profiling of Four Antibacterial Peptide Genes from the Bumblebee Bombus ignitus. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 150, 141–146. [Google Scholar] [CrossRef]
- Erler, S.; Popp, M.; Lattorff, H.M.G. Dynamics of Immune System Gene Expression upon Bacterial Challenge and Wounding in a Social Insect (Bombus terrestris). PLoS ONE 2011, 6, e18126. [Google Scholar] [CrossRef]
- Chan, Q.W.; Foster, L.J. Changes in Protein Expression during Honey Bee Larval Development. Genome Biol. 2008, 9, R156. [Google Scholar] [CrossRef]
- Erban, T.; Petrova, D.; Harant, K.; Jedelsky, P.L.; Titera, D. Two-Dimensional Gel Proteome Analysis of Honeybee, Apis mellifera, Worker Red-Eye Pupa Hemolymph. Apidologie 2014, 45, 53–72. [Google Scholar] [CrossRef]
- Feng, M.; Ramadan, H.; Han, B.; Fang, Y.; Li, J. Hemolymph Proteome Changes during Worker Brood Development Match the Biological Divergences between Western Honey Bees (Apis mellifera) and Eastern Honey Bees (Apis cerana). BMC Genom. 2014, 15, 563. [Google Scholar] [CrossRef]
- Erban, T.; Harant, K.; Kamler, M.; Markovic, M.; Titera, D. Detailed Proteome Mapping of Newly Emerged Honeybee Worker Hemolymph and Comparison with the Red-Eye Pupal Stage. Apidologie 2016, 47, 805–817. [Google Scholar] [CrossRef]
- Ararso, Z.; Ma, C.; Qi, Y.; Feng, M.; Han, B.; Hu, H.; Meng, L.; Li, J. Proteome Comparisons between Hemolymph of Two Honeybee Strains (Apis mellifera ligustica) Reveal Divergent Molecular Basis in Driving Hemolymph Function and High Royal Jelly Secretion. J. Proteome Res. 2018, 17, 402–419. [Google Scholar] [CrossRef]
- Cabbri, R.; Ferlizza, E.; Nanetti, A.; Monari, E.; Andreani, G.; Galuppi, R.; Isani, G. Biomarkers of Nutritional Status in Honeybee Haemolymph: Effects of Different Biotechnical Approaches for Varroa destructor Treatment and Wintering Phase. Apidologie 2018, 49, 606–618. [Google Scholar] [CrossRef]
- Słowińska, M.; Nynca, J.; Bąk, B.; Wilde, J.; Siuda, M.; Ciereszko, A. 2D-DIGE Proteomic Analysis Reveals Changes in Haemolymph Proteome of 1-Day-Old Honey Bee (Apis mellifera) Workers in Response to Infection with Varroa destructor Mites. Apidologie 2019, 50, 632–656. [Google Scholar] [CrossRef]
- Bogaerts, A.; Baggerman, G.; Vierstraete, E.; Schoofs, L.; Verleyen, P. The Hemolymph Proteome of the Honeybee: Gel-Based or Gel-Free? Proteomics 2009, 9, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.; Coffey, M.; Kavanagh, K. Proteomic Analysis of Summer and Winter Apis mellifera Workers Shows Reduced Protein Abundance in Winter Samples. J. Insect Physiol. 2022, 139, 104397. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, B.; Hoffmann, J. The Host Defense of Drosophila melanogaster. Annu. Rev. Immunol. 2007, 25, 697–743. [Google Scholar] [CrossRef]
- Arafah, K.; Voisin, S.N.; Masson, V.; Alaux, C.; Le Conte, Y.; Bocquet, M.; Bulet, P. MALDI–MS Profiling to Address Honey Bee Health Status under Bacterial Challenge through Computational Modeling. Proteomics 2019, 19, 1900268. [Google Scholar] [CrossRef]
- Raymann, K.; Coon, K.L.; Shaffer, Z.; Salisbury, S.; Moran, N.A. Pathogenicity of Serratia marcescens Strains in Honey Bees. mBio 2018, 9, e01649-18. [Google Scholar] [CrossRef] [PubMed]
- Liegeois, S.; Ferrandon, D. Sensing Microbial Infections in the Drosophila melanogaster Genetic Model Organism. Immunogenetics 2022, 74, 35–62. [Google Scholar] [CrossRef]
- Bulet, P.; Uttenweiler-Joseph, S.; Moniatte, M.; Van Dorsselaer, A.; Hoffmann, J.A. Differential Display of Peptides Induced during the Immune Response of Drosophila: A Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry Study. J. Protein Chem. 1998, 17, 528–529. [Google Scholar]
- Uttenweiler-Joseph, S.; Moniatte, M.; Lagueux, M.; Van Dorsselaer, A.; Hoffmann, J.A.; Bulet, P. Differential Display of Peptides Induced during the Immune Response of Drosophila: A Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry Study. Proc. Natl. Acad. Sci. USA 1998, 95, 11342–11347. [Google Scholar] [CrossRef]
- Clemmons, A.W.; Lindsay, S.A.; Wasserman, S.A. An Effector Peptide Family Required for Drosophila Toll-Mediated Immunity. PLoS Pathog. 2015, 11, e1004876. [Google Scholar] [CrossRef] [PubMed]
- Houdelet, C.; Bocquet, M.; Bulet, P. Matrix-assisted Laser Desorption/Ionization Mass Spectrometry Biotyping, an Approach for Deciphering and Assessing the Identity of the Honeybee Pathogen Nosema. Rapid Commun. Mass Spectrom. 2021, 35, e8980. [Google Scholar] [CrossRef] [PubMed]
- Houdelet, C.; Sinpoo, C.; Chantaphanwattana, T.; Voisin, S.N.; Bocquet, M.; Chantawannakul, P.; Bulet, P. Proteomics of Anatomical Sections of the Gut of Nosema-Infected Western Honeybee (Apis mellifera) Reveals Different Early Responses to Nosema spp. Isolates. J. Proteome Res. 2021, 20, 804–817. [Google Scholar] [CrossRef] [PubMed]
- Lessons in Immunity—1st Edition. Available online: https://www.elsevier.com/books/lessons-in-immunity/ballarin/978-0-12-803252-7 (accessed on 21 November 2022).
- Burritt, N.L.; Foss, N.J.; Neeno-Eckwall, E.C.; Church, J.O.; Hilger, A.M.; Hildebrand, J.A.; Warshauer, D.M.; Perna, N.T.; Burritt, J.B. Sepsis and Hemocyte Loss in Honey Bees (Apis mellifera) Infected with Serratia marcescens Strain Sicaria. PLoS ONE 2016, 11, e0167752. [Google Scholar] [CrossRef]
- Steele, M.I.; Motta, E.V.S.; Gattu, T.; Martinez, D.; Moran, N.A. The Gut Microbiota Protects Bees from Invasion by a Bacterial Pathogen. Microbiol. Spectr. 2021, 9, e0039421. [Google Scholar] [CrossRef]
- Maeda, H. Role of Microbial Proteases in Pathogenesis. Microbiol. Immunol. 1996, 40, 685–699. [Google Scholar] [CrossRef]
- Matsumoto, K.; Maeda, H.; Takata, K.; Kamata, R.; Okamura, R. Purification and Characterization of Four Proteases from a Clinical Isolate of Serratia marcescens Kums 3958. J. Bacteriol. 1984, 157, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Haddix, P.L.; Shanks, R.M.Q. Prodigiosin Pigment of Serratia marcescens Is Associated with Increased Biomass Production. Arch. Microbiol. 2018, 200, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Houdelet, C.; Arafah, K.; Bocquet, M.; Bulet, P. Molecular Histoproteomy by MALDI Mass Spectrometry Imaging to Uncover Markers of the Impact of Nosema on Apis mellifera. Proteomics 2022, 22, e2100224. [Google Scholar] [CrossRef]
- Wu, Q.; Patočka, J.; Kuča, K. Insect Antimicrobial Peptides, a Mini Review. Toxins 2018, 10, 461. [Google Scholar] [CrossRef]
- Bulet, P.; Stöcklin, R. Insect Antimicrobial Peptides: Structures, Properties and Gene Regulation. Protein Pept. Lett. 2005, 12, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Piqué, N.; Miñana-Galbis, D.; Merino, S.; Tomás, J.M. Virulence Factors of Erwinia amylovora: A Review. Int. J. Mol. Sci. 2015, 16, 12836–12854. [Google Scholar] [CrossRef] [PubMed]
- Agrios, G. Plant Pathology, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 9780080473789, ISBN 978-0-12-044565-3. [Google Scholar]
- Perombelon, M.C.M.; Kelman, A. Ecology of the Soft Rot Erwinias. Annu. Rev. Phytopathol. 1980, 18, 361–387. [Google Scholar] [CrossRef]
- Basset, A.; Khush, R.S.; Braun, A.; Gardan, L.; Boccard, F.; Hoffmann, J.A.; Lemaitre, B. The Phytopathogenic Bacteria Erwinia carotovora Infects Drosophila and Activates an Immune Response. Proc. Natl. Acad. Sci. USA 2000, 97, 3376–3381. [Google Scholar] [CrossRef]
- Wael, L.; Greef, M.; Laere, O. The Honeybee as a Possible Vector of Erwinia amylovora (Burr.) Wislow et al. Acta Hortic. 1990, 273, 107–114. [Google Scholar] [CrossRef]
- Alexandrova, M.; Porrini, C.; Bazzi, C.; Carpana, E.; Bigliardi, M.; Sabatini, A.G. Erwinia amylovora Longevity in Beehives, Beehive Products and Honeybees. Acta Hortic. 2002, 590, 201–205. [Google Scholar] [CrossRef]
- Bertin, Y.; Chaucheyras-Durand, F.; Robbe-Masselot, C.; Durand, A.; de la Foye, A.; Harel, J.; Cohen, P.S.; Conway, T.; Forano, E.; Martin, C. Carbohydrate Utilization by Enterohaemorrhagic Escherichia coli O157:H7 in Bovine Intestinal Content. Environ. Microbiol. 2013, 15, 610–622. [Google Scholar] [CrossRef]
- Brissac, T.; Ziveri, J.; Ramond, E.; Tros, F.; Kock, S.; Dupuis, M.; Brillet, M.; Barel, M.; Peyriga, L.; Cahoreau, E.; et al. Gluconeogenesis, an Essential Metabolic Pathway for Pathogenic Francisella: Gluconeogenesis in Francisella Virulence. Mol. Microbiol. 2015, 98, 518–534. [Google Scholar] [CrossRef]
- Lourenco, A.P.; Florecki, M.M.; Simoes, Z.L.P.; Evans, J.D. Silencing of Apis mellifera Dorsal Genes Reveals Their Role in Expression of the Antimicrobial Peptide Defensin-1. Insect Mol. Biol. 2018, 27, 577–589. [Google Scholar] [CrossRef]
- Schlüns, H.; Crozier, R.H. Relish Regulates Expression of Antimicrobial Peptide Genes in the Honeybee, Apis mellifera, Shown by RNA Interference: Bee Relish Regulates Antimicrobial Peptide Genes. Insect Mol. Biol. 2007, 16, 753–759. [Google Scholar] [CrossRef]
- Rutschmann, S.; Kilinc, A.; Ferrandon, D. Cutting Edge: The Toll Pathway Is Required for Resistance to Gram-Positive Bacterial Infections in Drosophila. J. Immunol. 2002, 168, 1542–1546. [Google Scholar] [CrossRef]
- Shen, Z.-J.; Liu, Y.-J.; Gao, X.-H.; Liu, X.-M.; Zhang, S.-D.; Li, Z.; Zhang, Q.-W.; Liu, X.-X. Molecular Identification of Two Thioredoxin Genes From Grapholita molesta and Their Function in Resistance to Emamectin Benzoate. Front. Physiol. 2018, 9, 1421. [Google Scholar] [CrossRef]
- Mucci, C.A.; Ramirez, L.; Giffoni, R.S.; Lamattina, L. Cold Stress Induces Specific Antioxidant Responses in Honey Bee Brood. Apidologie 2021, 52, 596–607. [Google Scholar] [CrossRef]
- Zaluski, R.; Bittarello, A.C.; Vieira, J.C.S.; Braga, C.P.; Padilha, P.D.M.; Fernandes, M.D.S.; Bovi, T.D.S.; Orsi, R.D.O. Modification of the Head Proteome of Nurse Honeybees (Apis mellifera) Exposed to Field-Relevant Doses of Pesticides. Sci. Rep. 2020, 10, 2190. [Google Scholar] [CrossRef]
- Corona, M.; Robinson, G.E. Genes Blackwell Publishing Ltd. of the Antioxidant System of the Honey Bee: Annotation and Phylogeny. Insect Mol. Biol. 2006, 15, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Muller, U.R.; Jutel, M.; Reimers, A.; Zumkehr, J.; Huber, C.; Kriegel, C.; Steiner, U.; Haeberli, G.; Akdis, M.; Helbling, A.; et al. Clinical and Immunologic Effects of H1 Antihistamine Preventive Medication during Honeybee Venom Immunotherapy. J. Allergy Clin. Immunol. 2008, 122, 1001–1007 e4. [Google Scholar] [CrossRef] [PubMed]
- Enayati, A.A.; Ranson, H.; Hemingway, J. Insect Glutathione Transferases and Insecticide Resistance. Insect Mol. Biol. 2005, 14, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Lumjuan, N.; Rajatileka, S.; Changsom, D.; Wicheer, J.; Leelapat, P.; Prapanthadara, L.; Somboon, P.; Lycett, G.; Ranson, H. The Role of the Aedes aegypti Epsilon Glutathione Transferases in Conferring Resistance to DDT and Pyrethroid Insecticides. Insect Biochem. Mol. Biol. 2011, 41, 203–209. [Google Scholar] [CrossRef]
- Rinehart, J.P.; Li, A.; Yocum, G.D.; Robich, R.M.; Hayward, S.A.L.; Denlinger, D.L. Up-Regulation of Heat Shock Proteins Is Essential for Cold Survival during Insect Diapause. Proc. Natl. Acad. Sci. USA 2007, 104, 11130–11137. [Google Scholar] [CrossRef]
- Wrońska, A.K.; Boguś, M.I. Heat Shock Proteins (HSP 90, 70, 60, and 27) in Galleria mellonella (Lepidoptera) Hemolymph Are Affected by Infection with Conidiobolus coronatus (Entomophthorales). PLoS ONE 2020, 15, e0228556. [Google Scholar] [CrossRef] [PubMed]
- Masson, V.; Arafah, K.; Voisin, S.; Bulet, P. Comparative Proteomics Studies of Insect Cuticle by Tandem Mass Spectrometry: Application of a Novel Proteomics Approach to the Pea Aphid Cuticular Proteins. Proteomics 2018, 18, 1700368. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | Pathway | Pathway ID | #Seqs |
|---|---|---|---|
| M. l. | Glycolysis/gluconeogenesis | ko00010 | 7 |
| M. l. | Glucose metabolism | R-DME-70326 | 5 |
| M. l. | Metabolism of carbohydrates | R-DME-71387 | 5 |
| M. l. | Gluconeogenesis | R-DME-70263 | 4 |
| M. l. | Innate Immune System | R-DME-168249 | 4 |
| M. l. | Glycolysis | R-DME-70171 | 4 |
| M. l. | Methane metabolism | ko00680 | 4 |
| M. l. | HIF-1 signaling pathway | ko04066 | 4 |
| M. l. | Carbon fixation in photosynthetic organisms | ko00710 | 4 |
| M. l. | Neutrophil degranulation | R-DME-6798695 | 3 |
| P. c. c. | Glycolysis/Gluconeogenesis | ko00010 | 7 |
| P. c. c. | Innate Immune System | R-DME-168249 | 6 |
| P. c. c. | Metabolism of carbohydrates | R-DME-71387 | 5 |
| P. c. c. | Glucose metabolism | R-DME-70326 | 5 |
| P. c. c. | Gluconeogenesis | R-DME-70263 | 4 |
| P. c. c. | Glycolysis | R-DME-70171 | 4 |
| P. c. c. | Neutrophil degranulation | R-DME-6798695 | 4 |
| P. c. c. | Methane metabolism | ko00680 | 4 |
| P. c. c. | HIF-1 signaling pathway | ko04066 | 4 |
| P. c. c. | Carbon fixation in photosynthetic organisms | ko00710 | 4 |
| S. m. | Innate Immune System | R-DME-168249 | 10 |
| S. m. | Metabolism of carbohydrates | R-DME-71387 | 8 |
| S. m. | Glycolysis/Gluconeogenesis | ko00010 | 8 |
| S. m. | Neutrophil degranulation | R-DME-6798695 | 7 |
| S. m. | Carbon fixation in photosynthetic organisms | ko00710 | 7 |
| S. m. | Platelet activation, signaling and aggregation | R-DME-76002 | 6 |
| S. m. | Glucose metabolism | R-DME-70326 | 6 |
| S. m. | Methane metabolism | ko00680 | 6 |
| S. m. | Response to elevated platelet cytosolic Ca2+ | R-DME-76005 | 5 |
| S. m. | Gluconeogenesis | R-DME-70263 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bournonville, L.; Askri, D.; Arafah, K.; Voisin, S.N.; Bocquet, M.; Bulet, P. Unraveling the Bombus terrestris Hemolymph, an Indicator of the Immune Response to Microbial Infections, through Complementary Mass Spectrometry Approaches. Int. J. Mol. Sci. 2023, 24, 4658. https://doi.org/10.3390/ijms24054658
Bournonville L, Askri D, Arafah K, Voisin SN, Bocquet M, Bulet P. Unraveling the Bombus terrestris Hemolymph, an Indicator of the Immune Response to Microbial Infections, through Complementary Mass Spectrometry Approaches. International Journal of Molecular Sciences. 2023; 24(5):4658. https://doi.org/10.3390/ijms24054658
Chicago/Turabian StyleBournonville, Lorène, Dalel Askri, Karim Arafah, Sébastien N. Voisin, Michel Bocquet, and Philippe Bulet. 2023. "Unraveling the Bombus terrestris Hemolymph, an Indicator of the Immune Response to Microbial Infections, through Complementary Mass Spectrometry Approaches" International Journal of Molecular Sciences 24, no. 5: 4658. https://doi.org/10.3390/ijms24054658
APA StyleBournonville, L., Askri, D., Arafah, K., Voisin, S. N., Bocquet, M., & Bulet, P. (2023). Unraveling the Bombus terrestris Hemolymph, an Indicator of the Immune Response to Microbial Infections, through Complementary Mass Spectrometry Approaches. International Journal of Molecular Sciences, 24(5), 4658. https://doi.org/10.3390/ijms24054658