
Zebra-Sphinx: Modeling Sphingolipidoses in Zebrafish



Abstract
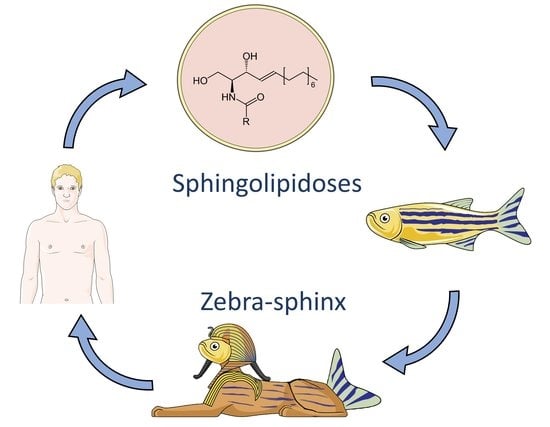
1. Introduction
2. Sphingolipidoses
2.1. Gaucher Disease
2.2. Fabry Disease
2.3. Niemann–Pick Disease
2.4. Krabbe Disease
2.5. Farber Lipogranulomatosis
2.6. GM1 and GM2 Gangliosidoses
2.6.1. GM1 Gangliosidosis
2.6.2. GM2 Gangliosidosis
AB Variant
Tay–Sachs Disease
Sandhoff Disease
2.7. Metachromatic Leukodystrophy
3. Sphingolipids in Zebrafish
3.1. Zebrafish Lipidomics
3.2. Sphingolipid Metabolizing Enzymes in Zebrafish
4. Zebrafish as an Animal Model for Sphingolipidoses: The “Zebra-Sphinx” Platform
4.1. Gaucher Disease
4.2. Fabry Disease
4.3. Niemann–Pick Disease
4.4. Krabbe Disease
4.5. Farber Lipogranulomatosis
4.6. GM2 Gangliosidoses
4.6.1. Tay–Sachs Disease
4.6.2. Sandhoff Disease
4.7. Metachromatic Leukodystrophy
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Thudichum, J.L.W. A Treatise on the Chemical Constitution of the Brain: Based Throughout upon Original Researches. Glasgow Med. J. 1884, 22, 363–364. [Google Scholar]
- Breslow, D.K. Sphingolipid homeostasis in the endoplasmic reticulum and beyond. Cold Spring Harb. Perspect. Biol. 2013, 5, a013326. [Google Scholar] [CrossRef]
- Futerman, A.H.; Riezman, H. The ins and outs of sphingolipid synthesis. Trends Cell Biol. 2005, 15, 312–318. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [CrossRef]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell. Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Grassi, S.; Chiricozzi, E.; Mauri, L.; Sonnino, S.; Prinetti, A. Sphingolipids and neuronal degeneration in lysosomal storage disorders. J. Neurochem. 2019, 148, 600–611. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers. 2018, 4, 27. [Google Scholar] [CrossRef]
- Abed Rabbo, M.; Khodour, Y.; Kaguni, L.S.; Stiban, J. Sphingolipid lysosomal storage diseases: From bench to bedside. Lipids. Health Dis. 2021, 20, 44. [Google Scholar] [CrossRef]
- Eckhardt, M. Pathology and current treatment of neurodegenerative sphingolipidoses. Neuromolecular Med. 2010, 12, 362–382. [Google Scholar] [CrossRef]
- Santos, R.; Amaral, O. Advances in Sphingolipidoses: CRISPR-Cas9 editing as an option for modelling and therapy. Int. J. Mol. Sci. 2019, 20, 5897. [Google Scholar] [CrossRef]
- Fernandez-Pereira, C. Therapeutic approaches in lysosomal storage diseases. Biomolecules 2021, 11, 1775. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Mistry, P.K. Therapies for lysosomal storage diseases: Principles, practice, and prospects for refinements based on evolving science. Mol. Genet. Metab. 2022, 137, 81–91. [Google Scholar] [CrossRef]
- Andrade-Campos, M.M. Identification of risk features for complication in Gaucher’s disease patients: A machine learning analysis of the Spanish registry of Gaucher disease. Orphanet. J. Rare Dis. 2020, 15, 256. [Google Scholar] [CrossRef]
- Cox, T.M.; Cachón-González, M.B. The cellular pathology of lysosomal diseases. J. Pathol. 2021, 226, 241–254. [Google Scholar] [CrossRef]
- Roh, J.; Subramanian, S.; Weinreb, N.J.; Kartha, R.V. Gaucher disease-more than just a rare lipid storage disease. J. Mol. Med. 2022, 100, 499–518. [Google Scholar] [CrossRef]
- Ługowska, A. Gene expression profile in patients with Gaucher disease indicates activation of inflammatory processes. Sci. Rep. 2019, 9, 6060. [Google Scholar] [CrossRef]
- Maor, G. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Di Fonzo, A.B. Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef]
- Alaei, M.; Jafari, N.; Rohani, F.; Ahmadabadi, F.; Azadi, R. Are There Neurological Symptoms in Type 1 of Gaucher Disease? Iran. J. Child. Neurol. 2018, 12, 99–106. [Google Scholar] [PubMed]
- Stone, W.L.; Basit, H.; Master, S.R. Gaucher disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK448080/ (accessed on 19 November 2022).
- Limgala, R.P.; Goker-Alpan, O. Effect of Substrate Reduction Therapy in Comparison to Enzyme Replacement Therapy on Immune Aspects and Bone Involvement in Gaucher Disease. Biomolecules 2020, 10, 526. [Google Scholar] [CrossRef]
- Istaiti, M. Upgrading the evidence for the use of ambroxol in Gaucher disease and GBA related Parkinson: Investigator initiated registry based on real life data. Am. J. Hematol. 2021, 96, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R. Chapter 17—Fabry disease. Handb. Clin. Neurol. 2015, 132, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Turkmen, K.; Baloglu, I. Fabry disease: Where are we now? Int. Urol. Nephrol. 2020, 52, 2113–2122. [Google Scholar] [CrossRef]
- Germain, D.P. Challenging the traditional approach for interpreting genetic variants: Lessons from Fabry disease. Clin. Genet. 2022, 101, 390–402. [Google Scholar] [CrossRef]
- Stamerra, C.A.; Del Pinto, R.; di Giosia, P.; Ferri, C.; Sahebkar, A. Anderson-Fabry Disease: From Endothelial Dysfunction to Emerging Therapies. Adv. Pharmacol. Pharm. Sci. 2021, 2021, 5548445. [Google Scholar] [CrossRef]
- Li, X. Fabry disease: Mechanism and therapeutics strategies. Front. Pharmacol. 2022, 13, 1025740. [Google Scholar] [CrossRef]
- Ivanova, M.M.; Changsila, E.; Iaonou, C.; Goker-Alpan, O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE. 2019, 14, e0210617. [Google Scholar] [CrossRef]
- Song, H.Y. reversal of the inflammatory responses in Fabry Patient iPSC-Derived cardiovascular endothelial cells by CRISPR/Cas9-Corrected mutation. Int. J. Mol. Sci. 2021, 22, 2381. [Google Scholar] [CrossRef]
- Kok, K. Fabry Disease: Molecular basis, pathophysiology, diagnostics and potential therapeutic directions. Biomolecules 2021, 11, 271. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Tapia, D.; Kimonis, V.E.; Lombardo, D.M. Regional strain pattern and correlation with cardiac magnetic resonance imaging in Fabry Disease. J. Cardiovasc. Echogr. 2021, 31, 131–136. [Google Scholar] [CrossRef]
- Aguiar, P. Biomarkers of Myocardial Fibrosis: Revealing the Natural History of Fibrogenesis in Fabry Disease Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e007124. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: Evidence for a better outcome with early treatment. Circulation 2009, 119, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Brand, E. Fabry disease: The current treatment landscape. Drugs 2021, 81, 635–645. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, S.J.; Hollak, C.E.M.; van Kuilenburg, A.B.P.; Langeveld, M. Developments in the treatment of Fabry disease. J. Inherit. Metab. Dis. 2020, 43, 908–921. [Google Scholar] [CrossRef]
- Torres, S. Lysosomal and mitochondrial liaisons in Niemann-Pick disease. Front. Physiol. 2017, 8, 982. [Google Scholar] [CrossRef]
- Hollak, C.E. Acid sphingomyelinase (Asm) deficiency patients in The Netherlands and Belgium: Disease spectrum and natural course in attenuated patients. Mol. Genet. Metab. 2012, 107, 526–533. [Google Scholar] [CrossRef]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef]
- McGovern, M.M.; Avetisyan, R.; Sanson, B.J.; Lidove, O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J. Rare Dis. 2017, 12, 41. [Google Scholar] [CrossRef]
- Aldosari, M.H. Liposome-targeted recombinant human acid sphingomyelinase: Production, formulation, and in vitro evaluation. Eur. J. Pharm. Biopharm. 2019, 137, 185–195. [Google Scholar] [CrossRef]
- Diaz, G.A. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genet. Med. 2021, 23, 1543–1550. [Google Scholar] [CrossRef]
- Sitarska, D.; Tylki-Szymańska, A.; Ługowska, A. Treatment trials in Niemann-Pick type C disease. Metab. Brain Dis. 2021, 36, 2215–2221. [Google Scholar] [CrossRef]
- Geberhiwot, T. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J. Rare Dis. 2018, 13, 50. [Google Scholar] [CrossRef] [PubMed]
- Gumus, E. Niemann-Pick disease type C in the newborn period: A single-center experience. Eur. J. Pediatr. 2017, 176, 1669–1676. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, B.H.; Lee, Y.S.; Kang, K.S. Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 2007, 360, 593–599. [Google Scholar] [CrossRef]
- Lee, S.E. Human iNSC-derived brain organoid model of lysosomal storage disorder in Niemann-Pick disease type C. Cell Death Dis. 2020, 11, 1059. [Google Scholar] [CrossRef]
- Bradbury, A.M.; Bongarzone, E.R.; Sands, M.S. Krabbe disease: New hope for an old disease. Neurosci Lett. 2021, 752, 135841. [Google Scholar] [CrossRef] [PubMed]
- Rafi, M.A. Krabbe disease: A personal perspective and hypothesis. Bioimpacts 2022, 12, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Feltri, M.L. Mechanisms of demyelination and neurodegeneration in globoid cell leukodystrophy. Glia 2021, 69, 2309–2331. [Google Scholar] [CrossRef]
- Hawkins-Salsbury, J.A. Psychosine, the cytotoxic sphingolipid that accumulates in globoid cell leukodystrophy, alters membrane architecture. J. Lipid. Res. 2013, 54, 3303–3311. [Google Scholar] [CrossRef]
- White, A.B. Psychosine accumulates in membrane microdomains in the brain of Krabbe patients, disrupting the raft architecture. J. Neurosci. 2009, 29, 6068–6077. [Google Scholar] [CrossRef] [PubMed]
- Belleri, M.; Ronca, R.; Coltrini, D.; Nico, B.; Ribatti, D.; Poliani, P.L.; Giacomini, A.; Alessi, P.; Marchesini, S.; Santos, M.B.; et al. Inhibition of angiogenesis by β-galactosylceramidase deficiency in globoid cell leukodystrophy. Brain. 2013, 136, 2859–2875. [Google Scholar] [CrossRef] [PubMed]
- Coltrini, D.; Chandran, A.M.K.; Belleri, M.; Poliani, P.L.; Cominelli, M.; Pagani, F.; Capra, M.; Calza, S.; Prioni, S.; Mauri, L.; et al. β-Galactosylceramidase deficiency causes upregulation of long pentraxin-3 in the central nervous system of Krabbe patients and Twitcher Mice. Int. J. Mol. Sci. 2022, 23, 9436. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.M. Consensus guidelines for newborn screening, diagnosis and treatment of infantile Krabbe disease. Orphanet J. Rare Dis. 2018, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Yoon, I.C.; Bascou, N.A.; Poe, M.D.; Szabolcs, P.; Escolar, M.L. Long-term neurodevelopmental outcomes of hematopoietic stem cell transplantation for late-infantile Krabbe disease. Blood 2021, 137, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Yan, C.; Ji, K.; Lin, P.; Chi, L.; Zhao, X.; Zhao, Y. Adult-onset Krabbe disease in two generations of a Chinese family. Ann. Transl. Med. 2018, 6, 174. [Google Scholar] [CrossRef]
- Elsea, S.H.; Solyom, A.; Martin, K.; Harmatz, P.; Mitchell, J.; Lampe, C.; Grant, C.; Selim, L.; Mungan, N.O.; Guelbert, N.; et al. ASAH1 pathogenic variants associated with acid ceramidase deficiency: Farber disease and spinal muscular atrophy with progressive myoclonic epilepsy. Hum. Mutat. 2020, 41, 1469–1487. [Google Scholar] [CrossRef]
- Sands, M.S. Farber disease: Understanding a fatal childhood disorder and dissecting ceramide biology. EMBO Mol. Med. 2013, 5, 799–801. [Google Scholar] [CrossRef]
- Ehlert, K.; Frosch, M.; Fehse, N.; Zander, A.; Roth, J.; Vormoor, J. Farber disease: Clinical presentation, pathogenesis and a new approach to treatment. Pediatr. Rheumatol. Online J. 2007, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Sandhoff, K.; Harzer, K. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Ganglioside Metabolism and Its Inherited Diseases. Methods Mol. Biol. 2018, 1804, 97–141. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, E.R.; Annunziata, I.; d’Azzo, A.; Platt, F.M.; Tifft, C.J.; Stepien, K.M. GM1 Gangliosidosis-A Mini-Review. Front. Genet. 2021, 12, 734878. [Google Scholar] [CrossRef] [PubMed]
- Nestrasil, I.; Ahmed, A.; Utz, J.M.; Rudser, K.; Whitley, C.B.; Jarnes-Utz, J.R. Distinct progression patterns of brain disease in infantile and juvenile gangliosidoses: Volumetric quantitative MRI study. Mol. Genet. Metab. 2018, 123, 97–104. [Google Scholar] [CrossRef]
- Jarnes Utz, J.R.; Kim, S.; King, K.; Ziegler, R.; Schema, L.; Redtree, E.S.; Whitley, C.B. Infantile gangliosidoses: Mapping a timeline of clinical changes. Mol. Genet. Metab. 2017, 121, 170–179. [Google Scholar] [CrossRef]
- Rha, A.K.; Maguire, A.S.; Martin, D.R. GM1 Gangliosidosis: Mechanisms and Management. Appl. Clin. Genet. 2021, 14, 209–233. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Walterfang, M.; Patterson, M.C. Miglustat in Niemann-Pick disease type C patients: A review. Orphanet J. Rare. Dis. 2018, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Hollak, C.E.; Boot, R.G.; Groener, J.E.; Maas, M. Substrate reduction therapy of glycosphingolipid storage disorders. J. Inherit. Metab. Dis. 2006, 29, 449–456. [Google Scholar] [CrossRef]
- Fischetto, R.; Palladino, V.; Mancardi, M.; Giacomini, T.; Palladino, S.; Gaeta, A.; Di Rocco, M.; Zampini, L.; Lassandro, G.; Favia, V.; et al. Substrate reduction therapy with Miglustat in pediatric patients with GM1 type 2 gangliosidosis delays neurological involvement: A multicenter experience. Mol. Genet. Genomic. Med. 2020, 8, e1371. [Google Scholar] [CrossRef]
- Leal, A.F.; Benincore-Flórez, E.; Solano-Galarza, D.; Garzón Jaramillo, R.G.; Echeverri-Peña, O.Y.; Suarez, D.A.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J. GM2 Gangliosidoses: Clinical Features, Pathophysiological Aspects, and Current Therapies. Int. J. Mol. Sci. 2020, 21, 6213. [Google Scholar] [CrossRef]
- Ganne, B.; Dauriat, B.; Richard, L.; Lamari, F.; Ghorab, K.; Magy, L.; Benkirane, M.; Perani, A.; Marquet, V.; Calvas, P.; et al. GM2 gangliosidosis AB variant: First case of late onset and review of the literature. Neurol. Sci. 2022, 43, 6517–6527. [Google Scholar] [CrossRef] [PubMed]
- Tropak, M.B.; Yonekawa, S.; Karumuthil-Melethil, S.; Thompson, P.; Wakarchuk, W.; Gray, S.J.; Walia, J.S.; Mark, B.L.; Mahuran, D. Construction of a hybrid β-hexosaminidase subunit capable of forming stable homodimers that hydrolyze GM2 ganglioside in vivo. Mol. Ther. Methods. Clin. Dev. 2016, 3, 15057. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.J.; Tăbăran, A.F.; Overn, P.; O’Sullivan, M.G.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; Whitley, C.B. A novel gene editing system to treat both Tay-Sachs and Sandhoff diseases. Gene Ther. 2020, 27, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Cataltepe, O.; Puri, A.; Batista, A.R.; Moser, R.; McKenna-Yasek, D.; Douthwright, C.; Gernoux, G.; Blackwood, M.; Mueller, C.; et al. AAV gene therapy for Tay-Sachs disease. Nat. Med. 2022, 28, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Kot, S.; Karumuthil-Melethil, S.; Woodley, E.; Zaric, V.; Thompson, P.; Chen, Z.; Lykken, E.; Keimel, J.G.; Kaemmerer, W.F.; Gray, S.J. Investigating Immune Responses to the scAAV9-. Int. J. Mol. Sci. 2021, 22, 6751. [Google Scholar] [CrossRef]
- Solovyeva, V.V.; Shaimardanova, A.A.; Chulpanova, D.S.; Kitaeva, K.V.; Chakrabarti, L.; Rizvanov, A.A. New Approaches to Tay-Sachs Disease Therapy. Front. Physiol. 2018, 9, 1663. [Google Scholar] [CrossRef]
- Ihsan Fazal, M.; Kacprzyk, R.; Timson, D.J. In silico analysis of the effects of disease-associated mutations of β-hexosaminidase A in Tay-Sachs disease. J. Genet. 2020, 99, 42. [Google Scholar] [CrossRef]
- Maegawa, G.H.; Stockley, T.; Tropak, M.; Banwell, B.; Blaser, S.; Kok, F.; Giugliani, R.; Mahuran, D.; Clarke, J.T. The natural history of juvenile or subacute GM2 gangliosidosis: 21 New cases and literature review of 134 previously reported. Pediatrics 2006, 118, e1550–e1562. [Google Scholar] [CrossRef]
- Májovská, J.; Hennig, A.; Nestrasil, I.; Schneider, S.A.; Jahnová, H.; Vaněčková, M.; Magner, M.; Dušek, P. Pontocerebellar atrophy is the hallmark neuroradiological finding in late-onset Tay-Sachs disease. Neurol. Sci. 2022, 43, 3273–3281. [Google Scholar] [CrossRef]
- Tim-Aroon, T.; Wichajarn, K.; Katanyuwong, K.; Tanpaiboon, P.; Vatanavicharn, N.; Sakpichaisakul, K.; Kongkrapan, A.; Eu-Ahsunthornwattana, J.; Thongpradit, S.; Moolsuwan, K.; et al. Infantile onset Sandhoff disease: Clinical manifestation and a novel common mutation in Thai patients. BMC Pediatr. 2021, 21, 22. [Google Scholar] [CrossRef]
- García Morales, L.; Mustelier Bécquer, R.G.; Pérez Joglar, L.; Zaldívar Vaillant, T. Sandhoff disease in the elderly: A case study. Amyotroph. Lateral Scler. Front. Degener 2022, 23, 137–138. [Google Scholar] [CrossRef]
- Masingue, M.; Dufour, L.; Lenglet, T.; Saleille, L.; Goizet, C.; Ayrignac, X.; Ory-Magne, F.; Barth, M.; Lamari, F.; Mandia, D.; et al. Natural History of Adult Patients with GM2 Gangliosidosis. Ann. Neurol. 2020, 87, 609–617. [Google Scholar] [CrossRef]
- Alonso-Pérez, J.; Casasús, A.; Gimenez-Muñoz, Á.; Duff, J.; Rojas-Garcia, R.; Illa, I.; Straub, V.; Töpf, A.; Díaz-Manera, J. Late onset Sandhoff disease presenting with lower motor neuron disease and stuttering. Neuromuscul. Disord. 2021, 31, 769–772. [Google Scholar] [CrossRef]
- Shaimardanova, A.A.; Chulpanova, D.S.; Solovyeva, V.V.; Mullagulova, A.I.; Kitaeva, K.V.; Allegrucci, C.; Rizvanov, A.A. Metachromatic Leukodystrophy: Diagnosis, Modeling, and Treatment Approaches. Front. Med. 2020, 7, 576221. [Google Scholar] [CrossRef]
- Hossain, M.A.; Hasegawa-Ogawa, M.; Manome, Y.; Igarashi, M.; Wu, C.; Suzuki, K.; Igarashi, J.; Iwamoto, T.; Okano, H.J.; Eto, Y. Generation and characterization of motor neuron progenitors and motor neurons using metachromatic leukodystrophy-induced pluripotent stem cells. Mol. Genet. Metab. Rep. 2022, 31, 100852. [Google Scholar] [CrossRef]
- Biffi, A.; Lucchini, G.; Rovelli, A.; Sessa, M. Metachromatic leukodystrophy: An overview of current and prospective treatments. Bone Marrow Transplant. 2008, 42 (Suppl. S2), S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Calbi, V.; Natali Sora, M.G.; Sessa, M.; Baldoli, C.; Rancoita, P.M.V.; Ciotti, F.; Sarzana, M.; Fraschini, M.; Zambon, A.A.; et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: Long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 2022, 399, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Streisinger, G.; Walker, C.; Dower, N.; Knauber, D.; Singer, F. Production of clones of homozygous diploid zebra fish (Brachydanio rerio). Nature 1981, 291, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Dudziak, K.; Nowak, M.; Sozoniuk, M. One Host-Multiple Applications: Zebrafish (Danio rerio) as Promising Model for Studying Human Cancers and Pathogenic Diseases. Int. J. Mol. Sci. 2022, 23, 10255. [Google Scholar] [CrossRef]
- Chia, K.; Klingseisen, A.; Sieger, D.; Priller, J. Zebrafish as a model organism for neurodegenerative disease. Front. Mol. Neurosci. 2022, 15, 940484. [Google Scholar] [CrossRef]
- Bowley, G.; Kugler, E.; Wilkinson, R.; Lawrie, A.; van Eeden, F.; Chico, T.J.A.; Evans, P.C.; Noël, E.S.; Serbanovic-Canic, J. Zebrafish as a tractable model of human cardiovascular disease. Br. J. Pharmacol. 2022, 179, 900–917. [Google Scholar] [CrossRef]
- Fontana, B.D.; Norton, W.H.J.; Parker, M.O. Modelling ADHD-Like Phenotypes in Zebrafish. Curr. Top. Behav. Neurosci. 2022, 57, 395–414. [Google Scholar] [CrossRef]
- Canzian, J.; Gonçalves, F.L.S.; Müller, T.E.; Franscescon, F.; Santos, L.W.; Adedara, I.A.; Rosemberg, D.B. Zebrafish as a potential non-traditional model organism in translational bipolar disorder research: Genetic and behavioral insights. Neurosci. Biobehav. Rev. 2022, 136, 104620. [Google Scholar] [CrossRef]
- Phillips, J.B.; Westerfield, M. Chapter 47—Zebrafish as a Model to Understand Human Genetic Diseases. In The Zebrafish in Biomedical Research: Biology, Husbandry, Diseases, and Research Applications; American College of Laboratory Animal Medicine: Chester, NH, USA, 2020; pp. 619–626. [Google Scholar] [CrossRef]
- Crouzier, L.; Richard, E.M.; Sourbron, J.; Lagae, L.; Maurice, T.; Delprat, B. Use of Zebrafish Models to Boost Research in Rare Genetic Diseases. Int. J. Mol. Sci. 2021, 22, 13356. [Google Scholar] [CrossRef]
- Fraher, D.; Sanigorski, A.; Mellett, N.A.; Meikle, P.J.; Sinclair, A.J.; Gibert, Y. Zebrafish Embryonic Lipidomic Analysis Reveals that the Yolk Cell Is Metabolically Active in Processing Lipid. Cell Rep. 2016, 14, 1317–1329. [Google Scholar] [CrossRef]
- Quinlivan, V.H.; Wilson, M.H.; Ruzicka, J.; Farber, S.A. An HPLC-CAD/fluorescence lipidomics platform using fluorescent fatty acids as metabolic tracers. J. Lipid. Res. 2017, 58, 1008–1020. [Google Scholar] [CrossRef]
- Zhang, T.; Peterson, R.T. Modeling Lysosomal Storage Diseases in the Zebrafish. Front. Mol. Biosci. 2020, 7, 82. [Google Scholar] [CrossRef]
- Xu, M.M.; Legradi, J.; Leonards, P. Using comprehensive lipid profiling to study effects of PFHxS during different stages of early zebrafish development. Sci. Total. Environ. 2022, 808, 151739. [Google Scholar] [CrossRef]
- Zhang, W.; Song, Y.; Chai, T.; Liao, G.; Zhang, L.; Jia, Q.; Qian, Y.; Qiu, J. Lipidomics perturbations in the brain of adult zebrafish (Danio rerio) after exposure to chiral ibuprofen. Sci. Total. Environ. 2020, 713, 136565. [Google Scholar] [CrossRef]
- Hachicho, N.; Reithel, S.; Miltner, A.; Heipieper, H.J.; Küster, E.; Luckenbach, T. Body Mass Parameters, Lipid Profiles and Protein Contents of Zebrafish Embryos and Effects of 2,4-Dinitrophenol Exposure. PLoS ONE 2015, 10, e0134755. [Google Scholar] [CrossRef]
- Liebisch, G.; Fahy, E.; Aoki, J.; Dennis, E.A.; Durand, T.; Ejsing, C.S.; Fedorova, M.; Feussner, I.; Griffiths, W.J.; Köfeler, H.; et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid. Res. 2020, 61, 1539–1555. [Google Scholar] [CrossRef]
- Pirro, V.; Guffey, S.C.; Sepúlveda, M.S.; Mahapatra, C.T.; Ferreira, C.R.; Jarmusch, A.K.; Cooks, R.G. Lipid dynamics in zebrafish embryonic development observed by DESI-MS imaging and nanoelectrospray-MS. Mol. Biosyst. 2016, 12, 2069–2079. [Google Scholar] [CrossRef]
- Dueñas, M.E.; Essner, J.J.; Lee, Y.J. 3D MALDI Mass Spectrometry Imaging of a Single Cell: Spatial Mapping of Lipids in the Embryonic Development of Zebrafish. Sci Rep. 2017, 7, 14946. [Google Scholar] [CrossRef]
- Xu, M.; Legradi, J.; Leonards, P. Evaluation of LC-MS and LCxLC-MS in analysis of zebrafish embryo samples for comprehensive lipid profiling. Anal. Bioanal. Chem. 2020, 412, 4313–4325. [Google Scholar] [CrossRef]
- Castanon, I.; Hannich, J.T.; Abrami, L.; Huber, F.; Dubois, M.; Muller, M.; van der Goot, F.G.; Gonzalez-Gaitan, M. Wnt-controlled sphingolipids modulate Anthrax Toxin Receptor palmitoylation to regulate oriented mitosis in zebrafish. Nat. Commun. 2020, 11, 3317. [Google Scholar] [CrossRef]
- da Silva, K.M.; Iturrospe, E.; van den Boom, R.; van de Lavoir, M.; Robeyns, R.; Vergauwen, L.; Knapen, D.; Cuykx, M.; Covaci, A.; van Nuijs, A.L.N. Lipidomics profiling of zebrafish liver through untargeted liquid chromatography-high resolution mass spectrometry. J. Sep. Sci. 2022, 45, 2935–2945. [Google Scholar] [CrossRef]
- Choi, J.; Leonard, S.W.; Kasper, K.; McDougall, M.; Stevens, J.F.; Tanguay, R.L.; Traber, M.G. Novel function of vitamin E in regulation of zebrafish (Danio rerio) brain lysophospholipids discovered using lipidomics. J. Lipid. Res. 2015, 56, 1182–1190. [Google Scholar] [CrossRef]
- Zhang, T.; Trauger, S.A.; Vidoudez, C.; Doane, K.P.; Pluimer, B.R.; Peterson, R.T. Parallel Reaction Monitoring reveals structure-specific ceramide alterations in the zebrafish. Sci. Rep. 2019, 9, 19939. [Google Scholar] [CrossRef]
- Zelnik, I.D.; Rozman, B.; Rosenfeld-Gur, E.; Ben-Dor, S.; Futerman, A.H. A Stroll Down the CerS Lane. Adv. Exp. Med. Biol. 2019, 1159, 49–63. [Google Scholar] [CrossRef]
- Brondolin, M.; Berger, S.; Reinke, M.; Tanaka, H.; Ohshima, T.; Fuβ, B.; Hoch, M. Identification and expression analysis of the zebrafish homologs of the ceramide synthase gene family. Dev. Dyn. 2013, 242, 189–200. [Google Scholar] [CrossRef]
- Mendelson, K.; Pandey, S.; Hisano, Y.; Carellini, F.; Das, B.C.; Hla, T.; Evans, T. The ceramide synthase 2b gene mediates genomic sensing and regulation of sphingosine levels during zebrafish embryogenesis. eLife 2017, 6, e21992. [Google Scholar] [CrossRef]
- Tomasello, D.L.; Kim, J.L.; Khodour, Y.; McCammon, J.M.; Mitalipova, M.; Jaenisch, R.; Futerman, A.H.; Sive, H. FAM57B is a modulator of ceramide synthesis that regulates sphingolipid homeostasis and synaptic composition in the developing brain. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- Lelieveld, L.T.; Gerhardt, S.; Maas, S.; Zwiers, K.C.; de Wit, C.; Beijk, E.H.; Ferraz, M.J.; Artola, M.; Meijer, A.H.; Tudorache, C.; et al. Consequences of excessive glucosylsphingosine in glucocerebrosidase-deficient zebrafish. J. Lipid Res. 2022, 63, 100199. [Google Scholar] [CrossRef]
- Bradford, Y.M.; Van Slyke, C.E.; Ruzicka, L.; Singer, A.; Eagle, A.; Fashena, D.; Howe, D.G.; Frazer, K.; Martin, R.; Paddock, H.; et al. Zebrafish information network, the knowledgebase for Danio rerio research. Genetics 2022, 220, iyac016. [Google Scholar] [CrossRef]
- Taniguchi, M.; Okazaki, T. The role of sphingomyelin and sphingomyelin synthases in cell death, proliferation and migration-from cell and animal models to human disorders. Biochim. Biophys. Acta 2014, 1841, 692–703. [Google Scholar] [CrossRef]
- Vacaru, A.M.; Tafesse, F.G.; Ternes, P.; Kondylis, V.; Hermansson, M.; Brouwers, J.F.; Somerharju, P.; Rabouille, C.; Holthuis, J.C. Sphingomyelin synthase-related protein SMSr controls ceramide homeostasis in the ER. J. Cell. Biol. 2009, 185, 1013–1027. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Reviews. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Keatinge, M.; Gegg, M.E.; Watson, L.; Mortiboys, H.; Li, N.; Dunning, M.; Ailani, D.; Bui, H.; van Rens, A.; Lefeber, D.J.; et al. Unexpected opposing biological effect of genetic risk factors for Parkinson’s disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yabu, T.; Imamura, S.; Yamashita, M.; Okazaki, T. Identification of Mg2+ -dependent neutral sphingomyelinase 1 as a mediator of heat stress-induced ceramide generation and apoptosis. J. Biol. Chem. 2008, 283, 29971–29982. [Google Scholar] [CrossRef]
- Yabu, T.; Shiba, H.; Shibasaki, Y.; Nakanishi, T.; Imamura, S.; Touhata, K.; Yamashita, M. Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase1 by JNK signaling. Cell Death Differ. 2015, 22, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Yabu, T.; Tomimoto, H.; Taguchi, Y.; Yamaoka, S.; Igarashi, Y.; Okazaki, T. Thalidomide-induced antiangiogenic action is mediated by ceramide through depletion of VEGF receptors, and is antagonized by sphingosine-1-phosphate. Blood 2005, 106, 125–134. [Google Scholar] [CrossRef]
- Yabu, T.; Shimuzu, A.; Yamashita, M. A novel mitochondrial sphingomyelinase in zebrafish cells. J. Biol. Chem. 2009, 284, 20349–20363. [Google Scholar] [CrossRef]
- Bravo, G.Á.; Cedeño, R.R.; Casadevall, M.P.; Ramió-Torrentà, L. Sphingosine-1-Phosphate (S1P) and S1P Signaling Pathway Modulators, from Current Insights to Future Perspectives. Cells 2022, 11, 2058. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, K.; Zygmunt, T.; Torres-Vázquez, J.; Evans, T.; Hla, T. Sphingosine 1-phosphate receptor signaling regulates proper embryonic vascular patterning. J. Biol. Chem. 2013, 288, 2143–2156. [Google Scholar] [CrossRef]
- Hisano, Y.; Inoue, A.; Okudaira, M.; Taimatsu, K.; Matsumoto, H.; Kotani, H.; Ohga, R.; Aoki, J.; Kawahara, A. Maternal and Zygotic Sphingosine Kinase 2 Are Indispensable for Cardiac Development in Zebrafish. J. Biol. Chem. 2015, 290, 14841–14851. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Ren, K.; Zeng, Y.Z.; Zheng, Z.; Yi, G.H. Biological function of SPNS2: From zebrafish to human. Mol. Immunol. 2018, 103, 55–62. [Google Scholar] [CrossRef]
- Elsaid, H.O.A.; Furriol, J.; Blomqvist, M.; Diswall, M.; Leh, S.; Gharbi, N.; Anonsen, J.H.; Babickova, J.; Tøndel, C.; Svarstad, E.; et al. Reduced α-galactosidase A activity in zebrafish (Danio rerio) mirrors distinct features of Fabry nephropathy phenotype. Mol Genet. Metab. Rep. 2022, 31, 100851. [Google Scholar] [CrossRef] [PubMed]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene ’knockdown’ in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Xiao, A.; Zhou, M.; Zhu, Z.; Lin, S.; Zhang, B. Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol. 2011, 29, 699–700. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Cade, L.; Khayter, C.; Reyon, D.; Peterson, R.T.; Joung, J.K.; Yeh, J.R. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat. Biotechnol. 2011, 29, 697–698. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.; Sun, C.; Gao, L.; Zhu, D.; Xu, X.; Zhu, X.; Xiong, J.W.; Xi, J.J. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013, 23, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Kaini, P.; Sander, J.D.; Joung, J.K.; Peterson, R.T.; Yeh, J.R. Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PLoS ONE 2013, 8, e68708. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Akhmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, e98186. [Google Scholar] [CrossRef]
- Zancan, I.; Bellesso, S.; Costa, R.; Salvalaio, M.; Stroppiano, M.; Hammond, C.; Argenton, F.; Filocamo, M.; Moro, E. Glucocerebrosidase deficiency in zebrafish affects primary bone ossification through increased oxidative stress and reduced Wnt/β-catenin signaling. Hum. Mol. Genet. 2015, 24, 1280–1294. [Google Scholar] [CrossRef] [PubMed]
- Keatinge, M.; Bui, H.; Menke, A.; Chen, Y.C.; Sokol, A.M.; Bai, Q.; Ellett, F.; Da Costa, M.; Burke, D.; Gegg, M.; et al. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum. Mol. Genet. 2015, 24, 6640–6652. [Google Scholar] [CrossRef]
- Lelieveld, L.T.; Mirzaian, M.; Kuo, C.L.; Artola, M.; Ferraz, M.J.; Peter, R.E.A.; Akiyama, H.; Greimel, P.; van den Berg, R.; Overkleeft, H.S.; et al. Role of β-glucosidase 2 in aberrant glycosphingolipid metabolism: Model of glucocerebrosidase deficiency in zebrafish. J. Lipid. Res. 2019, 60, 1851–1867. [Google Scholar] [CrossRef]
- Schwend, T.; Loucks, E.J.; Snyder, D.; Ahlgren, S.C. Requirement of Npc1 and availability of cholesterol for early embryonic cell movements in zebrafish. J. Lipid. Res. 2011, 52, 1328–1344. [Google Scholar] [CrossRef]
- Tseng, W.C.; Loeb, H.E.; Pei, W.; Tsai-Morris, C.H.; Xu, L.; Cluzeau, C.V.; Wassif, C.A.; Feldman, B.; Burgess, S.M.; Pavan, W.J.; et al. Modeling Niemann-Pick disease type C1 in zebrafish: A robust platform for in vivo screening of candidate therapeutic compounds. Dis. Model. Mech. 2018, 11, dmm034165. [Google Scholar] [CrossRef]
- Lin, Y.; Cai, X.; Wang, G.; Ouyang, G.; Cao, H. Model construction of Niemann-Pick type C disease in zebrafish. Biol. Chem. 2018, 399, 903–910. [Google Scholar] [CrossRef]
- Wiweger, M.; Majewski, L.; Adamek-Urbanska, D.; Wasilewska, I.; Kuznicki, J. npc2-Deficient Zebrafish Reproduce Neurological and Inflammatory Symptoms of Niemann-Pick Type C Disease. Front. Cell. Neuro. 2021, 15, 647860. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.C.; Johnson Escauriza, A.J.; Tsai-Morris, C.H.; Feldman, B.; Dale, R.K.; Wassif, C.A.; Porter, F.D. The role of Niemann-Pick type C2 in zebrafish embryonic development. Development 2021, 148, dev194258. [Google Scholar] [CrossRef] [PubMed]
- Zizioli, D.; Guarienti, M.; Tobia, C.; Gariano, G.; Borsani, G.; Bresciani, R.; Ronca, R.; Giacopuzzi, E.; Preti, A.; Gaudenzi, G.; et al. Molecular cloning and knockdown of galactocerebrosidase in zebrafish: New insights into the pathogenesis of Krabbe’s disease. Biochim. Biophys. Acta 2014, 1842, 665–675. [Google Scholar] [CrossRef]
- Zhou, J.; Tawk, M.; Tiziano, F.D.; Veillet, J.; Bayes, M.; Nolent, F.; Garcia, V.; Servidei, S.; Bertini, E.; Castro-Giner, F.; et al. Spinal muscular atrophy associated with progressive myoclonic epilepsy is caused by mutations in ASAH1. Am. J. Hum. Genet. 2012, 91, 5–14. [Google Scholar] [CrossRef]
- Berg, R.D.; Levitte, S.; O’Sullivan, M.P.; O’Leary, S.M.; Cambier, C.J.; Cameron, J.; Takaki, K.K.; Moens, C.B.; Tobin, D.M.; Keane, J.; et al. Lysosomal Disorders Drive Susceptibility to Tuberculosis by Compromising Macrophage Migration. Cell 2016, 165, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Kalén, M.; Wallgard, E.; Asker, N.; Nasevicius, A.; Athley, E.; Billgren, E.; Larson, J.D.; Wadman, S.A.; Norseng, E.; Clark, K.J.; et al. Combination of reverse and chemical genetic screens reveals angiogenesis inhibitors and targets. Chem. Biol. 2009, 16, 432–441. [Google Scholar] [CrossRef]
- Kuil, L.E.; López Martí, A.; Carreras Mascaro, A.; van den Bosch, J.C.; van den Berg, P.; van der Linde, H.C.; Schoonderwoerd, K.; Ruijter, G.J.G.; van Ham, T.J. Hexb enzyme deficiency leads to lysosomal abnormalities in radial glia and microglia in zebrafish brain development. Glia 2019, 67, 1705–1718. [Google Scholar] [CrossRef]
- Matsui, H.; Ito, J.; Matsui, N.; Uechi, T.; Onodera, O.; Kakita, A. Cytosolic dsDNA of mitochondrial origin induces cytotoxicity and neurodegeneration in cellular and zebrafish models of Parkinson’s disease. Nat. Commun. 2021, 12, 3101. [Google Scholar] [CrossRef]
- Marques, A.R.; Aten, J.; Ottenhoff, R.; van Roomen, C.P.; Herrera Moro, D.; Claessen, N.; Vinueza Veloz, M.F.; Zhou, K.; Lin, Z.; Mirzaian, M.; et al. Reducing GBA2 Activity Ameliorates Neuropathology in Niemann-Pick Type C Mice. PLoS ONE 2015, 10, e0135889. [Google Scholar] [CrossRef]
- Mistry, P.K.; Liu, J.; Sun, L.; Chuang, W.L.; Yuen, T.; Yang, R.; Lu, P.; Zhang, K.; Li, J.; Keutzer, J.; et al. Glucocerebrosidase 2 gene deletion rescues type 1 Gaucher disease. Proc. Natl. Acad. Sci. USA 2014, 111, 4934–4939. [Google Scholar] [CrossRef]
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; International Parkinson’s Disease Genomics Consortium (IPDGC); Heutink, P.; Shulman, J.M. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. [Google Scholar] [CrossRef]
- Toyooka, K. Chapter 37—Fabry disease. Handb. Clin. Neurol. 2013, 115, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Okamoto, R.; Kawano, T.; Iwasaki, T. Development of Organelle Replacement Therapy Using a Stearyl-Polyhistidine Peptide against Lysosomal Storage Disease Cells. Molecules 2019, 24, 2995. [Google Scholar] [CrossRef]
- Spiegel, R.; Raas-Rothschild, A.; Reish, O.; Regev, M.; Meiner, V.; Bargal, R.; Sury, V.; Meir, K.; Nadjari, M.; Hermann, G.; et al. The clinical spectrum of fetal Niemann-Pick type C. Am. J. Med. Genet. A 2009, 149A, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Flanagan-Steet, H.; Sias, C.; Steet, R. Altered chondrocyte differentiation and extracellular matrix homeostasis in a zebrafish model for mucolipidosis II. Am. J. Pathol. 2009, 175, 2063–2075. [Google Scholar] [CrossRef]
- Lu, P.N.; Moreland, T.; Christian, C.J.; Lund, T.C.; Steet, R.A.; Flanagan-Steet, H. Inappropriate cathepsin k secretion promotes its enzymatic activation driving heart and valve malformation. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Qian, Y.; van Meel, E.; Flanagan-Steet, H.; Yox, A.; Steet, R.; Kornfeld, S. Analysis of mucolipidosis ii/iii gnptab missense mutations identifies domains of udp-glcnac:Lysosomal enzyme glcnac-1-phosphotransferase involved in catalytic function and lysosomal enzyme recognition. J. Biol. Chem. 2015, 290, 3045–3056. [Google Scholar] [CrossRef]
- Moro, E.; Tomanin, R.; Friso, A.; Modena, N.; Tiso, N.; Scarpa, M.; Argenton, F. A novel functional role of iduronate-2-sulfatase in zebrafish early development. Matrix Biol. 2010, 29, 43–50. [Google Scholar] [CrossRef]
- Costa, R.; Urbani, A.; Salvalaio, M.; Bellesso, S.; Cieri, D.; Zancan, I.; Filocamo, M.; Bonaldo, P.; Szabò, I.; Tomanin, R.; et al. Perturbations in cell signaling elicit early cardiac defects in mucopolysaccharidosis type II. Hum. Mol. Genet. 2017, 26, 1643–1655. [Google Scholar] [CrossRef]
- Bellesso, S.; Salvalaio, M.; Lualdi, S.; Tognon, E.; Costa, R.; Braghetta, P.; Giraudo, C.; Stramare, R.; Rigon, L.; Filocamo, M.; et al. Fgf signaling deregulation is associated with early developmental skeletal defects in animal models for mucopolysaccharidosis type II (mpsII). Hum. Mol. Genet. 2018, 27, 2262–2275. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lin, H.Y.; Chuang, C.K.; Zhang, P.H.; Tu, R.Y.; Lin, S.P.; Tsai, H.J. Effect of mutated ids overexpression on ids enzyme activity and developmental phenotypes in zebrafish embryos: A valuable index for assessing critical point-mutations associated with mucopolysaccharidosis type II occurrence in humans. Diagnostics 2020, 10, 854. [Google Scholar] [CrossRef]
- White, A.B.; Galbiati, F.; Givogri, M.I.; Lopez Rosas, A.; Qiu, X.; van Breemen, R.; Bongarzone, E.R. Persistence of psychosine in brain lipid rafts is a limiting factor in the therapeutic recovery of a mouse model for Krabbe disease. J. Neurosci. Res. 2011, 89, 352–364. [Google Scholar] [CrossRef]
- MacMillan, C.J.; Furlong, S.J.; Doucette, C.D.; Chen, P.L.; Hoskin, D.W.; Easton, A.S. Bevacizumab diminishes experimental autoimmune encephalomyelitis by inhibiting spinal cord angiogenesis and reducing peripheral T-cell responses. J. Neuropathol. Exp. Neurol. 2012, 71, 983–999. [Google Scholar] [CrossRef]
- Cachon-Gonzalez, M.B.; Zaccariotto, E.; Cox, T.M. Genetics and Therapies for GM2 Gangliosidosis. Curr. Gene Ther. 2018, 18, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Wiley, D.S.; Redfield, S.E.; Zon, L.I. Chemical screening in zebrafish for novel biological and therapeutic discovery. Methods Cell. Biol. 2017, 138, 651–679. [Google Scholar] [CrossRef]
- Patton, E.E.; Zon, L.I.; Langenau, D.M. Zebrafish disease models in drug discovery: From preclinical modelling to clinical trials. Nat. Rev. Drug Discov. 2021, 20, 611–628. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | OMIM | Affected Gene | Deficient Protein | Main Accumulated Metabolite |
|---|---|---|---|---|
| Gaucher | #230800 (type I) #230900 (type II) #231000 (type III) | GBA1 | Acid β-glucocerebrosidase | Glucosylceramide |
| Fabry | #301500 | GLA | α-Galactosidase A | Globotriaosylceramide |
| Niemann–Pick | #257200 (type A) #607616 (type B) | SMPD1 | Acid sphingomyelinase | Sphingomyelin |
| #257220 (type C1) | NPC1 | NPC intracellular cholesterol transporter 1 | Cholesterol | |
| #607625 (type C2) | NPC2 | NPC intracellular cholesterol transporter 2 | ||
| Krabbe | #245200 | GALC | β-Galactosylceramidase | β-Galactosylsphingosine |
| Farber lipogranulomatosis | #228000 | ASAH1 | Acid ceramidase | Ceramide |
| GM1 gangliosidosis | #230500 (type I) #230600 (type II) #230650 (type III) | GBL1 | β-Galactosidase | GM1 ganglioside |
| GM2 gangliosidosis | #272750 (AB variant) | GM2A | GM2 activator protein | GM2 ganglioside |
| #272800 (Tay-Sachs) | HEXA | Hexosaminidase α-subunit | ||
| #268800 (Sandhoff) | HEXB | Hexosaminidase β-subunit | ||
| Metachromatic leukodystrophy | #250100 | ARSA | Arylsulfatase A | Sulfo-galactosylceramide |
| Disease | Human Gene | Zebrafish Orthologous | Zebrafish Model |
|---|---|---|---|
| Gaucher | GBA1 | gba1 | MO [138] Spontaneous mutation [138] TALEN [139] CRISPR/Cas9 [117,140] |
| Fabry | GLA | gla | CRISPR/Cas9 [131] |
| Niemann–Pick | SMPD1 NPC1 NPC2 | smpd1 npc1 npc2 | CRISPR/Cas9 [122] MO [141] CRISPR/Cas9 [142,143,144,145] |
| Krabbe | GALC | GALCa GALCb | MO [146] |
| Farber lipogranulomatosis | ASAH1 | asah1a asah1b | MO [147] CRISPR/Cas9 [111] |
| GM2 gangliosidosis | HEXA HEXB | hexa hexb | MO [148,149] CRISPR/Cas9 [150] |
| Metachromatic leukodystrophy | ARSA | arsa | MO [148] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mignani, L.; Guerra, J.; Corli, M.; Capoferri, D.; Presta, M. Zebra-Sphinx: Modeling Sphingolipidoses in Zebrafish. Int. J. Mol. Sci. 2023, 24, 4747. https://doi.org/10.3390/ijms24054747
Mignani L, Guerra J, Corli M, Capoferri D, Presta M. Zebra-Sphinx: Modeling Sphingolipidoses in Zebrafish. International Journal of Molecular Sciences. 2023; 24(5):4747. https://doi.org/10.3390/ijms24054747
Chicago/Turabian StyleMignani, Luca, Jessica Guerra, Marzia Corli, Davide Capoferri, and Marco Presta. 2023. "Zebra-Sphinx: Modeling Sphingolipidoses in Zebrafish" International Journal of Molecular Sciences 24, no. 5: 4747. https://doi.org/10.3390/ijms24054747
APA StyleMignani, L., Guerra, J., Corli, M., Capoferri, D., & Presta, M. (2023). Zebra-Sphinx: Modeling Sphingolipidoses in Zebrafish. International Journal of Molecular Sciences, 24(5), 4747. https://doi.org/10.3390/ijms24054747