
Variant Landscape of 15 Genes Involved in Corneal Dystrophies: Report of 30 Families and Comprehensive Analysis of the Literature

Abstract
:1. Introduction
2. Results
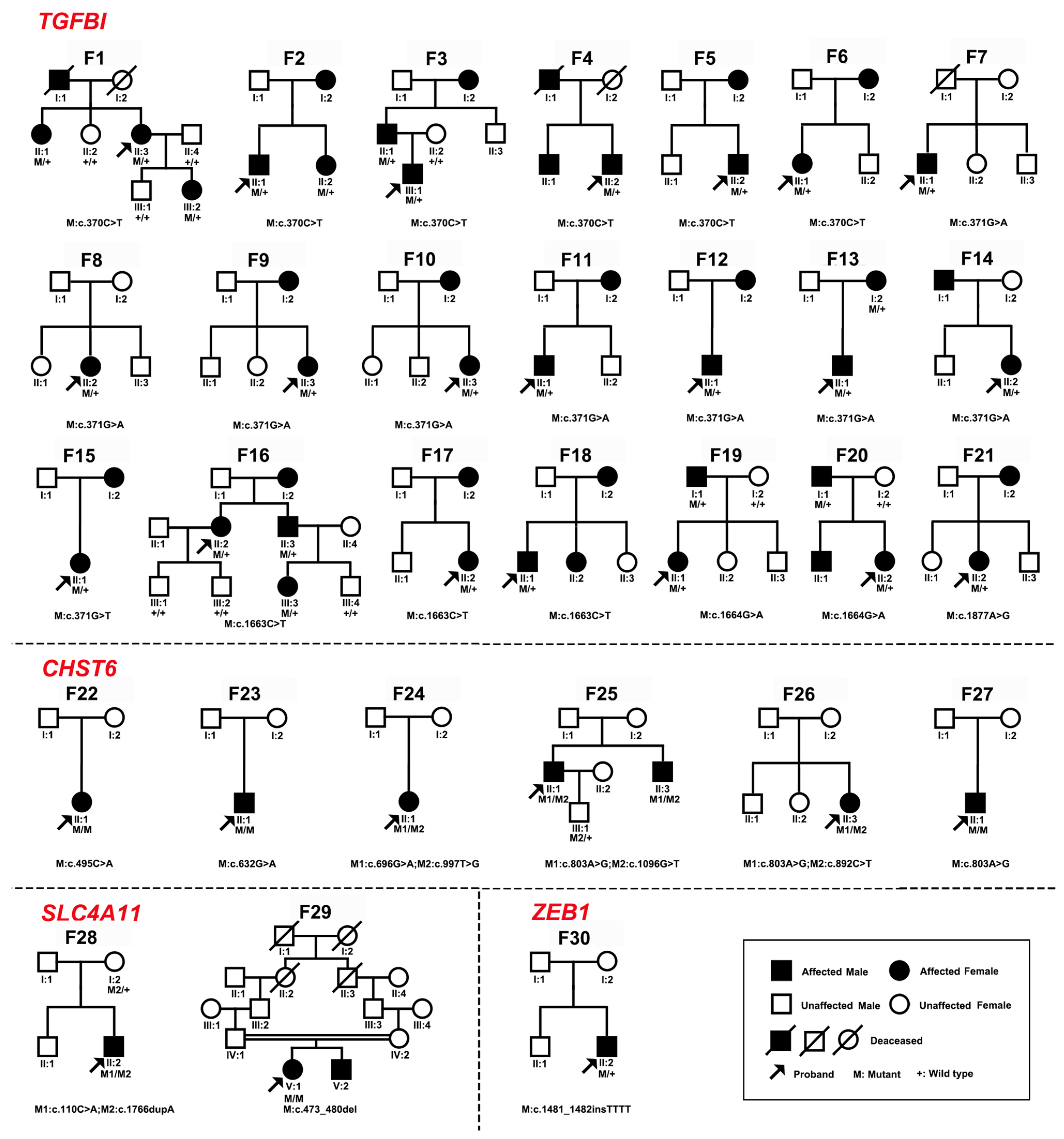
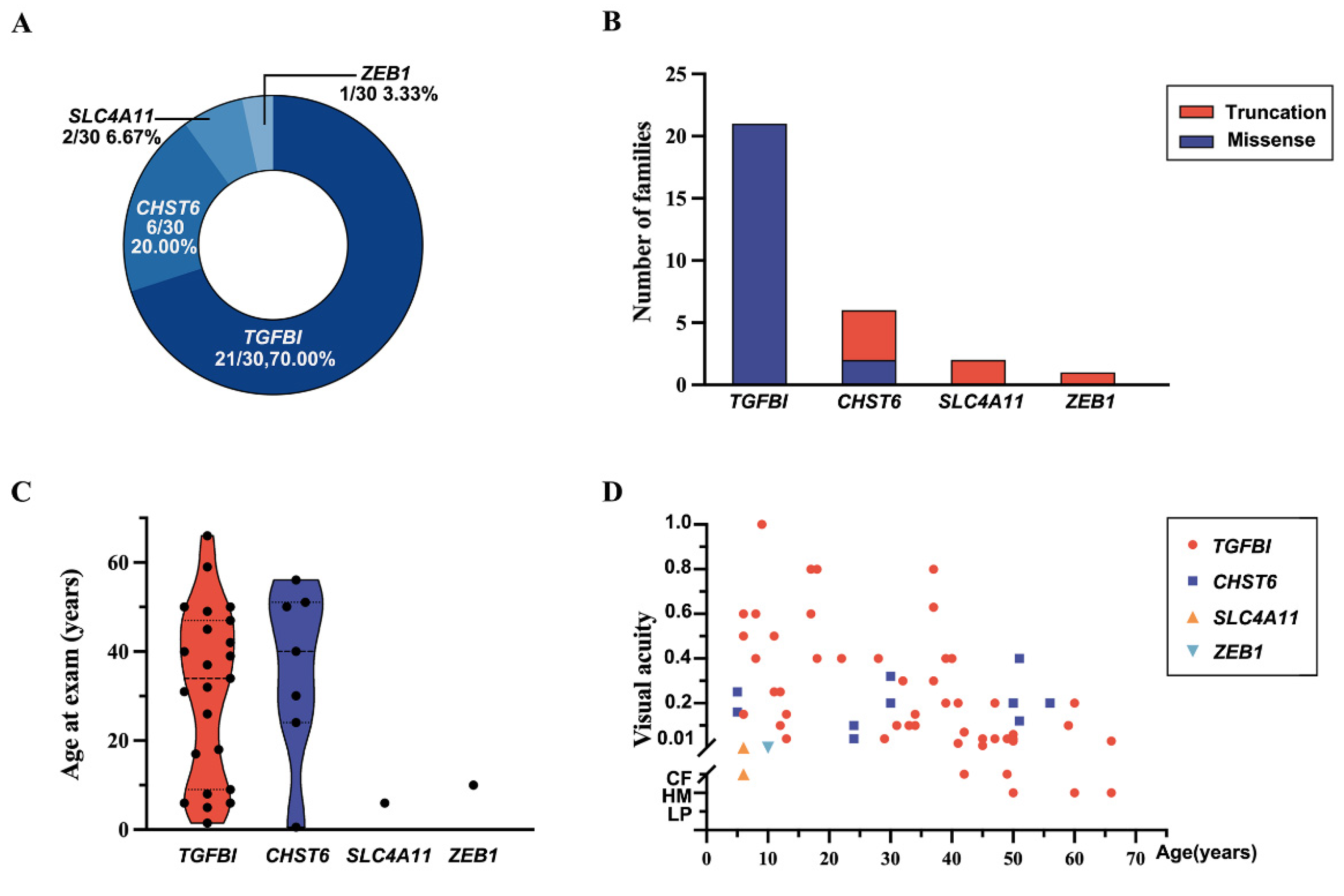
2.1. Genes and Pathogenic or Likely Pathogenic Variants of CDs in Our In-House Database
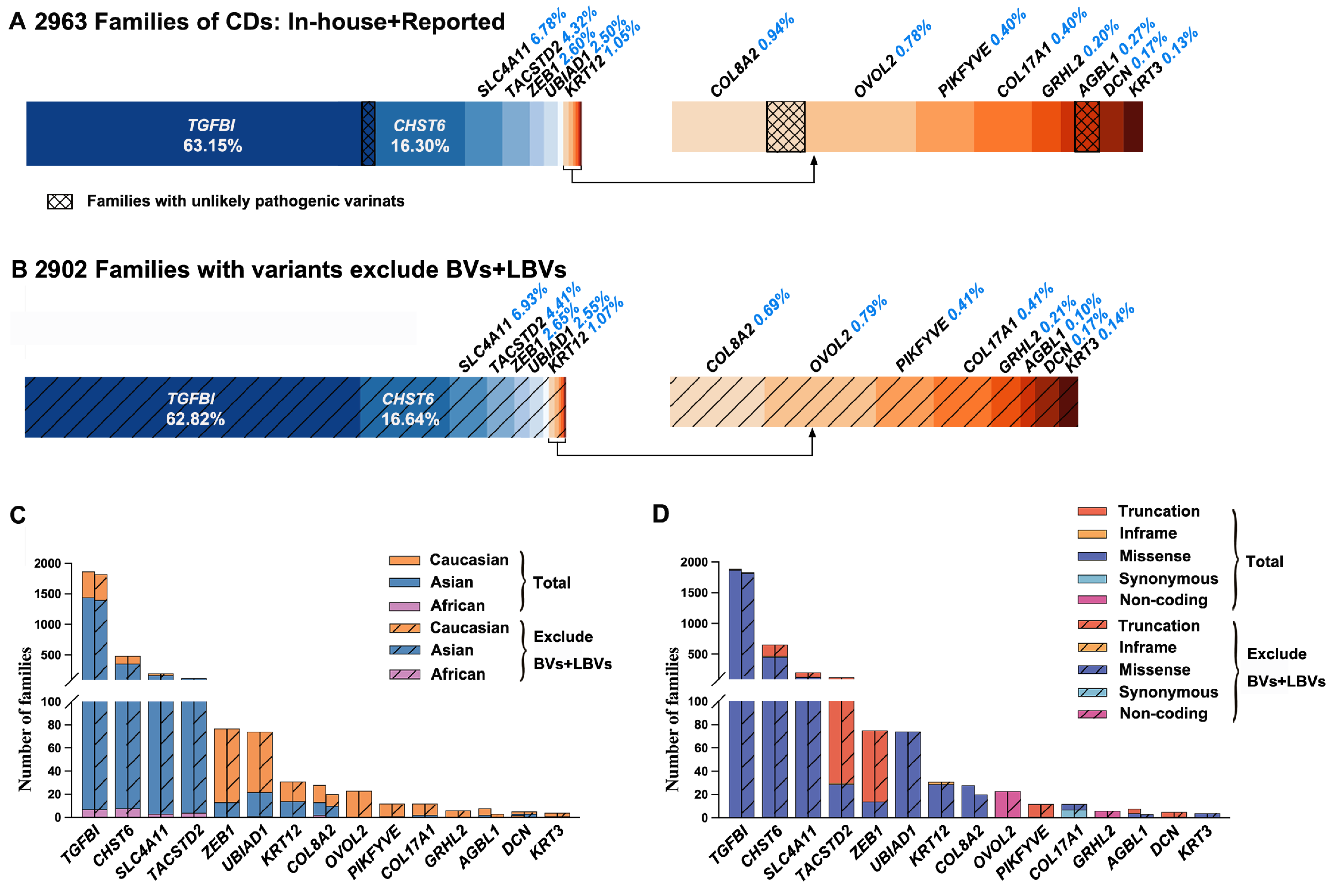
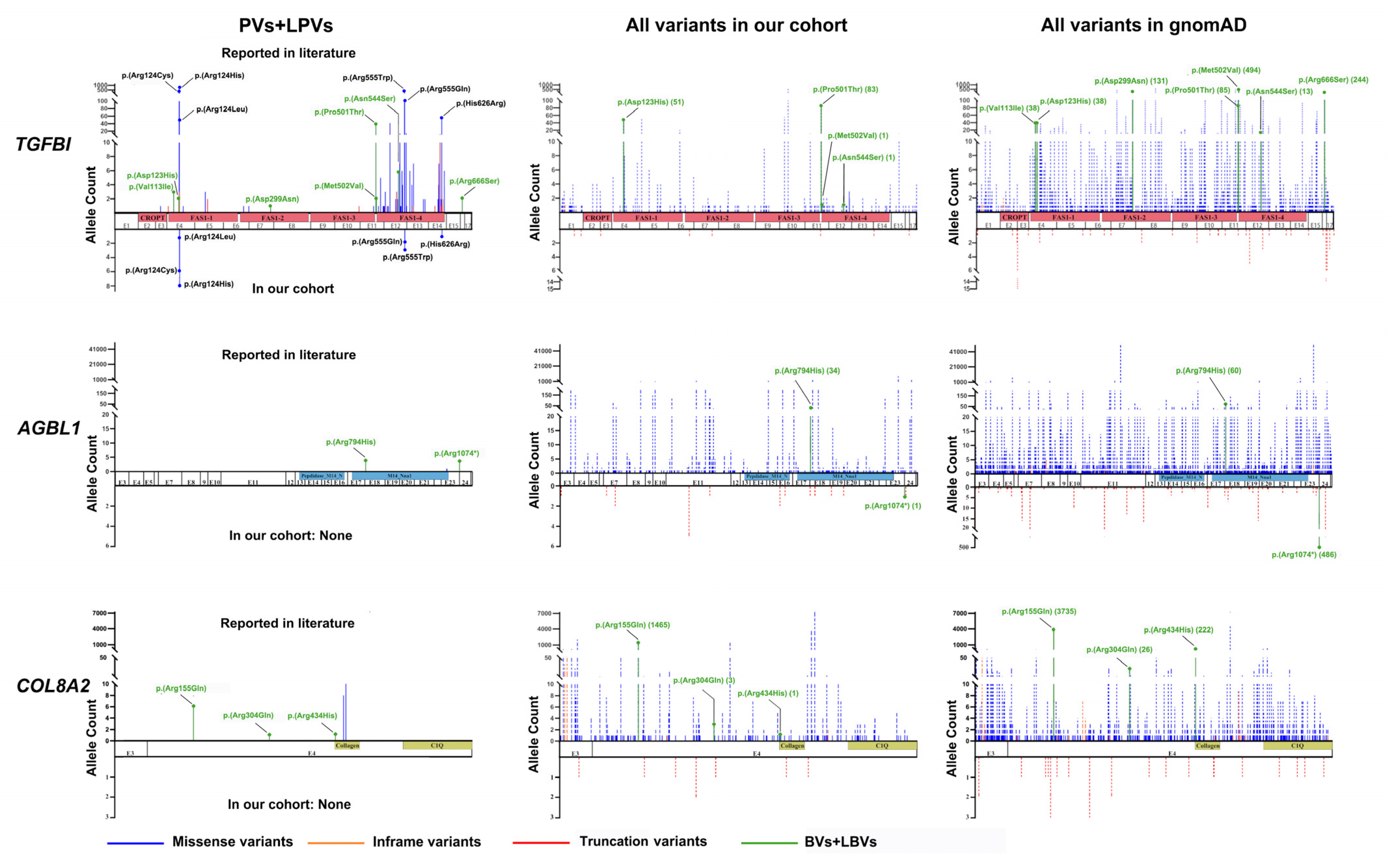
2.2. Genes and Variants of CDs in the Published Literature
2.3. Misinterpreted Variants in the Published Literature
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Genes and Variants of CDs in In-House Database
4.3. Analysis of Clinical Information
4.4. Review of Genes and Variants of CDs
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Musch, D.C.; Niziol, L.M.; Stein, J.D.; Kamyar, R.M.; Sugar, A. Prevalence of corneal dystrophies in the United States: Estimates from claims data. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6959–6963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.S.; Møller, H.U.; Aldave, A.J.; Seitz, B.; Bredrup, C.; Kivelä, T.; Munier, F.L.; Rapuano, C.J.; Nischal, K.K.; Kim, E.K.; et al. IC3D classification of corneal dystrophies—Edition 2. Cornea 2015, 34, 117–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.S.; Møller, H.U.; Lisch, W.; Kinoshita, S.; Aldave, A.J.; Belin, M.W.; Kivelä, T.; Busin, M.; Munier, F.L.; Seitz, B.; et al. The IC3D classification of the corneal dystrophies. Cornea 2008, 27 (Suppl. 2), S1–S83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munier, F.L.; Korvatska, E.; Djemaï, A.; Le Paslier, D.; Zografos, L.; Pescia, G.; Schorderet, D.F. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat. Genet. 1997, 15, 247–251. [Google Scholar] [CrossRef]
- Akama, T.O.; Nishida, K.; Nakayama, J.; Watanabe, H.; Ozaki, K.; Nakamura, T.; Dota, A.; Kawasaki, S.; Inoue, Y.; Maeda, N.; et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat. Genet. 2000, 26, 237–241. [Google Scholar] [CrossRef]
- Vithana, E.N.; Morgan, P.; Sundaresan, P.; Ebenezer, N.D.; Tan, D.T.; Mohamed, M.D.; Anand, S.; Khine, K.O.; Venkataraman, D.; Yong, V.H.; et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat. Genet. 2006, 38, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, M.; Kurahashi, H.; Tanaka, T.; Nishida, K.; Shimomura, Y.; Tano, Y.; Nakamura, Y. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nat. Genet. 1999, 21, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Moroi, S.E.; Gokhale, P.A.; Schteingart, M.T.; Sugar, A.; Downs, C.A.; Shimizu, S.; Krafchak, C.; Fuse, N.; Elner, S.G.; Elner, V.M.; et al. Clinicopathologic correlation and genetic analysis in a case of posterior polymorphous corneal dystrophy. Am. J. Ophthalmol. 2003, 135, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.; Dubé, M.P.; Marcadier, J.; Jiang, H.; Federico, A.; George, S.; Seamone, C.; Andrews, D.; Dubord, P.; Holland, S.; et al. Mutations in the UBIAD1 gene, encoding a potential prenyltransferase, are causal for Schnyder crystalline corneal dystrophy. PLoS ONE 2007, 2, e685. [Google Scholar] [CrossRef] [Green Version]
- Irvine, A.D.; Corden, L.D.; Swensson, O.; Swensson, B.; Moore, J.E.; Frazer, D.G.; Smith, F.J.; Knowlton, R.G.; Christophers, E.; Rochels, R.; et al. Mutations in cornea-specific keratin K3 or K12 genes cause Meesmann’s corneal dystrophy. Nat. Genet. 1997, 16, 184–187. [Google Scholar] [CrossRef]
- Biswas, S.; Munier, F.L.; Yardley, J.; Hart-Holden, N.; Perveen, R.; Cousin, P.; Sutphin, J.E.; Noble, B.; Batterbury, M.; Kielty, C.; et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum. Mol. Genet. 2001, 10, 2415–2423. [Google Scholar] [CrossRef] [Green Version]
- Davidson, A.E.; Liskova, P.; Evans, C.J.; Dudakova, L.; Nosková, L.; Pontikos, N.; Hartmannová, H.; Hodaňová, K.; Stránecký, V.; Kozmík, Z.; et al. Autosomal-Dominant Corneal Endothelial Dystrophies CHED1 and PPCD1 Are Allelic Disorders Caused by Non-coding Mutations in the Promoter of OVOL2. Am. J. Hum. Genet. 2016, 98, 75–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Tiab, L.; Jiao, X.; Munier, F.L.; Zografos, L.; Frueh, B.E.; Sergeev, Y.; Smith, J.; Rubin, B.; Meallet, M.A.; et al. Mutations in PIP5K3 are associated with François-Neetens mouchetée fleck corneal dystrophy. Am. J. Hum. Genet. 2005, 77, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, F.; Byström, B.; Davidson, A.E.; Backman, L.J.; Kellgren, T.G.; Tuft, S.J.; Koskela, T.; Rydén, P.; Sandgren, O.; Danielson, P.; et al. Mutations in collagen, type XVII, alpha 1 (COL17A1) cause epithelial recurrent erosion dystrophy (ERED). Hum. Mutat. 2015, 36, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Liskova, P.; Dudakova, L.; Evans, C.J.; Rojas Lopez, K.E.; Pontikos, N.; Athanasiou, D.; Jama, H.; Sach, J.; Skalicka, P.; Stranecky, V.; et al. Ectopic GRHL2 Expression Due to Non-coding Mutations Promotes Cell State Transition and Causes Posterior Polymorphous Corneal Dystrophy 4. Am. J. Hum. Genet. 2018, 102, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riazuddin, S.A.; Vasanth, S.; Katsanis, N.; Gottsch, J.D. Mutations in AGBL1 cause dominant late-onset Fuchs corneal dystrophy and alter protein-protein interaction with TCF4. Am. J. Hum. Genet. 2013, 93, 758–764. [Google Scholar] [CrossRef] [Green Version]
- Bredrup, C.; Knappskog, P.M.; Majewski, J.; Rødahl, E.; Boman, H. Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Investig. Ophthalmol. Vis. Sci. 2005, 46, 420–426. [Google Scholar] [CrossRef]
- Evans, C.J.; Davidson, A.E.; Carnt, N.; Rojas López, K.E.; Veli, N.; Thaung, C.M.; Tuft, S.J.; Hardcastle, A.J. Genotype-Phenotype Correlation for TGFBI Corneal Dystrophies Identifies p.(G623D) as a Novel Cause of Epithelial Basement Membrane Dystrophy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5407–5414. [Google Scholar] [CrossRef] [Green Version]
- Jun, I.; Ji, Y.W.; Choi, S.I.; Lee, B.R.; Min, J.S.; Kim, E.K. Compound heterozygous mutations in TGFBI cause a severe phenotype of granular corneal dystrophy type 2. Sci. Rep. 2021, 11, 6986. [Google Scholar] [CrossRef]
- Matthaei, M.; Hribek, A.; Clahsen, T.; Bachmann, B.; Cursiefen, C.; Jun, A.S. Fuchs Endothelial Corneal Dystrophy: Clinical, Genetic, Pathophysiologic, and Therapeutic Aspects. Annu. Rev. Vis. Sci. 2019, 5, 151–175. [Google Scholar] [CrossRef]
- Romano, V.; Parekh, M.; Virgili, G.; Coco, G.; Leon, P.; Islein, K.; Ponzin, D.; Ferrari, S.; Fasolo, A.; Yu, A.C.; et al. Gender Matching Did Not Affect 2-year Rejection or Failure Rates Following DSAEK for Fuchs Endothelial Corneal Dystrophy. Am. J. Ophthalmol. 2022, 235, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Barut Selver, O.; Karaca, I.; Palamar, M.; Egrilmez, S.; Yagci, A. Graft Failure and Repeat Penetrating Keratoplasty. Exp. Clin. Transpl. 2021, 19, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Chao-Shern, C.; DeDionisio, L.A.; Jang, J.H.; Chan, C.C.; Thompson, V.; Christie, K.; Nesbit, M.A.; McMullen, C.B.T. Evaluation of TGFBI corneal dystrophy and molecular diagnostic testing. Eye 2019, 33, 874–881. [Google Scholar] [CrossRef] [Green Version]
- Weiss, J.S.; Willoughby, C.E.; Abad-Morales, V.; Turunen, J.A.; Lisch, W. Update on the Corneal Dystrophies-Genetic Testing and Therapy. Cornea 2022, 41, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.S. Corneal dystrophies: Molecular genetics to therapeutic intervention--Fifth ARVO/Pfizer Ophthalmics Research Institute Conference. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5391–5402. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Fujiki, K.; Murakami, A.; Kato, T.; Chen, L.Z.; Onoe, H.; Nakayasu, K.; Sakurai, M.; Takahashi, M.; Sugiyama, K.; et al. Analysis of COL8A2 gene mutation in Japanese patients with Fuchs’ endothelial dystrophy and posterior polymorphous dystrophy. Jpn. J. Ophthalmol. 2004, 48, 195–198. [Google Scholar] [CrossRef]
- Soumittra, N.; Loganathan, S.K.; Madhavan, D.; Ramprasad, V.L.; Arokiasamy, T.; Sumathi, S.; Karthiyayini, T.; Rachapalli, S.R.; Kumaramanickavel, G.; Casey, J.R.; et al. Biosynthetic and functional defects in newly identified SLC4A11 mutants and absence of COL8A2 mutations in Fuchs endothelial corneal dystrophy. J. Hum. Genet. 2014, 59, 444–453. [Google Scholar] [CrossRef]
- Hanany, M.; Sharon, D. Allele frequency analysis of variants reported to cause autosomal dominant inherited retinal diseases question the involvement of 19% of genes and 10% of reported pathogenic variants. J. Med. Genet. 2019, 56, 536–542. [Google Scholar] [CrossRef]
- Wang, Y.; Xiao, X.; Li, X.; Yi, Z.; Jiang, Y.; Zhang, F.; Zhou, L.; Li, S.; Jia, X.; Sun, W.; et al. Genetic and clinical landscape of ARR3-associated MYP26: The most common cause of Mendelian early-onset high myopia with a unique inheritance. Br. J. Ophthalmol. 2022. [Google Scholar] [CrossRef]
- Wang, J.; Xiao, X.; Li, S.; Jiang, H.; Sun, W.; Wang, P.; Zhang, Q. Landscape of pathogenic variants in six pre-mRNA processing factor genes for retinitis pigmentosa based on large in-house data sets and database comparisons. Acta Ophthalmol. 2022, 100, e1412–e1425. [Google Scholar] [CrossRef]
- Lechner, J.; Dash, D.P.; Muszynska, D.; Hosseini, M.; Segev, F.; George, S.; Frazer, D.G.; Moore, J.E.; Kaye, S.B.; Young, T.; et al. Mutational spectrum of the ZEB1 gene in corneal dystrophies supports a genotype-phenotype correlation. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3215–3223. [Google Scholar] [CrossRef] [Green Version]
- Mazzotta, C.; Traversi, C.; Raiskup, F.; Rizzo, C.L.; Renieri, A. First identification of a triple corneal dystrophy association: Keratoconus, epithelial basement membrane corneal dystrophy and fuchs’ endothelial corneal dystrophy. Case Rep. Ophthalmol. 2014, 5, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Abad-Morales, V.; Barbany, M.; Gris, O.; Güell, J.L.; Pomares, E. Coexistence of Meesmann Corneal Dystrophy and a Pseudo-Unilateral Lattice Corneal Dystrophy in a Patient With a Novel Pathogenic Variant in the Keratin K3 Gene: A Case Report. Cornea 2021, 40, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, D.; Li, Y.; Fan, Y.; Chen, H.; Hong, J.; Xu, J. Novel Mutations Associated With Various Types of Corneal Dystrophies in a Han Chinese Population. Front. Genet. 2019, 10, 881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Marchite, C.; Sánchez-Sánchez, F.; López-Sánchez, E.; Escribano, J. R124C and R555W TGFBI mutations in Spanish families with autosomal-dominant corneal dystrophies. Mol. Vis. 2007, 13, 1390–1396. [Google Scholar]
- Song, X.; Cai, L.; Li, Y.; Zhu, J.; Jin, P.; Chen, L.; Ma, F. Identification and characterization of transforming growth factor β induced gene (TGFBIG) from Branchiostoma belcheri: Insights into evolution of TGFBI family. Genomics 2014, 103, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Morand, S.; Buchillier, V.; Maurer, F.; Bonny, C.; Arsenijevic, Y.; Munier, F.L.; Schorderet, D.F. Induction of apoptosis in human corneal and HeLa cells by mutated BIGH3. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2973–2979. [Google Scholar] [CrossRef] [PubMed]
- Runager, K.; Basaiawmoit, R.V.; Deva, T.; Andreasen, M.; Valnickova, Z.; Sørensen, C.S.; Karring, H.; Thøgersen, I.B.; Christiansen, G.; Underhaug, J.; et al. Human phenotypically distinct TGFBI corneal dystrophies are linked to the stability of the fourth FAS1 domain of TGFBIp. J. Biol. Chem. 2011, 286, 4951–4958. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, N.S.; Poulsen, E.T.; Lukassen, M.V.; Chao Shern, C.; Mogensen, E.H.; Weberskov, C.E.; DeDionisio, L.; Schauser, L.; Moore, T.C.B.; Otzen, D.E.; et al. Biochemical mechanisms of aggregation in TGFBI-linked corneal dystrophies. Prog. Retin. Eye Res. 2020, 77, 100843. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, C.; Zheng, T.; Zhang, Y.; Liu, H.; Wang, X.; Tang, X.; Zhao, B.; Liu, P. Torin 1 alleviates impairment of TFEB-mediated lysosomal biogenesis and autophagy in TGFBI (p.G623_H626del)-linked Thiel-Behnke corneal dystrophy. Autophagy 2022, 18, 765–782. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Li, M.; Guo, X.; Li, S.; Xiao, X.; Jia, X.; Liu, X.; Zhang, Q. Mutation analysis of seven known glaucoma-associated genes in Chinese patients with glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3594–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Li, S.; Sun, W.; Xiao, X.; Jia, X.; Liu, M.; Xu, L.; Long, Y.; Zhang, Q. An Ophthalmic Targeted Exome Sequencing Panel as a Powerful Tool to Identify Causative Mutations in Patients Suspected of Hereditary Eye Diseases. Transl. Vis. Sci. Technol. 2019, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Patch, A.M.; Ellard, S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet. Test Mol. Biomark. 2010, 14, 533–537. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Position | Nucleotide Change | Effect | Number of | Alleles in GnomAD | HGMD | ACMG Rank | ACMG Evidence | |
|---|---|---|---|---|---|---|---|---|---|
| Families | Alleles | ||||||||
| A. TGFBI (Chr:5 NM_000358.2; pLI = 0) | |||||||||
| 1 | 135382095 | c.370C>T | p.(Arg124Cys) | 6 | 6 | 2/280,458 | DM | P | PS3+PM1+PM2+PP1+PP4 |
| 2 | 135382096 | c.371G>A | p.(Arg124His) | 8 | 8 | 10/249,054 | DM | P | PS3+PM1+PM2+PM5+PP1+PP4 |
| 3 | 135382096 | c.371G>T | p.(Arg124Leu) | 1 | 1 | / | DM | P | PS4+PM1+PM5+PP1+PP4 |
| 4 | 135392469 | c.1663C>T | p.(Arg555Trp) | 3 | 3 | / | DM | P | PS3+PS4+PM1+PP1+PP4 |
| 5 | 135392470 | c.1664G>A | p.(Arg555Gln) | 2 | 2 | / | DM | P | PS3+PS4+PM1+PP1+PP4 |
| 6 | 135396596 | c.1877A>G | p.(His626Arg) | 1 | 1 | / | DM | P | PS3+PS4+PP1+PP4 |
| B. CHST6 (Chr:16 NM_021615.4; pLI = 0) | |||||||||
| 1 | 75513232 | c.495C>A | p.(Cys165*) | 1 | 2 | / | DM | P | PVS1+PS4+PM4+PP1+PP4 |
| 2 | 75513095 | c.632G>A | p.(Arg211Gln) | 1 | 2 | 1/240,952 | DM | LP | PS4+PP1+PP2+PP3+PP4 |
| 3 | 75513031 | c.696G>A | p.(Trp232*) | 2 | 2 | 1/237,152 | DM | P | PVS1+PS4+PM4+PP1+PP4 |
| 4 | 75512924 | c.803A>G | p.(Tyr268Cys) | 23 | 25 | 20/275,774 | DM | LP | PM3+PP1+PP2+PP3+PP4 |
| 5 | 75512835 | c.892C>T | p.(Gln298*) | 2 | 2 | 4/250,212 | DM | P | PVS1+PM2+PM4+PP1+PP4 |
| 6 | 75512730 | c.997T>G | p.(Trp333Gly) | 2 | 2 | / | DM? | LP | PS4+PP1+PP2+PP3+ PP4 |
| 7 * | 75512631 | c.1096G>T | p.(Glu366*) | 1 | 1 | / | / | P | PVS1+PS4+PM4 |
| C. SLC4A11 (Chr:20 NM_032034.3; pLI = 0) | |||||||||
| 1 * | 3218216 | c.110C>A | p.(Ser37*) | 1 | 1 | 4/251,490 | / | P | PVS1+PM2+PM4 |
| 2 | 3214820_3214827 | c.473_480del | p.(Arg158Glnfs*4) | 1 | 2 | / | DM | P | PVS1+PS4+PM4 |
| 3 * | 3210194 | c.1766dupA | p.(Tyr589*) | 1 | 1 | / | / | P | PVS1+PS4+PM4 |
| D. ZEB1 (Chr:10 NM_030751.5; pLI = 0.97) | |||||||||
| 1 * | 31809744_31809745 | c.1481_1482insTTTT | p.(Lys494Asnfs*12) | 1 | 1 | / | / | P | PVS1+PS4+PM4 |
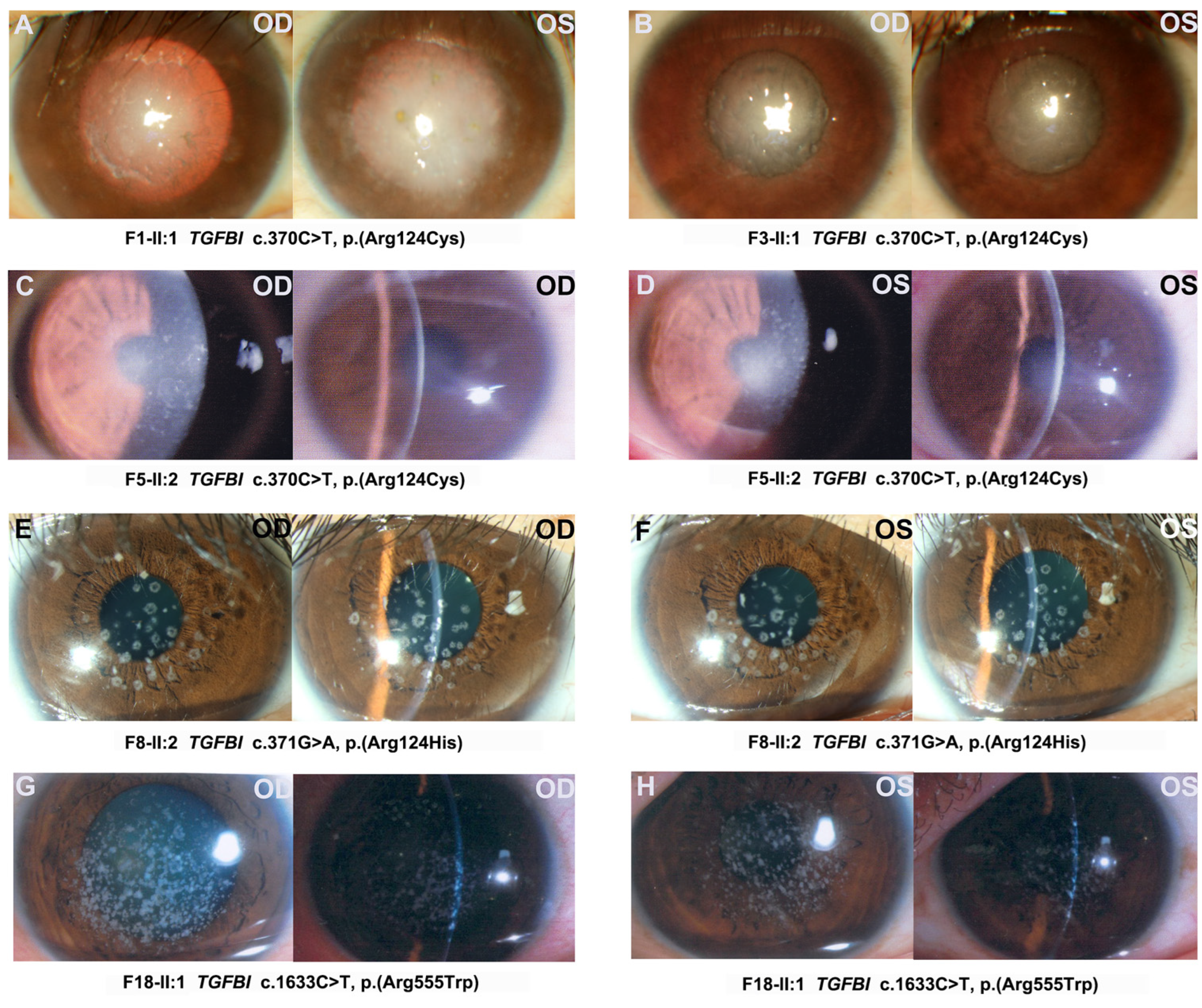
| Patient ID | Genotype | Protein | Gender | Age at Exam | First Symptom | Visual Acuity | Optometry | Phenotype |
|---|---|---|---|---|---|---|---|---|
| (Years) | (OD; OS) | (OD; OS(DS)) | ||||||
| A. TGFBI (NM_000358.2) | ||||||||
| F1-II:3 | c.[370C>T];[370=] | p.[(Arg124Cys)];[(Arg124=)] | F | 49 | Poor vision | 0.04; CF | NA; NA | LCD |
| F2-II:1 | c.[370C>T];[370=] | p.[(Arg124Cys)];[(Arg124=)] | M | 42 | Poor vision | CF; 0.07 | NA; NA | LCD |
| F2-II:2 | c.[370C>T];[370=] | p.[(Arg124Cys)];[(Arg124=)] | F | 31 | Poor vision | 0.1; 0.1 | NA; NA | LCD |
| F3-III:1 | c.[370C>T];[370=] | p.[(Arg124Cys)];[(Arg124=)] | M | 9 | Ocular pain | 1; 1 | NA; NA | LCD |
| F4-II:2 | c.[370C>T];[370=] | p.[(Arg124Cys)];[(Arg124=)] | M | 50 | Poor vision | 0.04; CF | NA; NA | LCD |
| F5-II:2 | c.[370C>T];[370=] | p.[(Arg124Cys)];[(Arg124=)] | M | 18 | Ocular pain | 0.4; 0.8 | NA; NA | LCD |
| F6-II:1 | c.[370C>T];[370=] | p.[(Arg124Cys)];[(Arg124=)] | F | 32 | Poor vision | 0.3; 0.3 | NA; NA | LCD |
| F7-II:1 | c.[371G>A];[371=] | p.[(Arg124His)];[(Arg124=)] | M | 59 | Poor vision | 0.1; 0.1 | 0.62; 0.75 | CD+glaucoma |
| F8-II:2 | c.[371G>A];[371=] | p.[(Arg124His)];[(Arg124=)] | F | 29 | Poor vision | 0.04; 0.04 | −7.00; −6.50 | GCD2+HMy |
| F9-II:3 | c.[371G>A];[371=] | p.[(Arg124His)];[(Arg124=)] | F | 47 | Poor vision | 0.2; 0.02 | −16.00; −10.00 | GCD2+HMy |
| F10-II:3 | c.[371G>A];[371=] | p.[(Arg124His)];[(Arg124=)] | F | 66 | Poor vision | HM; 0.03 | NA; −9.00 | CD+HMy |
| F11-II:1 | c.[371G>A];[371=] | p.[(Arg124His)];[(Arg124=)] | M | 37 | Corneal opacities | 0.63; 0.63 | −1.25; −1.75 | CD |
| F12-II:1 | c.[371G>A];[371=] | p.[(Arg124His)];[(Arg124=)] | M | 12 | None* | 0.3; 0.6 | −0.25; −0.25 | CD+glaucoma |
| F13-II:2 | c.[371G>A];[371=] | p.[(Arg124His)];[(Arg124=)] | M | 5 | Ocular pain | 0.16; 0.25 | 0.75; 0.75 | CD |
| F14-II:2 | c.[371G>A];[371=] | p.[(Arg124His)];[(Arg124=)] | F | 50 | Poor vision | 0.03; 0.06 | −7.62; −7.25 | CD+HMy |
| F15-II:1 | c.[371G>T];[371=] | p.[(Arg124Leu)];[(Arg124=)] | F | 1.5 | None* | NA; NA | −1.00; −0.75 | CD+strabismus |
| F16-II:2 | c.[1663C>T];[1663=] | p.[(Arg555Trp)];[(Arg555=)] | F | 45 | Poor vision | 0.02; 0.04 | NA; NA | GCD1 |
| F17-II:2 | c.[1663C>T];[1663=] | p.[(Arg555Trp)];[(Arg555=)] | F | 17 | Poor vision; ocular pain | 0.6; 0.8 | NA; NA | GCD1 |
| F18-II:1 | c.[1663C>T];[1663=] | p.[(Arg555Trp)];[(Arg555=)] | M | 40 | Poor vision | 0.4; 0.4 | NA; NA | GCD1 |
| F19-II:1 | c.[1664G>A];[1664=] | p.[(Arg555Gln)];[(Arg555=)] | F | 8 | Ocular pain | 0.4; 0.6 | −2.75; −1.00 | CD |
| F20-II:2 | c.[1664G>A];[1664=] | p.[(Arg555Gln)];[(Arg555=)] | F | 6 | Ocular pain | 0.6; 0.5 | NA; NA | CD |
| F20-I:1 | c.[1664G>A];[1664=] | p.[(Arg555Gln)];[(Arg555=)] | M | 39 | Ocular pain | 0.4; 0.2 | NA; NA | CD |
| F21-II:2 | c.[1877A>G];[1877=] | p.[(His626Arg)];[(His626=)] | F | 34 | Poor vision | 0.1; 0.15 | −0.25; −0.75 | LCD |
| B. CHST6 (NM_021615.4) | ||||||||
| F22-II:1 | c.[495C>A];[495C>A] | p.[(Cys165*)];[(Cys165*)] | F | 44 | Poor vision | NA; NA | NA; NA | CD |
| F23-II:1 | c.[632G>A];[632G>A] | p.[(Arg211Gln)];[(Arg211Gln)] | M | 30 | Poor vision | 0.32; 0.2 | NA; NA | CD |
| F24-II:1 | c.[696G>A];[997T>G] | p.[(Trp232*)];[(Trp333Gly)] | F | 24 | Poor vision | 0.1; 0.04 | NA; NA | CD |
| F25-II:1 | c.[803A>G];[1096G>T] | p.[(Tyr268Cys)];[(Glu366*)] | M | 56 | Poor vision | 0.2; 0.2 | NA; NA | MCD |
| F25-II:3 | c.[803A>G];[1096G>T] | p.[(Tyr268Cys)];[(Glu366*)] | M | 50 | Poor vision | 0.2; 0.2 | NA; NA | MCD |
| F26-II:3 | c.[803A>G];[892C>T] | p.[(Tyr268Cys)];[(Gln298*)] | F | 51 | Poor vision; ocular pain | 0.12; 0.4 | NA; NA | MCD |
| F27-II:1 | c.[803A>G];[803A>G] | p.[(Tyr268Cys)];[(Tyr268Cys)] | M | 0.5 | Corneal opacities | NA; NA | NA; NA | CD |
| C. SLC4A11 (NM_032034.3) | ||||||||
| F28-II:2 | c.[110C>A]; [1766dupA] | p.[(Ser37*)];[(Tyr589*)] | M | 6 | Poor vision | 0.06; 0.12 | NA; NA | CD |
| D. ZEB1 (NM_030751.5) | ||||||||
| F30-II:2 | c.[1481_1482insTTTT];[1481=] | p.[(Lys494Asnfs*12)];[(Lys494=)] | M | 10 | Poor vision | NA; NA | −8.00; −7.50 | CD+HMy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, D.; Wang, J.; Wang, Y.; Jiang, Y.; Li, S.; Xiao, X.; Wang, P.; Zhang, Q. Variant Landscape of 15 Genes Involved in Corneal Dystrophies: Report of 30 Families and Comprehensive Analysis of the Literature. Int. J. Mol. Sci. 2023, 24, 5012. https://doi.org/10.3390/ijms24055012
Zhu D, Wang J, Wang Y, Jiang Y, Li S, Xiao X, Wang P, Zhang Q. Variant Landscape of 15 Genes Involved in Corneal Dystrophies: Report of 30 Families and Comprehensive Analysis of the Literature. International Journal of Molecular Sciences. 2023; 24(5):5012. https://doi.org/10.3390/ijms24055012
Chicago/Turabian StyleZhu, Di, Junwen Wang, Yingwei Wang, Yi Jiang, Shiqiang Li, Xueshan Xiao, Panfeng Wang, and Qingjiong Zhang. 2023. "Variant Landscape of 15 Genes Involved in Corneal Dystrophies: Report of 30 Families and Comprehensive Analysis of the Literature" International Journal of Molecular Sciences 24, no. 5: 5012. https://doi.org/10.3390/ijms24055012