
KIT D816V Mast Cells Derived from Induced Pluripotent Stem Cells Recapitulate Systemic Mastocytosis Transcriptional Profile
 , , , , , , and
, , , , , , and
Abstract
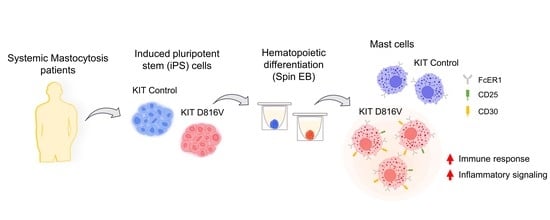
1. Introduction
2. Results
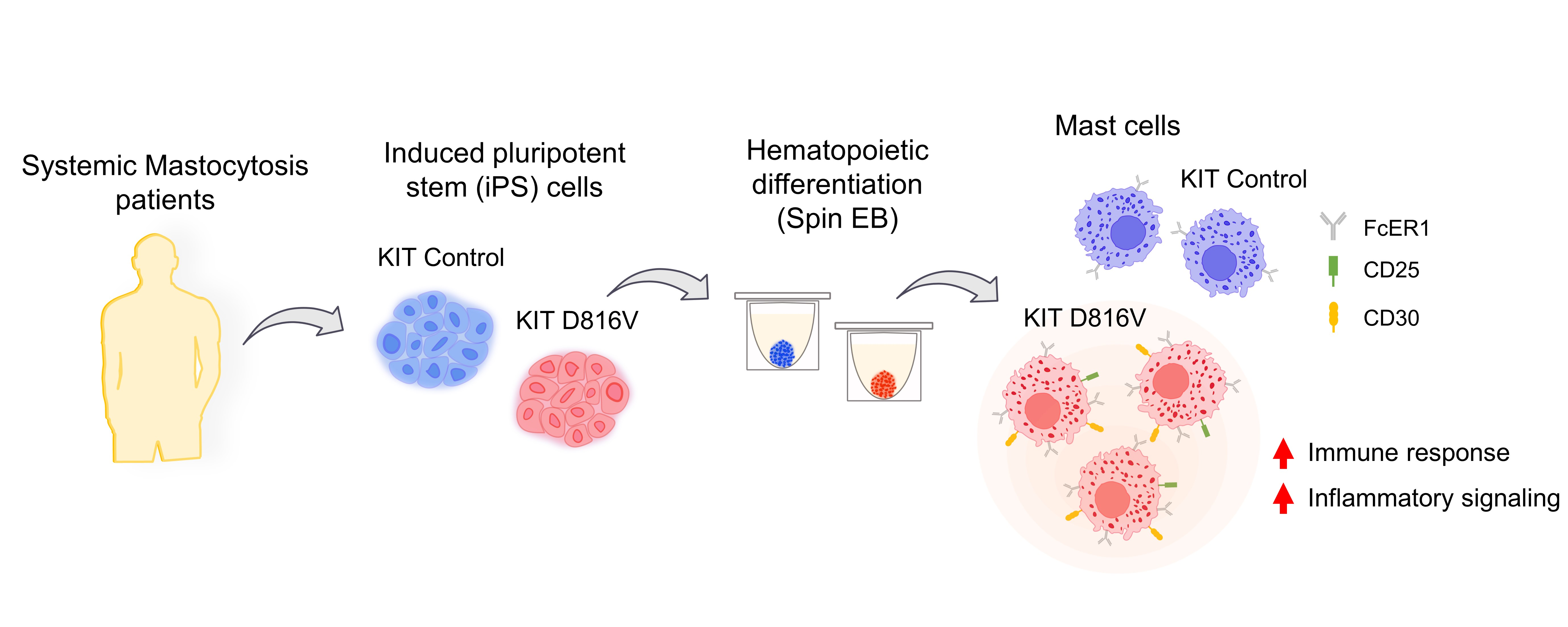
2.1. MCs Are Efficiently Differentiated from KIT D816V iPS Cells in a Feeder-Free Spin EB Culture
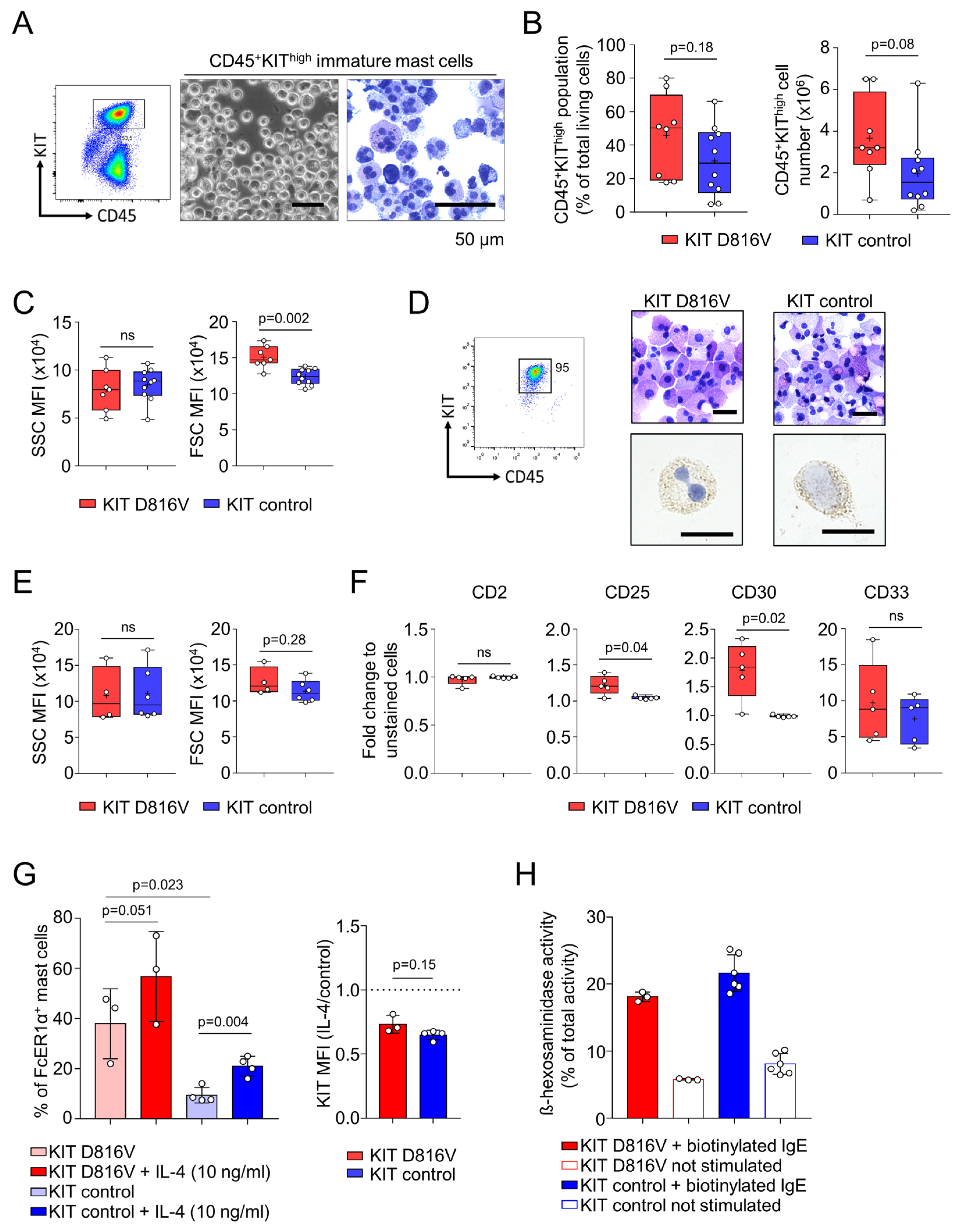
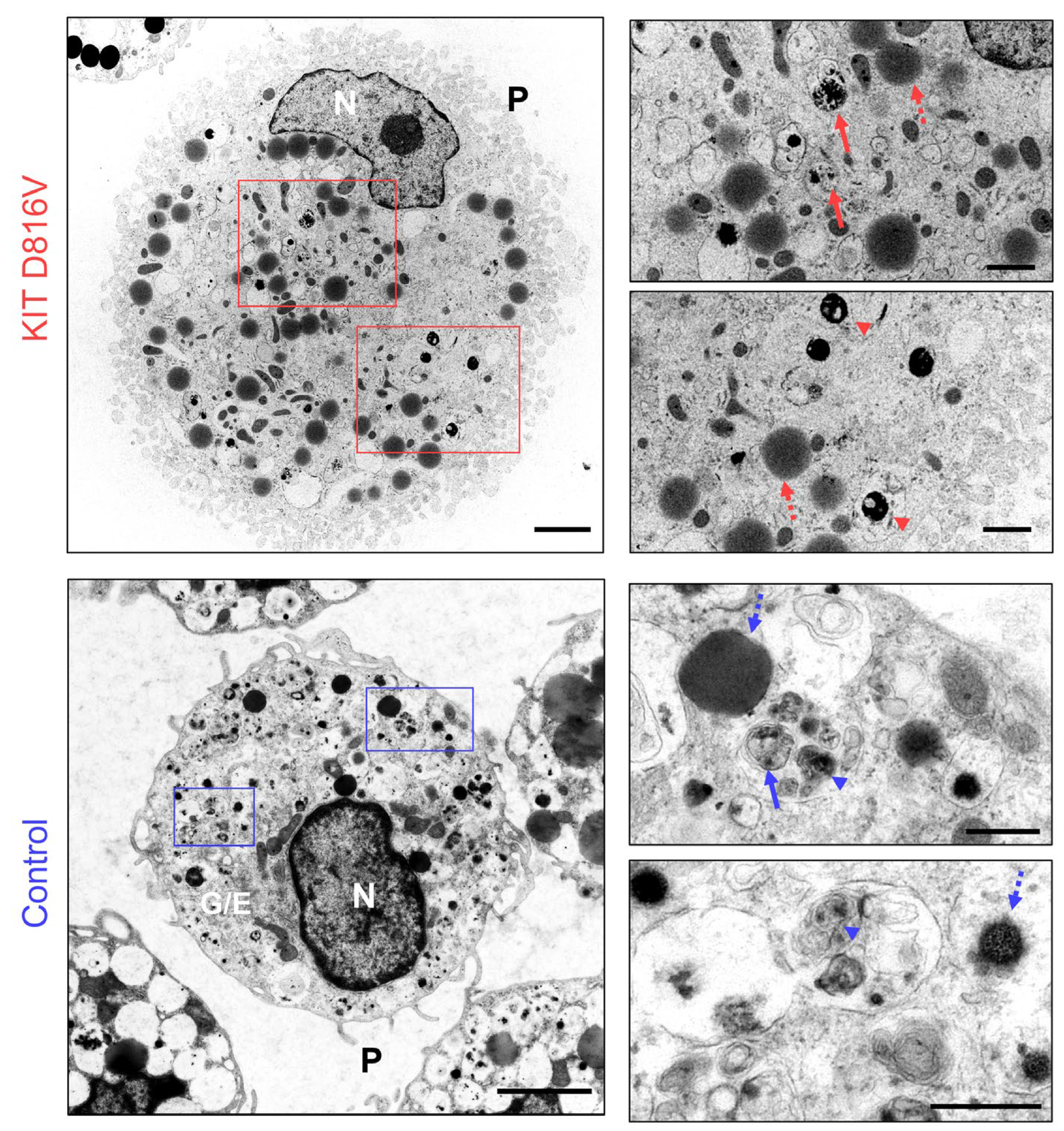
2.2. KIT D816V iPS Cell-Derived MCs Recapitulate Features of SM MCs

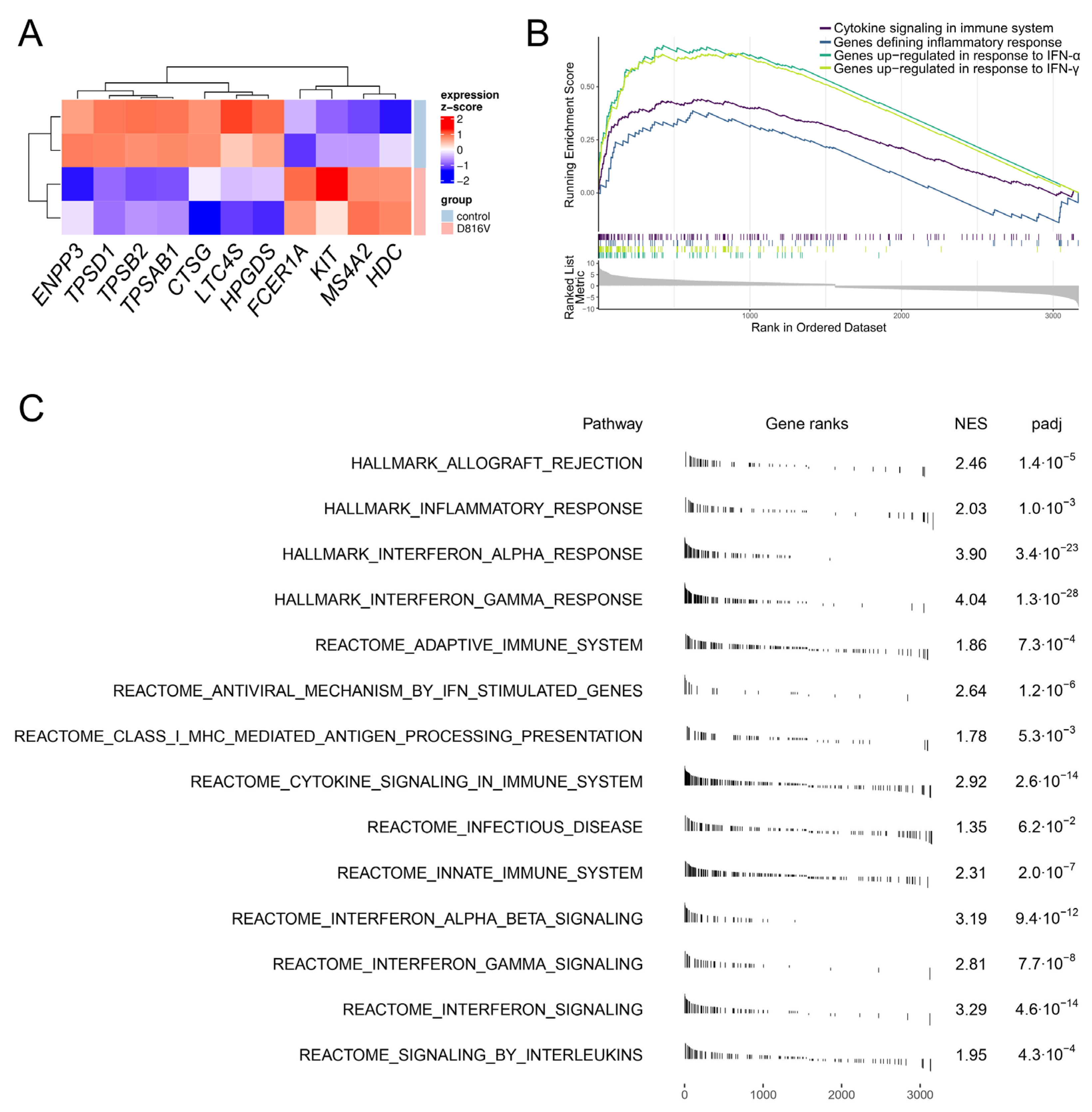
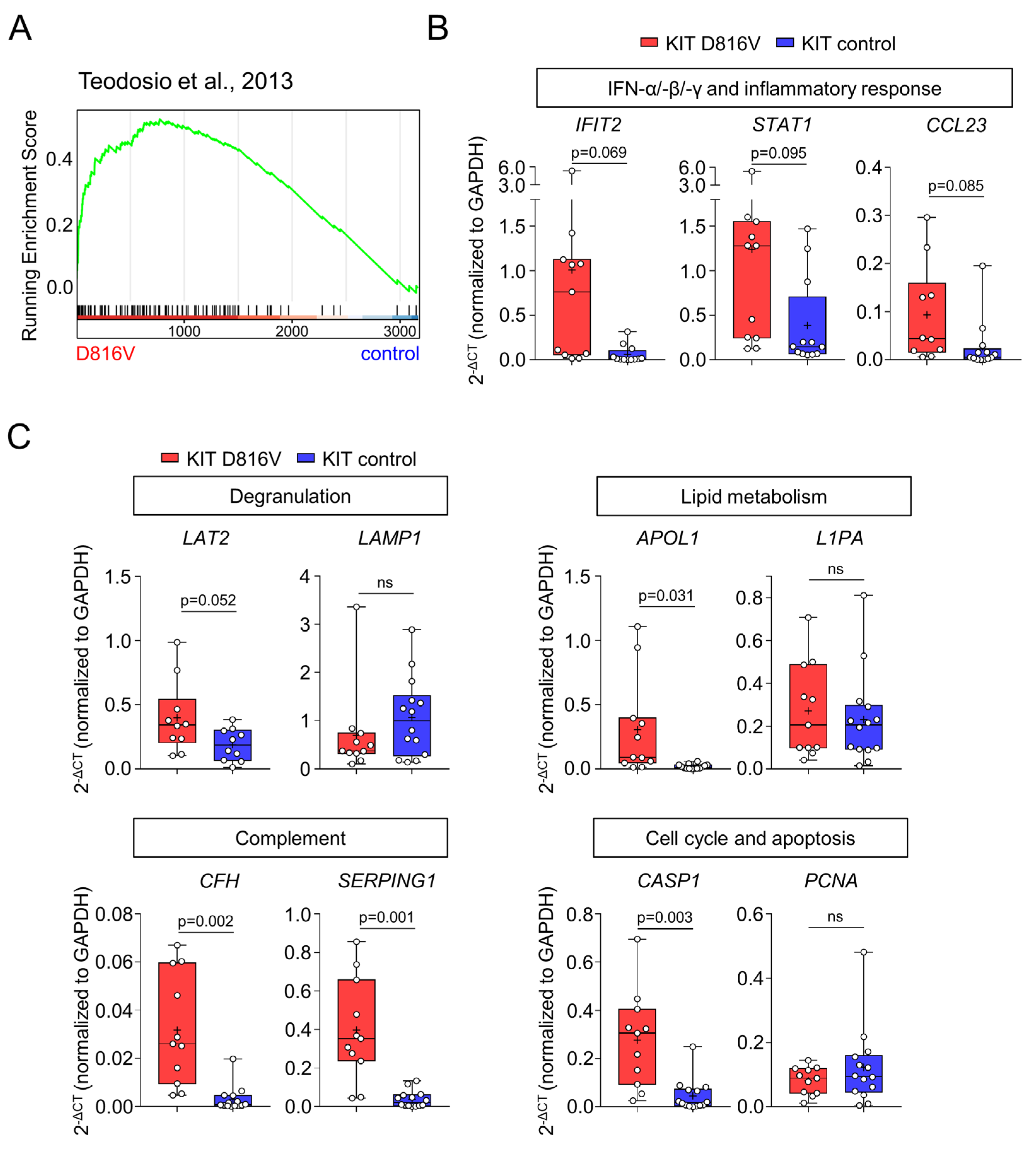
2.3. KIT D816V iPS Cell-Derived MCs Recapitulate the Gene Expression Profile of SM MCs
3. Discussion
4. Materials and Methods
4.1. The iPS Cell Lines and iPS Cell Culture

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Diagnosis | KIT Mutational Profile | iPSC Line | Additional Mutations in Each iPSC Line | Reference |
|---|---|---|---|---|---|
| 1 | ASM | D816V | iPSC_KIT D816V #1 | … | Toledo et al., 2021 [33] |
| iPSC_KIT D816V #2 | NFE2 (p.Glu261Alafs*3) | ||||
| iPSC_KIT D816V #3 | NFE2 (p.Glu261Alafs*3) | ||||
| iPSC_control #1 | NRAS (p.Gly12Asp), TET2 (p.Cys973Alafs*34) | ||||
| iPSC_control #2 | NRAS (p.Gly12Asp), TET2 (p.Cys973Alafs*34) | ||||
| 2 | MCL | D816V | iPSC_KIT D816V #4 | … | Toledo et al., 2021 [33] |
| iPSC_control #3 | … | ||||
| 3 | MCL | S476I | iPSC_control #4 | ... | Atakhanov et al., 2022 [32] |
| iPSC_control #5 |
4.2. The iPS Cell Differentiation toward MCs
| Spin EB (96-Well Format) | Hematopoietic Progenitors (10 cm Dish) | Mast Cell Maturation (6-Well Plate) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Component | Final Concentration | Cat. N° | Brand | Day 0–2 | Day 2–7 | Day 8–14 | Day 0–14 | Day 0–60 | |
| BASAL MEDIUM | IMDM | 50% | 12440-053 | Gibco | |||||
| Ham’s F-12 | 50% | 11765-054 | Gibco | ||||||
| Chemically defined lipid concentrate | 1% | 11905-031 | Gibco | ||||||
| GlutaMAX | 2 mM | 35050-038 | Gibco | ||||||
| 1-thioglycerol | 400 µM | M1753 | Sigma | ||||||
| BSA detox. | 0.5% | A8022 | Sigma | ||||||
| Albiomin 20% | 0.5% | B235258 | Biotest | ||||||
| L-ascorbic acid | 50 µg/ml | 72132 | Stemcell Technologies | ||||||
| h-Transferrin | 6 µg/ml | T0665 | Sigma | ||||||
| SUPPLEMENTS | Y-27632 | 10 µM | Ab120129 | Abcam | |||||
| BMP-4 | 10 ng/ml | 130-111-168 | Miltenyi | ||||||
| bFGF-2 | 10 ng/ml | 100-18B | Peprotech | ||||||
| VEGF | 10 ng/ml | 100-20 | Peprotech | ||||||
| SCF | 50 ng/ml | 130-095-745 | Miltenyi | ||||||
| IL-3 | 30 ng/ml | 200-03 | Peprotech | ||||||
| Flt3L | 50 ng/ml | 300-19 | Peprotech | ||||||
| hyper-IL-6 | 10 ng/ml | Fisher et al., 1997 [38] | |||||||
4.3. Magnetic Activated Cell Sorting (MACS)
4.4. Fluorescence Activated Cell Sorting (FACS)
4.5. Histochemistry and Immunohistochemistry
4.6. IL-4 Stimulation of MC
4.7. MC Degranulation Assay
4.8. Global Gene Expression Analysis
4.9. RT-qPCR
4.10. Transmission Electron Microscopy
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cardamone, C.; Parente, R.; De Feo, G.; Triggiani, M. Mast Cells as Effector Cells of Innate Immunity and Regulators of Adaptive Immunity. Immunol. Lett. 2016, 178, 10–14. [Google Scholar] [CrossRef]
- John, S.A.L.; Rathore, A.P.S.; Ginhoux, F. New Perspectives on the Origins and Heterogeneity of Mast Cells. Nat. Rev. Immunol. 2022, 23, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.S.; Maurer, M.; Metcalfe, D.D.; Pejler, G.; Sagi-Eisenberg, R.; Nilsson, G. The Ingenious Mast Cell: Contemporary Insights into Mast Cell Behavior and Function. Allergy 2022, 77, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Wernersson, S.; Pejler, G. Mast Cell Secretory Granules: Armed for Battle. Nat. Rev. Immunol. 2014, 14, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Gentek, R.; Ghigo, C.; Hoeffel, G.; Bulle, M.J.; Msallam, R.; Gautier, G.; Launay, P.; Chen, J.; Ginhoux, F.; Bajénoff, M. Hemogenic Endothelial Fate Mapping Reveals Dual Developmental Origin of Mast Cells. Immunity 2018, 48, 1160–1171.e5. [Google Scholar] [CrossRef]
- Li, Z.; Liu, S.; Xu, J.; Zhang, X.; Han, D.; Liu, J.; Xia, M.; Yi, L.; Shen, Q.; Xu, S.; et al. Adult Connective Tissue-Resident Mast Cells Originate from Late Erythro-Myeloid Progenitors. Immunity 2018, 49, 640–653.e5. [Google Scholar] [CrossRef]
- Wu, C.; Boey, D.; Bril, O.; Grootens, J.; Vijayabaskar, M.S.; Sorini, C.; Ekoff, M.; Wilson, N.K.; Ungerstedt, J.S.; Nilsson, G.; et al. Single-Cell Transcriptomics Reveals the Identity and Regulators of Human Mast Cell Progenitors. Blood Adv. 2022, 6, 4439–4449. [Google Scholar] [CrossRef]
- Dahlin, J.S.; Malinovschi, A.; Öhrvik, H.; Sandelin, M.; Janson, C.; Alving, K.; Hallgren, J. Lin− CD34hi CD117int/Hi FcεRI+ Cells in Human Blood Constitute a Rare Population of Mast Cell Progenitors. Blood 2016, 127, 383–391. [Google Scholar] [CrossRef]
- Plum, T.; Wang, X.; Rettel, M.; Krijgsveld, J.; Feyerabend, T.B.; Rodewald, H.R. Human Mast Cell Proteome Reveals Unique Lineage, Putative Functions, and Structural Basis for Cell Ablation. Immunity 2020, 52, 404–416.e5. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Hartmann, K.; Alvarez-Twose, I.; Brockow, K.; Hermine, O.; Niedoszytko, M.; Schwaab, J.; Lyons, J.J.; Carter, M.C.; et al. Updated Diagnostic Criteria and Classification of Mast Cell Disorders: A Consensus Proposal. HemaSphere 2021, 5, e646. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Metcalfe, D.D. Mastocytosis: 2016 Updated WHO Classification and Novel Emerging Treatment Concepts. Blood 2017, 129, 1420–1427. [Google Scholar] [CrossRef] [PubMed]
- Hoermann, G.; Sotlar, K.; Jawhar, M.; Kristensen, T.; Bachelot, G.; Nedoszytko, B.; Carter, M.C.; Horny, H.-P.; Bonadonna, P.; Sperr, W.R.; et al. Standards of Genetic Testing in the Diagnosis and Prognostication of Systemic Mastocytosis in 2022: Recommendations of the EU-US Cooperative Group. J. Allergy Clin. Immunol. Pract. 2022, 10, 1953–1963. [Google Scholar] [CrossRef]
- Arock, M.; Sotlar, K.; Akin, C.; Broesby-Olsen, S.; Hoermann, G.; Escribano, L.; Kristensen, T.K.; Kluin-Nelemans, H.C.; Hermine, O.; Dubreuil, P.; et al. KIT Mutation Analysis in Mast Cell Neoplasms: Recommendations of the European Competence Network on Mastocytosis. Leukemia 2015, 29, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Grootens, J.; Ungerstedt, J.S.; Ekoff, M.; Rönnberg, E.; Klimkowska, M.; Amini, R.-M.; Arock, M.; Söderlund, S.; Mattsson, M.; Nilsson, G.; et al. Single-Cell Analysis Reveals the KIT D816V Mutation in Haematopoietic Stem and Progenitor Cells in Systemic Mastocytosis. EBioMedicine 2019, 43, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Montero, A.C.; Jara-Acevedo, M.; Teodosio, C.; Sanchez, M.L.; Nunez, R.; Prados, A.; Aldanondo, I.; Sanchez, L.; Dominguez, M.; Botana, L.M.; et al. KIT Mutation in Mast Cells and Other Bone Marrow Hematopoietic Cell Lineages in Systemic Mast Cell Disorders: A Prospective Study of the Spanish Network on Mastocytosis (REMA) in a Series of 113 Patients. Blood 2006, 108, 2366–2372. [Google Scholar] [CrossRef]
- Kristensen, T.; Broesby-Olsen, S.; Vestergaard, H.; Bindslev-Jensen, C.; Møller, M.B. Circulating KIT D816V Mutation-Positive Non-Mast Cells in Peripheral Blood Are Characteristic of Indolent Systemic Mastocytosis. Eur. J. Haematol. 2012, 89, 42–46. [Google Scholar] [CrossRef]
- Sotlar, K.; Colak, S.; Bache, A.; Berezowska, S.; Krokowski, M.; Bültmann, B.; Valent, P.; Horny, H.P. Variable Presence of KITD816V in Clonal Haematological Non-Mast Cell Lineage Diseases Associated with Systemic Mastocytosis (SM-AHNMD). J. Pathol. 2010, 220, 586–595. [Google Scholar] [CrossRef]
- Jawhar, M.; Schwaab, J.; Schnittger, S.; Meggendorfer, M.; Pfirrmann, M.; Sotlar, K.; Horny, H.P.; Metzgeroth, G.; Kluger, S.; Naumann, N.; et al. Additional Mutations in SRSF2, ASXL1 and/or RUNX1 Identify a High-Risk Group of Patients with KIT D816V+ Advanced Systemic Mastocytosis. Leukemia 2016, 30, 136–143. [Google Scholar] [CrossRef]
- Valent, P.; Akin, C.; Hartmann, K.; Reiter, A.; Gotlib, J.; Sotlar, K.; Sperr, W.R.; Degenfeld-Schonburg, L.; Smiljkovic, D.; Triggiani, M.; et al. Drug-Induced Mast Cell Eradication: A Novel Approach to Treat Mast Cell Activation Disorders? J. Allergy Clin. Immunol. 2022, 149, 1866–1874. [Google Scholar] [CrossRef]
- Gotlib, J.; Kluin-Nelemans, H.C.; George, T.I.; Akin, C.; Sotlar, K.; Hermine, O.; Awan, F.T.; Hexner, E.; Mauro, M.J.; Sternberg, D.W.; et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N. Engl. J. Med. 2016, 374, 2530–2541. [Google Scholar] [CrossRef]
- Reiter, A.; Gotlib, J.; Álvarez-Twose, I.; Radia, D.H.; Lübke, J.; Bobbili, P.J.; Wang, A.; Norregaard, C.; Dimitrijevic, S.; Sullivan, E.; et al. Efficacy of Avapritinib versus Best Available Therapy in the Treatment of Advanced Systemic Mastocytosis. Leukemia 2022, 36, 2108–2120. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, A.; Gunawan, M.; Yong, K.S.M.; Bist, P.; Tan, W.W.S.; Tan, S.Y.; Liu, M.; Huang, E.K.; Fan, Y.; Chan, J.K.Y.; et al. A Humanized Mouse Model to Study Mast Cells Mediated Cutaneous Adverse Drug Reactions. J. Leukoc. Biol. 2020, 107, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Imanishi, M.; Fujikura, D.; Sugiyama, M.; Tanimoto, K.; Mochiji, Y.; Takahashi, Y.; Hiura, K.; Watanabe, M.; Kashimoto, T.; et al. New Inducible Mast Cell-Deficient Mouse Model (Mcpt5/Cma1DTR). Biochem. Biophys. Res. Commun. 2021, 551, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Bryce, P.J.; Falahati, R.; Kenney, L.L.; Leung, J.; Bebbington, C.; Tomasevic, N.; Krier, R.A.; Hsu, C.L.; Shultz, L.D.; Greiner, D.L.; et al. Humanized Mouse Model of Mast Cell-Mediated Passive Cutaneous Anaphylaxis and Passive Systemic Anaphylaxis. J. Allergy Clin. Immunol. 2016, 138, 769. [Google Scholar] [CrossRef]
- Akula, S.; Paivandy, A.; Fu, Z.; Thorpe, M.; Pejler, G.; Hellman, L. How Relevant Are Bone Marrow-Derived Mast Cells (BMMCs) as Models for Tissue Mast Cells? A Comparative Transcriptome Analysis of BMMCs and Peritoneal Mast Cells. Cells 2020, 9, 2118. [Google Scholar] [CrossRef]
- Egawa, N.; Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; et al. Drug Screening for ALS Using Patient-Specific Induced Pluripotent Stem Cells. Sci. Transl. Med. 2012, 4, 145ra104. [Google Scholar] [CrossRef]
- Yoshikubo, T.; Inoue, T.; Noguchi, M.; Okabe, H. Differentiation and Maintenance of Mast Cells from CD34+ Human Cord Blood Cells. Exp. Hematol. 2006, 34, 320–329. [Google Scholar] [CrossRef]
- Goossens, J.; Ieven, T.; Dilissen, E.; Jonckheere, A.-C.; Cremer, J.; Dupont, L.; Bullens, D. Human Mast Cell Differentiation Optimization to Study MRGPRX2-Induced Activation in Vitro. ERJ Open Res. 2022, 8, 63. [Google Scholar] [CrossRef]
- Rowe, R.G.; Daley, G.Q. Induced Pluripotent Stem Cells in Disease Modelling and Drug Discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef]
- Sharma, A.; Sances, S.; Workman, M.J.; Svendsen, C.N. Multi-Lineage Human IPSC-Derived Platforms for Disease Modeling and Drug Discovery. Cell Stem Cell 2020, 26, 309–329. [Google Scholar] [CrossRef]
- Satoh, T.; Toledo, M.A.S.; Boehnke, J.; Olschok, K.; Flosdorf, N.; Götz, K.; Küstermann, C.; Sontag, S.; Seré, K.; Koschmieder, S.; et al. Human DC3 Antigen Presenting Dendritic Cells From Induced Pluripotent Stem Cells. Front. Cell Dev. Biol. 2021, 9, 667304. [Google Scholar] [CrossRef] [PubMed]
- Atakhanov, S.; Christen, D.; Rolles, B.; Schüler, H.M.; Panse, J.; Chatain, N.; Koschmieder, S.; Brümmendorf, T.H.; Toledo, M.A.S.; Zenke, M. Towards Personalized Medicine with IPS Cell Technology: A Case Report of Advanced Systemic Mastocytosis with Associated Eosinophilia. Ann. Hematol. 2022, 101, 2533–2536. [Google Scholar] [CrossRef]
- Toledo, M.A.S.; Gatz, M.; Sontag, S.; Gleixner, K.V.; Eisenwort, G.; Feldberg, K.; Hamouda, A.E.I.; Kluge, F.; Guareschi, R.; Rossetti, G.; et al. Nintedanib Targets KIT D816V Neoplastic Cells Derived from Induced Pluripotent Stem Cells of Systemic Mastocytosis. Blood 2021, 137, 2070–2084. [Google Scholar] [CrossRef] [PubMed]
- Olschok, K.; Han, L.; de Toledo, M.A.S.; Böhnke, J.; Graßhoff, M.; Costa, I.G.; Theocharides, A.; Maurer, A.; Schüler, H.M.; Buhl, E.M.; et al. CALR Frameshift Mutations in MPN Patient-Derived IPSCs Accelerate Maturation of Megakaryocytes. Stem Cell Rep. 2021, 16, 2768–2783. [Google Scholar] [CrossRef] [PubMed]
- Sürün, D.; Schneider, A.; Mircetic, J.; Neumann, K.; Lansing, F.; Paszkowski-Rogacz, M.; Hänchen, V.; Lee-Kirsch, M.A.; Buchholz, F. Efficient Generation and Correction of Mutations in Human IPS Cells Utilizing MRNAs of CRISPR Base Editors and Prime Editors. Genes 2020, 11, 511. [Google Scholar] [CrossRef]
- Boehnke, J.; Atakhanov, S.; Toledo, M.A.S.; Schüler, H.M.; Sontag, S.; Chatain, N.; Koschmieder, S.; Brümmendorf, T.H.; Kramann, R.; Zenke, M. CRISPR/Cas9 Mediated CXCL4 Knockout in Human IPS Cells of Polycythemia Vera Patient with JAK2 V617F Mutation. Stem Cell Res. 2021, 55, 102490. [Google Scholar] [CrossRef]
- Sontag, S.; Förster, M.; Qin, J.; Wanek, P.; Mitzka, S.; Schüler, H.M.; Koschmieder, S.; Rose-John, S.; Seré, K.; Zenke, M. Modelling IRF8 Deficient Human Hematopoiesis and Dendritic Cell Development with Engineered IPS Cells. Stem Cells 2017, 35, 898–908. [Google Scholar] [CrossRef]
- Fischer, M.; Goldschmitt, J.; Peschel, C.; Brakenhoff, J.P.G.; Kallen, K.J.; Wollmer, A.; Grötzinger, J.; Rose-John, S.I. A Bioactive Designer Cytokine for Human Hematopoietic Progenitor Cell Expansion. Nat. Biotechnol. 1997, 15, 142–145. [Google Scholar] [CrossRef]
- Jamur, M.C.; Grodzki, A.C.G.; Moreno, A.N.; de Mello, L.d.F.; Pastor, M.V.D.; Berenstein, E.H.; Siraganian, R.P.; Oliver, C. Identification and Isolation of Rat Bone Marrow-Derived Mast Cells Using the Mast Cell-Specific Monoclonal Antibody AA4. J. Histochem. Cytochem. 2001, 49, 219–228. [Google Scholar] [CrossRef]
- Dasilva-Freire, N.; Mayado, A.; Teodosio, C.; Jara-Acevedo, M.; Álvarez-Twose, I.; Matito, A.; Sánchez-Muñoz, L.; Caldas, C.; Henriques, A.; Muñoz-González, J.I.; et al. Bone Marrow Mast Cell Antibody-Targetable Cell Surface Protein Expression Profiles in Systemic Mastocytosis. Int. J. Mol. Sci. 2019, 20, 552. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Sayama, K.; Yano, K.; Lantz, C.S.; Noben-Trauth, N.; Ra, C.; Costa, J.J.; Galli, S.J. IgE Enhances Fcε Receptor I Expression and IgE-Dependent Release of Histamine and Lipid Mediators from Human Umbilical Cord Blood-Derived Mast Cells: Synergistic Effect of IL-4 and IgE on Human Mast Cell Fcε Receptor I Expression and Mediator Release. J. Immunol. 1999, 162, 5455–5465. [Google Scholar] [CrossRef]
- Xia, H.-Z.; Du, Z.; Craig, S.; Klisch, G.; Noben-Trauth, N.; Kochan, J.P.; Huff, T.H.; Irani, A.-M.A.; Schwartz2, L.E. Effect of Recombinant Human IL-4 on Tryptase, Chymase, and Fc Epsilon Receptor Type I Expression in Recombinant Human Stem Cell Factor-Dependent Fetal Liver-Derived Human Mast Cells. J. Immunol. 1997, 159, 2911–2921. [Google Scholar] [CrossRef]
- Nilsson, G.; Miettinen, U.; Ishizaka, T.; Ashman, L.K.; Irani, A.M.; Schwartz, L.B. Interleukin-4 Inhibits the Expression of Kit and Tryptase During Stem Cell Factor-Dependent Development of Human Mast Cells From Fetal Liver Cells. Blood 1994, 84, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, A.M.; Tepper, R.I.; Weller, P.F.; Morgan, E.S.; Estrella, P.; Monahan- Earley, R.A.; Galli, S.J. Piecemeal Degranulation of Mast Cells in the Inflammatory Eyelid Lesions of Interleukin-4 Transgenic Mice. Evidence of Mast Cell Histamine Release In Vivo by Diamine Oxidase-Gold Enzyme-Affinity Ultrastructural Cytochemistry. Blood 1994, 83, 3600–3612. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, A.M.; Schleimer, R.P.; Lichtenstein, L.M. Human Mast Cells Synthesize New Granules During Recovery From Degranulation. In Vitro Studies With Mast Cells Purified From Human Lungs. Blood 1988, 71, 76–85. [Google Scholar] [CrossRef]
- Motakis, E.; Guhl, S.; Ishizu, Y.; Itoh, M.; Kawaji, H.; De Hoon, M.; Lassmann, T.; Carninci, P.; Hayashizaki, Y.; Zuberbier, T.; et al. Redefinition of the Human Mast Cell Transcriptome by Deep-CAGE Sequencing. Blood 2014, 123, e58–e67. [Google Scholar] [CrossRef]
- Roy, S.; Chompunud Na Ayudhya, C.; Thapaliya, M.; Deepak, V.; Ali, H. Multifaceted MRGPRX2: New Insight into the Role of Mast Cells in Health and Disease. J. Allergy Clin. Immunol. 2021, 148, 293–308. [Google Scholar] [CrossRef]
- Teodosio, C.; García-Montero, A.C.; Jara-Acevedo, M.; Sánchez-Muñoz, L.; Pedreira, C.E.; Álvarez-Twose, I.; Matarraz, S.; Morgado, J.M.; Bárcena, P.; Matito, A.; et al. Gene Expression Profile of Highly Purified Bone Marrow Mast Cells in Systemic Mastocytosis. J. Allergy Clin. Immunol. 2013, 131, 1213–1224, 1224.e1-4. [Google Scholar] [CrossRef]
- Ikuno, T.; Ito, S.; Inoue, T. Human Induced Pluripotent Stem Cell-Derived Mast Cells Useful for in Vitro Mast Cell Activation Assay Exhibiting Phenotypes and Morphological Characteristics of Human Mast Cells. J. Toxicol. Sci. 2019, 44, 789–797. [Google Scholar] [CrossRef]
- Igarashi, A.; Ebihara, Y.; Kumagai, T.; Hirai, H.; Nagata, K.; Tsuji, K. Mast Cells Derived from Human Induced Pluripotent Stem Cells Are Useful for Allergen Tests. Allergol. Int. 2018, 67, 234–242. [Google Scholar] [CrossRef]
- Jackson, C.W.; Pratt, C.M.; Rupprecht, C.P.; Pattanaik, D.; Krishnaswamy, G. Mastocytosis and Mast Cell Activation Disorders: Clearing the Air. Int. J. Mol. Sci. 2021, 22, 11270. [Google Scholar] [CrossRef] [PubMed]
- Caslin, H.L.; Kiwanuka, K.N.; Haque, T.T.; Taruselli, M.T.; MacKnight, H.P.; Paranjape, A.; Ryan, J.J. Controlling Mast Cell Activation and Homeostasis: Work Influenced by Bill Paul That Continues Today. Front. Immunol. 2018, 9, 868. [Google Scholar] [CrossRef]
- Hundley, T.R.; Gilfillan, A.M.; Tkaczyk, C.; Andrade, M.V.; Metcalfe, D.D.; Beaven, M.A. Kit and FcϵRI Mediate Unique and Convergent Signals for Release of Inflammatory Mediators from Human Mast Cells. Blood 2004, 104, 2410–2417. [Google Scholar] [CrossRef] [PubMed]
- Tobío, A.; Bandara, G.; Morris, D.A.; Kim, D.-K.; O’Connell, M.P.; Komarow, H.D.; Carter, M.C.; Smrz, D.; Metcalfe, D.D.; Olivera, A. Oncogenic D816V-KIT Signaling in Mast Cells Causes Persistent IL-6 Production. Haematologica 2020, 105, 124–135. [Google Scholar] [CrossRef]
- Kuehn, H.S.; Radinger, M.; Gilfillan, A.M. Measuring Mast Cell Mediator Release. Curr. Protoc. Immunol. 2010, 91, 7.38.1–7.38.9. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]




| Antibody | Specificity | Company | Cat. N° | Dilution | Assay |
|---|---|---|---|---|---|
| CD2-FITC | human | BD Biosciences | 556608 | 1:50 | FACS |
| CD25-PE | human | BD Biosciences | 567214 | 1:100 | FACS |
| CD30-PE | human | BD Biosciences | 550041 | 1:100 | FACS |
| CD33-APC | human | Miltenyi Biotec | 130-113-345 | 1:100 | FACS |
| CD45-APCVio770 | human | Miltenyi Biotec | 130-113-677 | 1:200 | FACS |
| CD117 (KIT)-PE-Cy7 | human | eBioscience | 25-1178-42 | 1:100 | FACS |
| FcER1a-PE | human | BD Biosciences | 566608 | 1:100 | FACS |
| Tryptase | human | Dako | M7052 | 1:200 | IH |
| Oligo Name | GC-Content | Tm | Sequence (5′->3′) | Length [mer] |
|---|---|---|---|---|
| APOL1_FRW | 55 | 62.1 | GCTTTGCTGAGAGTCTCTGTCC | 22 |
| APOL1_REV | 52 | 62.4 | GGGCTTACTTTGAGGATCTCCAG | 23 |
| CASP1_FRW | 55 | 59.4 | GCTTTCTGCTCTTCCACACC | 20 |
| CASP1_REV | 50 | 57.3 | TCCTCCACATCACAGGAACA | 20 |
| CCL23_FRW | 50 | 57.3 | ATGCTTGTTACTGCCCTTGG | 20 |
| CCL23_REV | 55 | 59.4 | GGGTCATCTGAGGACCAATC | 20 |
| CFH_FRW | 45 | 58.4 | CTGATCGCAAGAAAGACCAGTA | 22 |
| CFH_REV | 45 | 58.4 | TGGTAGCACTGAACGGAATTAG | 22 |
| GAPDH_FRW | 56 | 56 | GAAGGTGAAGGTCGGAGT | 18 |
| GAPDH_REV | 45 | 58 | GAAGATGGTGATGGGATTTC | 20 |
| IFIT2_FRW | 52.6 | 65 | AGCGAAGGTGGCTTTGAGA | 19 |
| IFIT2_REV | 55 | 65 | GAGGGTCAATGGCGTTCTGA | 20 |
| LAMP1_FRW | 50 | 57.3 | TGAAAAATGGCAACGGGACC | 20 |
| LAMP1_REV | 55 | 59.4 | ATGAGCTGGACGCTGTAACG | 20 |
| LAT2_FRW | 55 | 59.4 | CAGCATCCATCCATCAGTGG | 20 |
| LAT2_REV | 58 | 58.8 | TTCTTGGTCTGCCCTAGGC | 19 |
| LIPA_FRW | 55 | 59.4 | CTAGAATCTGCCAGCAAGCC | 20 |
| LIPA_REV | 45 | 55.2 | TGTGCCTTAACCGAATTCCT | 20 |
| MRGPRX2_FRW | 52 | 59.8 | AGTCCCAGGAAAGCACTTCTC | 21 |
| MRGPRX2_REV | 55 | 59.4 | AGGGTCTCCTTGCCACAAAG | 20 |
| PCNA_FRW | 42 | 59.3 | GAACTGGTTCATTCATCTCTATGG | 24 |
| PCNA_REV | 35 | 58.5 | TGTCACAGACAAGTAATGTCGATAAA | 26 |
| SERPING1_FRW | 55 | 59.4 | TGAAGCTCTACCACGCCTTC | 20 |
| SERPING1_REV | 55 | 59.4 | TGGTGGACACAGGTGAAGTC | 20 |
| STAT1_FRW | 55 | 59.4 | TGTATGCCATCCTCGAGAGC | 20 |
| STAT1_REV | 55 | 59.4 | AGACATCCTGCCACCTTGTG | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Toledo, M.A.S.; Fu, X.; Maié, T.; Buhl, E.M.; Götz, K.; Schmitz, S.; Kaiser, A.; Boor, P.; Braunschweig, T.; Chatain, N.; et al. KIT D816V Mast Cells Derived from Induced Pluripotent Stem Cells Recapitulate Systemic Mastocytosis Transcriptional Profile. Int. J. Mol. Sci. 2023, 24, 5275. https://doi.org/10.3390/ijms24065275
de Toledo MAS, Fu X, Maié T, Buhl EM, Götz K, Schmitz S, Kaiser A, Boor P, Braunschweig T, Chatain N, et al. KIT D816V Mast Cells Derived from Induced Pluripotent Stem Cells Recapitulate Systemic Mastocytosis Transcriptional Profile. International Journal of Molecular Sciences. 2023; 24(6):5275. https://doi.org/10.3390/ijms24065275
Chicago/Turabian Stylede Toledo, Marcelo A. S., Xuhuang Fu, Tiago Maié, Eva M. Buhl, Katrin Götz, Susanne Schmitz, Anne Kaiser, Peter Boor, Till Braunschweig, Nicolas Chatain, and et al. 2023. "KIT D816V Mast Cells Derived from Induced Pluripotent Stem Cells Recapitulate Systemic Mastocytosis Transcriptional Profile" International Journal of Molecular Sciences 24, no. 6: 5275. https://doi.org/10.3390/ijms24065275
APA Stylede Toledo, M. A. S., Fu, X., Maié, T., Buhl, E. M., Götz, K., Schmitz, S., Kaiser, A., Boor, P., Braunschweig, T., Chatain, N., Costa, I. G., Brümmendorf, T. H., Koschmieder, S., & Zenke, M. (2023). KIT D816V Mast Cells Derived from Induced Pluripotent Stem Cells Recapitulate Systemic Mastocytosis Transcriptional Profile. International Journal of Molecular Sciences, 24(6), 5275. https://doi.org/10.3390/ijms24065275