Author Contributions
Conceptualization, M.H. and D.S.; methodology, M.H.; formal analysis, M.H.; investigation, M.H.; resources, M.H. and D.S.; data curation, M.H.; writing—original draft preparation, M.H. and D.S.; writing—review and editing, N.K., F.C., S.-Y.H., T.W.F., S.-L.H., M.M. and S.S.; visualization, Y.H.; supervision, M.H.; project administration, M.H. All authors have read and agreed to the published version of the manuscript.
Figure 1.
Induction of S100A15 expression in oral mucosa by both gram− and gram+ oral bacterial pathogens. (A–D) RT-PCR expression of S100A15 by the stimulation of GF (A) and KB (B) cells with lipopolysaccharides (LPS) at a concentration of 100 ng/mL for regulated time intervals up to 72 h; expression of S100A15 by the stimulation of GF (C) and KB (D) cells with lipoteichoic acid (LTA) at a concentration of 50 ng/mL for regulated time intervals up to 72 h. Total RNAs were extracted from treated cells at different time intervals and subjected to RT-PCR analysis as described under material and methods. The RT-PCR products were analyzed on 2% agarose stained with ethidium bromide. The leader DNA 1.0 kbp was used as a DNA marker. Actin was used as an internal control. Data are representative of three independent experiments performed separately. (E,F) qRT-PCR expression of S100A15 by the stimulation of GF and KB cells with either heat-inactivated P. gingivalis (E) or heat-inactivated S. sanguinis (F) microbial pathogens (MPs) for regulated time intervals up to 72 h. Total RNA was extracted from treated cells at different time points and subjected to qRT-PCR analysis as described under material and methods. The represented data are the mean ± SD of three independent experiments performed in duplicate.
Figure 1.
Induction of S100A15 expression in oral mucosa by both gram− and gram+ oral bacterial pathogens. (A–D) RT-PCR expression of S100A15 by the stimulation of GF (A) and KB (B) cells with lipopolysaccharides (LPS) at a concentration of 100 ng/mL for regulated time intervals up to 72 h; expression of S100A15 by the stimulation of GF (C) and KB (D) cells with lipoteichoic acid (LTA) at a concentration of 50 ng/mL for regulated time intervals up to 72 h. Total RNAs were extracted from treated cells at different time intervals and subjected to RT-PCR analysis as described under material and methods. The RT-PCR products were analyzed on 2% agarose stained with ethidium bromide. The leader DNA 1.0 kbp was used as a DNA marker. Actin was used as an internal control. Data are representative of three independent experiments performed separately. (E,F) qRT-PCR expression of S100A15 by the stimulation of GF and KB cells with either heat-inactivated P. gingivalis (E) or heat-inactivated S. sanguinis (F) microbial pathogens (MPs) for regulated time intervals up to 72 h. Total RNA was extracted from treated cells at different time points and subjected to qRT-PCR analysis as described under material and methods. The represented data are the mean ± SD of three independent experiments performed in duplicate.
![Ijms 24 05348 g001]()
Figure 2.
Inhibition of induced expression of S100A15 by neutralization of toll-like receptors. (A) GF and KB cells were pre-treated with anti-TLR2, anti-TLR4, or control IgG 1 h prior to the stimulation with either heat-inactivated P. gingivalis or heat-inactivated S. sanguinis for 48 h. Total RNA was extracted from treated and control cells and subsequently subjected to qRT-PCR analysis to assess the relative transcription levels of S100A15. (B) GF and KB cells were pre-treated with anti-TLR2, anti-TLR4, or control IgG 1 h prior to stimulation with either LPS (100 ng/mL) or LTA (50 ng/mL) for 48 h. Total RNAs were extracted from treated and control cells and subsequently subjected to qRT-PCR analysis to assess the relative transcription levels of S100A15. Densitometric quantification of S100A15 mRNA over actin mRNA transcript. The data were normalized to the level of treated and untreated control cells in each sample. Each bar represents the mean ± SD of three independent experiments performed in duplicate, * p < 0.05.
Figure 2.
Inhibition of induced expression of S100A15 by neutralization of toll-like receptors. (A) GF and KB cells were pre-treated with anti-TLR2, anti-TLR4, or control IgG 1 h prior to the stimulation with either heat-inactivated P. gingivalis or heat-inactivated S. sanguinis for 48 h. Total RNA was extracted from treated and control cells and subsequently subjected to qRT-PCR analysis to assess the relative transcription levels of S100A15. (B) GF and KB cells were pre-treated with anti-TLR2, anti-TLR4, or control IgG 1 h prior to stimulation with either LPS (100 ng/mL) or LTA (50 ng/mL) for 48 h. Total RNAs were extracted from treated and control cells and subsequently subjected to qRT-PCR analysis to assess the relative transcription levels of S100A15. Densitometric quantification of S100A15 mRNA over actin mRNA transcript. The data were normalized to the level of treated and untreated control cells in each sample. Each bar represents the mean ± SD of three independent experiments performed in duplicate, * p < 0.05.
![Ijms 24 05348 g002]()
Figure 3.
Induction of ASK1, JNK, and p38 pathways in oral mucosa by both gram− and gram+ bacterial pathogens. Both GF and KB cells were stimulated with either heat-inactivated P. gingivalis (2 × 104 cells/mL) or S. sanguinis (2 × 104 cells/mL) microbial pathogens (MPs) for 48 h (A,B). Total protein lysate and nuclear extract were prepared to perform kinase assays, Western blot, and EMSA. Western blot demonstrates the induction of TRAF6 expression and phosphorylation of ASK1 activity in both GF and KB cells by stimulation with either heat-inactivated P. gingivalis (A) or heat-inactivated S. sanguinis (B) when compared with control cells. Also, Western blot analysis of the total lysates of treated and control cells using anti-phospho-ASK1, phospho- JNK, phospho-p38, TRAF6, ASK1, JNK, or p38 antibodies demonstrate the induction of JNK and p38 phosphorylation without any marked alteration to the expression level of ASK1, JNK, or p38 by stimulation with either heat-inactivated P. gingivalis (A) or heat-inactivated S. sanguinis(B), when compared with control cells. Actin was used as an internal control for loading and transfer. (C) Analysis of band intensity on films is presented as the relative ratio of TRAF6 expression to the expression of actin, the relative ratio of phospho-ASK1, Phospho-JNK, and Phospho-p38 to the expression of ASK1, JNK, and p38 expression. The data were normalized to the level of Actin expression in treated and untreated control cells in each sample. Each bar represents the mean ± SD of three independent experiments performed separately, * p < 0.05. (D,E) The DNA-binding activity of the transcription factors AP-1 and ATF-2 were investigated by EMSA using nuclear extracts of treated and control cells. The stimulation of both GF and KB cells with either heat-inactivated P. gingivalis (D) or heat-inactivated S. sanguinis (E) induced the DNA-binding activity of the transcription factor AP-1 when compared with control cells. Also, the stimulation of both GF and KB cells with either heat-inactivated P. gingivalis (F) or heat-inactivated S. sanguinis (G) induced the DNA-binding of the transcription factor ATF-2 when compared with control cells. (H,I) Analysis of band intensity of the transcription factors AP-1 and ATF-2. The data were normalized to the level of the binding activity of the transcription factors in control cells in each sample. Each bar represents the mean ± SD of three independent experiments performed separately, * p < 0.05.
Figure 3.
Induction of ASK1, JNK, and p38 pathways in oral mucosa by both gram− and gram+ bacterial pathogens. Both GF and KB cells were stimulated with either heat-inactivated P. gingivalis (2 × 104 cells/mL) or S. sanguinis (2 × 104 cells/mL) microbial pathogens (MPs) for 48 h (A,B). Total protein lysate and nuclear extract were prepared to perform kinase assays, Western blot, and EMSA. Western blot demonstrates the induction of TRAF6 expression and phosphorylation of ASK1 activity in both GF and KB cells by stimulation with either heat-inactivated P. gingivalis (A) or heat-inactivated S. sanguinis (B) when compared with control cells. Also, Western blot analysis of the total lysates of treated and control cells using anti-phospho-ASK1, phospho- JNK, phospho-p38, TRAF6, ASK1, JNK, or p38 antibodies demonstrate the induction of JNK and p38 phosphorylation without any marked alteration to the expression level of ASK1, JNK, or p38 by stimulation with either heat-inactivated P. gingivalis (A) or heat-inactivated S. sanguinis(B), when compared with control cells. Actin was used as an internal control for loading and transfer. (C) Analysis of band intensity on films is presented as the relative ratio of TRAF6 expression to the expression of actin, the relative ratio of phospho-ASK1, Phospho-JNK, and Phospho-p38 to the expression of ASK1, JNK, and p38 expression. The data were normalized to the level of Actin expression in treated and untreated control cells in each sample. Each bar represents the mean ± SD of three independent experiments performed separately, * p < 0.05. (D,E) The DNA-binding activity of the transcription factors AP-1 and ATF-2 were investigated by EMSA using nuclear extracts of treated and control cells. The stimulation of both GF and KB cells with either heat-inactivated P. gingivalis (D) or heat-inactivated S. sanguinis (E) induced the DNA-binding activity of the transcription factor AP-1 when compared with control cells. Also, the stimulation of both GF and KB cells with either heat-inactivated P. gingivalis (F) or heat-inactivated S. sanguinis (G) induced the DNA-binding of the transcription factor ATF-2 when compared with control cells. (H,I) Analysis of band intensity of the transcription factors AP-1 and ATF-2. The data were normalized to the level of the binding activity of the transcription factors in control cells in each sample. Each bar represents the mean ± SD of three independent experiments performed separately, * p < 0.05.
![Ijms 24 05348 g003]()
Figure 4.
ASK1 mediates the activation of both the JNK and p38 pathways in oral mucosa by stimulation with bacterial pathogens. (A–F) Both GF and KB cells were pre-treated with the inhibitor of ASK1 (thioredoxin) 1 h prior to stimulation with either heat-inactivated P. gingivalis (2 × 104 cells/mL) or heat-activated S. sanguinis (2 × 104 cells/mL) microbial pathogens (MPs) for 48 h. Total protein lysate and nuclear extract were then prepared for Western blot analysis and EMSA assay. (A,B) Western blot analysis of the total lysate of treated and control cells using anti-phospho-JNK, phospho-p38, JNK, or p38 antibodies demonstrate the inhibition of inactivated P. gingivalis (A) and S. sanguinis (B)-induced phosphorylation of both JNK and p38 proteins by the inhibitor of ASK1 in both GF and KB cells when compared with control cells. JNK, p38, and actin were used as internal controls for loading and transfer. (C,D) Analysis of band intensity on films is presented as the relative ratio of Phospho-JNK and Phospho-p38 to the expression of JNK and p38 expression. Bars represent the mean ± SD from three independent experiments performed separately, * p < 0.05. (E,F) Pre-treatment of both GF and KB cells with ASK1 inhibitor suppressed the induced DNA-binding activity of the transcription factors AP-1 (C,D) and ATF-2 (E,H) by the stimulation of both GF and KB cells with either heat-inactivated P. gingivalis (E,G) or heat-inactivated S. sanguinis (F,H) bacterial pathogens. (I) Analysis of band intensity of the transcription factors AP-1 and ATF-2. Bars represent mean ± SD from three independent experiments performed separately, * p < 0.05.
Figure 4.
ASK1 mediates the activation of both the JNK and p38 pathways in oral mucosa by stimulation with bacterial pathogens. (A–F) Both GF and KB cells were pre-treated with the inhibitor of ASK1 (thioredoxin) 1 h prior to stimulation with either heat-inactivated P. gingivalis (2 × 104 cells/mL) or heat-activated S. sanguinis (2 × 104 cells/mL) microbial pathogens (MPs) for 48 h. Total protein lysate and nuclear extract were then prepared for Western blot analysis and EMSA assay. (A,B) Western blot analysis of the total lysate of treated and control cells using anti-phospho-JNK, phospho-p38, JNK, or p38 antibodies demonstrate the inhibition of inactivated P. gingivalis (A) and S. sanguinis (B)-induced phosphorylation of both JNK and p38 proteins by the inhibitor of ASK1 in both GF and KB cells when compared with control cells. JNK, p38, and actin were used as internal controls for loading and transfer. (C,D) Analysis of band intensity on films is presented as the relative ratio of Phospho-JNK and Phospho-p38 to the expression of JNK and p38 expression. Bars represent the mean ± SD from three independent experiments performed separately, * p < 0.05. (E,F) Pre-treatment of both GF and KB cells with ASK1 inhibitor suppressed the induced DNA-binding activity of the transcription factors AP-1 (C,D) and ATF-2 (E,H) by the stimulation of both GF and KB cells with either heat-inactivated P. gingivalis (E,G) or heat-inactivated S. sanguinis (F,H) bacterial pathogens. (I) Analysis of band intensity of the transcription factors AP-1 and ATF-2. Bars represent mean ± SD from three independent experiments performed separately, * p < 0.05.
![Ijms 24 05348 g004]()
Figure 5.
Induction of NF-κB pathway by both gram− and gram+ bacterial pathogens in oral mucosa-derived cells. Both GF and KB cells were stimulated with either heat-inactivated P. gingivalis (2 × 104 cells/mL) or heat-inactivated S. sanguinis (2 × 104 cells/mL) microbial pathogens (MPs) for 48 h. Then, total protein lysate and nuclear extract were prepared to perform kinase assays, Western blot, and EMSA. Kinase assay demonstrates the activation of IKK by both in vitro phosphorylation of the IκBα substrate and NF-κB inhibition in both GF and KB cells by stimulation with either P. gingivalis (A) or S. sanguinis(B) when compared to control cells. Data are representative of three independent experiments performed separately. Western blot analysis of cytoplasmic protein of treated and control cells using anti-IκBα, IKKα, and actin antibodies. Western blot analysis of the protein stability of IκBα in the cytoplasmic protein of both cells demonstrates degradation of IκBα protein, evidence NF-κB activation in both GF and KB cells when stimulated with either heat-inactivated P. gingivalis (A) or heat-inactivated S. sanguinis (B) bacterial pathogens. IKKα and actin were used as internal controls for loading and transfer. (C) Analysis of band intensity on films is presented as the relative ratio of phospho-IκBα to the expression of IKK α and the degradation of IκBα to the expression level of actin. Bars represent the mean ± SD from three independent experiments performed separately, * p < 0.05. The DNA-binding activity of the transcription factors NF-κB was investigated by EMSA using nuclear extracts of treated and control cells. Stimulation of both GF and KB cells with heat-inactivated P. gingivalis (D) or heat-inactivated S. sanguinis (E) induced the DNA-binding activity of the transcription factor NF-κB when compared with those of control cells. (F) Analysis of band intensity of the transcription factor of NF-κB. Bars represent mean ± SD from three independent experiments performed separately, * p < 0.05.
Figure 5.
Induction of NF-κB pathway by both gram− and gram+ bacterial pathogens in oral mucosa-derived cells. Both GF and KB cells were stimulated with either heat-inactivated P. gingivalis (2 × 104 cells/mL) or heat-inactivated S. sanguinis (2 × 104 cells/mL) microbial pathogens (MPs) for 48 h. Then, total protein lysate and nuclear extract were prepared to perform kinase assays, Western blot, and EMSA. Kinase assay demonstrates the activation of IKK by both in vitro phosphorylation of the IκBα substrate and NF-κB inhibition in both GF and KB cells by stimulation with either P. gingivalis (A) or S. sanguinis(B) when compared to control cells. Data are representative of three independent experiments performed separately. Western blot analysis of cytoplasmic protein of treated and control cells using anti-IκBα, IKKα, and actin antibodies. Western blot analysis of the protein stability of IκBα in the cytoplasmic protein of both cells demonstrates degradation of IκBα protein, evidence NF-κB activation in both GF and KB cells when stimulated with either heat-inactivated P. gingivalis (A) or heat-inactivated S. sanguinis (B) bacterial pathogens. IKKα and actin were used as internal controls for loading and transfer. (C) Analysis of band intensity on films is presented as the relative ratio of phospho-IκBα to the expression of IKK α and the degradation of IκBα to the expression level of actin. Bars represent the mean ± SD from three independent experiments performed separately, * p < 0.05. The DNA-binding activity of the transcription factors NF-κB was investigated by EMSA using nuclear extracts of treated and control cells. Stimulation of both GF and KB cells with heat-inactivated P. gingivalis (D) or heat-inactivated S. sanguinis (E) induced the DNA-binding activity of the transcription factor NF-κB when compared with those of control cells. (F) Analysis of band intensity of the transcription factor of NF-κB. Bars represent mean ± SD from three independent experiments performed separately, * p < 0.05.
![Ijms 24 05348 g005]()
Figure 6.
Bacterial pathogens-induced expression of S100A15 in oral mucosa-derived cells is mediated by the MAP kinase and NF-κB pathways. (A) GF and KB cells were pre-treated with Thioredoxin 1 h prior to stimulation with either heat-inactivated gram− P. gingivalis or heat-inactivated gram+ S. sanguinis for 48 h. Total RNA was extracted from treated and control cells and subsequently subjected to qRT-PCR analysis to assess the relative transcription levels of S100A15. (B) GF and KB cells were pre-treated with inhibitors of JNK (SP600125), p38 (SB-203580), or NF-κB (Bay11-7082) 1 h prior to stimulation with either heat-inactivated P. gingivalis or heat-inactivated S. sanguinis for 48 h. Total RNA was extracted from treated and control cells and subsequently subjected to qRT-PCR analysis to assess the relative transcription levels of S100A15. (C) GF and KB cells were pre-treated with SP600125, SB-203580, or Bay11-7082 1 h prior to stimulation with heat-inactivated P. gingivalis or heat-inactivated S. sanguinis for 48 h. Total RNA was extracted from treated and control cells and subsequently subjected to qRT-PCR analysis to assess relative transcription levels of S100A15. Densitometric quantification of S100A15 mRNA over actin mRNA transcript. Each bar represents the mean ± SD of three independent experiments performed in duplicate, * p < 0.05.
Figure 6.
Bacterial pathogens-induced expression of S100A15 in oral mucosa-derived cells is mediated by the MAP kinase and NF-κB pathways. (A) GF and KB cells were pre-treated with Thioredoxin 1 h prior to stimulation with either heat-inactivated gram− P. gingivalis or heat-inactivated gram+ S. sanguinis for 48 h. Total RNA was extracted from treated and control cells and subsequently subjected to qRT-PCR analysis to assess the relative transcription levels of S100A15. (B) GF and KB cells were pre-treated with inhibitors of JNK (SP600125), p38 (SB-203580), or NF-κB (Bay11-7082) 1 h prior to stimulation with either heat-inactivated P. gingivalis or heat-inactivated S. sanguinis for 48 h. Total RNA was extracted from treated and control cells and subsequently subjected to qRT-PCR analysis to assess the relative transcription levels of S100A15. (C) GF and KB cells were pre-treated with SP600125, SB-203580, or Bay11-7082 1 h prior to stimulation with heat-inactivated P. gingivalis or heat-inactivated S. sanguinis for 48 h. Total RNA was extracted from treated and control cells and subsequently subjected to qRT-PCR analysis to assess relative transcription levels of S100A15. Densitometric quantification of S100A15 mRNA over actin mRNA transcript. Each bar represents the mean ± SD of three independent experiments performed in duplicate, * p < 0.05.
![Ijms 24 05348 g006]()
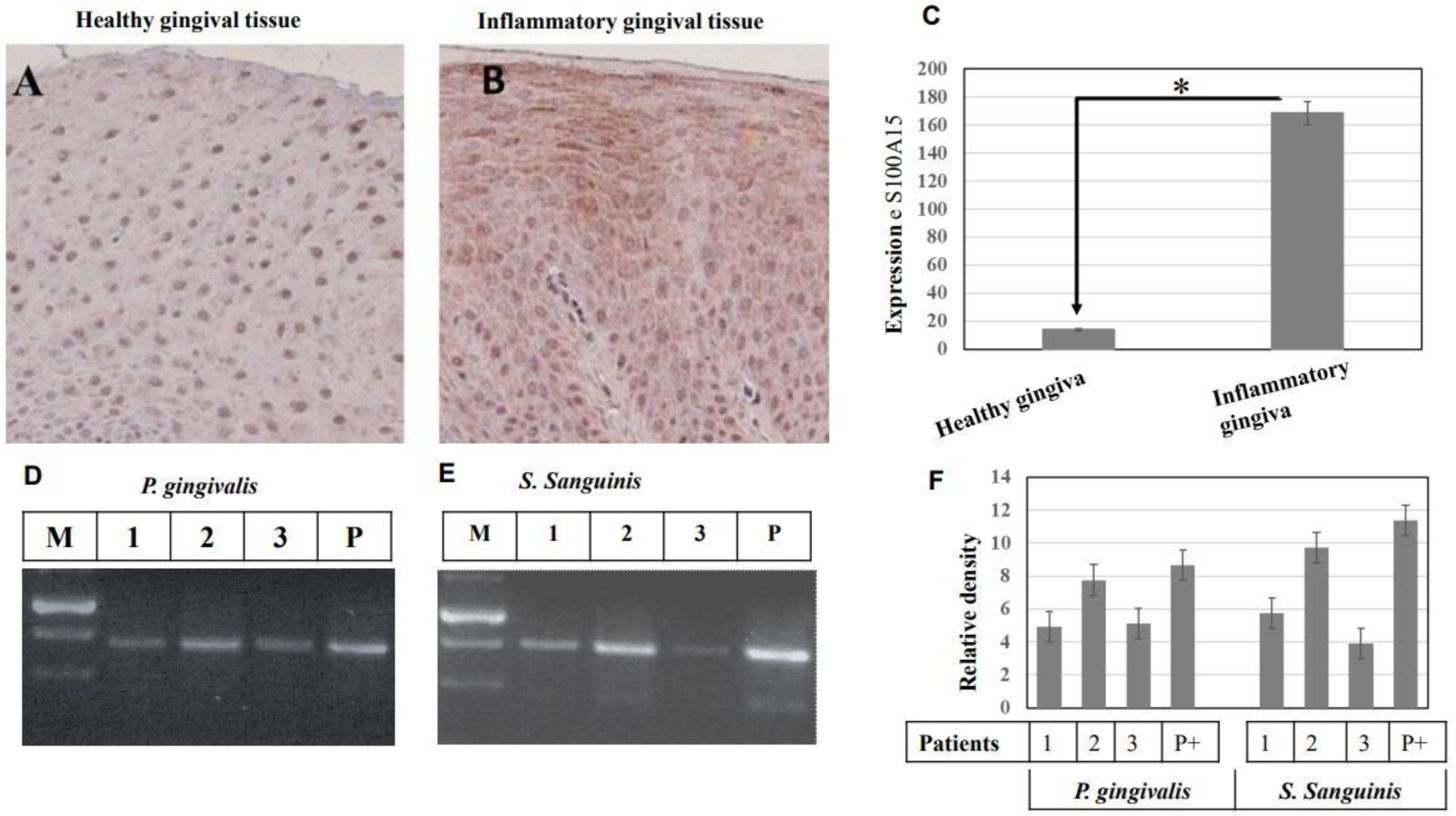
Figure 7.
Immunohistochemical analysis of S100A15 protein in oral mucosa of patients with inflammatory gingiva. (A), Gingival tissues of healthy individuals (n = 10) were stained with anti-S100A15 antibodies. (B) Gingival tissues of patients with inflammatory gingiva (n = 10) were stained with anti-S100A15 antibody. Original magnification × 100. Scale bar indicates 100 μm. (C) Statistical Analysis of S100A15 expression in gingival tissues of patients with inflammatory gingiva (n = 10) as a relative ratio to the expression of S100A15 in gingival tissues of healthy individuals. Bars represent mean ± SD from inflammatory (n = 10) and healthy (n = 10) gingival tissues, * p < 0.05. (D,E) Detection of oral bacterial pathogens (gram− P. gingivalis and gram+ S. sanguinis) in the saliva of patients with inflammatory gingiva by conventional PCR. Detection of both P. gingivalis and S. sanguinis in the saliva of patients with inflammatory gingiva by PCR. The PCR was performed using specific primers to amplify a 480 bp fragment of the 16S rRNA gene as a marker for P. gingivalis (D) and specific primers to amplify a 473 bp fragment of the UDP-N-acetylglucosamine-like protein gene as a marker for S. sanguinis (E). PCR products were separated on 2% agarose and stained with ethidium bromide. (F) Analysis of band intensity as the relative ratio of the amplified DNA fragment of 16S rRNA gene, the marker of P. gingivalis, and the amplified of the UDP-N-acetylglucosamine-the marker of S. sanguinis to the corresponding positive control. Bars represent the mean ± SD from three blots, * p < 0.05.
Figure 7.
Immunohistochemical analysis of S100A15 protein in oral mucosa of patients with inflammatory gingiva. (A), Gingival tissues of healthy individuals (n = 10) were stained with anti-S100A15 antibodies. (B) Gingival tissues of patients with inflammatory gingiva (n = 10) were stained with anti-S100A15 antibody. Original magnification × 100. Scale bar indicates 100 μm. (C) Statistical Analysis of S100A15 expression in gingival tissues of patients with inflammatory gingiva (n = 10) as a relative ratio to the expression of S100A15 in gingival tissues of healthy individuals. Bars represent mean ± SD from inflammatory (n = 10) and healthy (n = 10) gingival tissues, * p < 0.05. (D,E) Detection of oral bacterial pathogens (gram− P. gingivalis and gram+ S. sanguinis) in the saliva of patients with inflammatory gingiva by conventional PCR. Detection of both P. gingivalis and S. sanguinis in the saliva of patients with inflammatory gingiva by PCR. The PCR was performed using specific primers to amplify a 480 bp fragment of the 16S rRNA gene as a marker for P. gingivalis (D) and specific primers to amplify a 473 bp fragment of the UDP-N-acetylglucosamine-like protein gene as a marker for S. sanguinis (E). PCR products were separated on 2% agarose and stained with ethidium bromide. (F) Analysis of band intensity as the relative ratio of the amplified DNA fragment of 16S rRNA gene, the marker of P. gingivalis, and the amplified of the UDP-N-acetylglucosamine-the marker of S. sanguinis to the corresponding positive control. Bars represent the mean ± SD from three blots, * p < 0.05.
![Ijms 24 05348 g007]()
Figure 8.
Proposed model for the mechanisms whereby the gram− and gram+ bacterial pathogens induce the expression of the antimicrobial protein, S100A15, in the oral mucosa. The colonization of gram− and gram+ bacterial pathogens result in the activation of both TLR4 and TLR2, respectively. As a consequence, TRAF6 plays a central role in mediating TLR4 and TLR2-induced signaling to NF-kB/ASK1-JNK-AP-1/ASK1-p38-ATF-2 pathways to enhance the transcriptional activation of S100A5, which subsequently functions as an antimicrobial protein to protect oral mucosa from bacterial pathogens.
Figure 8.
Proposed model for the mechanisms whereby the gram− and gram+ bacterial pathogens induce the expression of the antimicrobial protein, S100A15, in the oral mucosa. The colonization of gram− and gram+ bacterial pathogens result in the activation of both TLR4 and TLR2, respectively. As a consequence, TRAF6 plays a central role in mediating TLR4 and TLR2-induced signaling to NF-kB/ASK1-JNK-AP-1/ASK1-p38-ATF-2 pathways to enhance the transcriptional activation of S100A5, which subsequently functions as an antimicrobial protein to protect oral mucosa from bacterial pathogens.



 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}