
miRNAs in Uremic Cardiomyopathy: A Comprehensive Review
 , , , and
, , , and
Abstract
:1. Introduction
2. General Mechanisms Leading to Cardiac Alterations in CKD
3. miRNAs in CKD-Induced Pathological Cardiac Remodeling
4. miRNAs, UCM and the Na/K-ATPase Signaling Pathway
5. miRNAs in the RAS-Mediated Enhancement of Fibrosis in UCM
6. miRNAs Interplay with Circulating Hormones to Exacerbate UCM
7. Clinical Studies of miRNAs in CKD/ESKD Individuals
8. Overview and Future Directions for Studies on miRNAs in UCM
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jankowski, J.; Floege, J.; Fliser, D.; Bohm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, M.; Coppolino, G.; De Nicola, L.; Serra, R.; Garofalo, C.; Andreucci, M.; Bolignano, D. Unraveling Cardiovascular Risk in Renal Patients: A New Take on Old Tale. Front. Cell Dev. Biol. 2019, 7, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M. Cause of Death in Patients with Reduced Kidney Function. J. Am. Soc. Nephrol. 2015, 26, 2504–2511. [Google Scholar] [CrossRef] [Green Version]
- Drummond, C.A.; Crotty Alexander, L.E.; Haller, S.T.; Fan, X.; Xie, J.X.; Kennedy, D.J.; Liu, J.; Yan, Y.; Hernandez, D.-A.; Mathew, D.P.; et al. Cigarette smoking causes epigenetic changes associated with cardiorenal fibrosis. Physiol. Genom. 2016, 48, 950–960. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, M.; Serra, R.; Michael, A.; Bolignano, D.; Coppolino, G.; Ielapi, N.; Serraino, G.F.; Mastroroberto, P.; Locatelli, F.; De Nicola, L.; et al. Smoking habit as a risk amplifier in chronic kidney disease patients. Sci. Rep. 2021, 11, 14778. [Google Scholar] [CrossRef] [PubMed]
- de Albuquerque Suassuna, P.G.; Sanders-Pinheiro, H.; de Paula, R.B. Uremic Cardiomyopathy: A New Piece in the Chronic Kidney Disease-Mineral and Bone Disorder Puzzle. Front. Med. 2018, 5, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.K.; Rai, R.; Flevaris, P.; Vaughan, D.E. Epigenetics in Reactive and Reparative Cardiac Fibrogenesis: The Promise of Epigenetic Therapy. J. Cell. Physiol. 2017, 232, 1941–1956. [Google Scholar] [CrossRef]
- Ardekani, A.M.; Naeini, M.M. The Role of MicroRNAs in Human Diseases. Avicenna J. Med. Biotechnol. 2010, 2, 161–179. [Google Scholar]
- Schimmel, K.; Jung, M.; Foinquinos, A.; José, G.S.; Beaumont, J.; Bock, K.; Grote-Levi, L.; Xiao, K.; Bär, C.; Pfanne, A.; et al. Natural Compound Library Screening Identifies New Molecules for the Treatment of Cardiac Fibrosis and Diastolic Dysfunction. Circulation 2020, 141, 751–767. [Google Scholar] [CrossRef]
- Ben-Nun, D.; Buja, L.M.; Fuentes, F. Prevention of heart failure with preserved ejection fraction (HFpEF): Reexamining microRNA-21 inhibition in the era of oligonucleotide-based therapeutics. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2020, 49, 107243. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Gao, X.M.; Winbanks, C.E.; Boey, E.J.; Tham, Y.K.; Kiriazis, H.; Gregorevic, P.; Obad, S.; Kauppinen, S.; Du, X.J.; et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. USA 2012, 109, 17615–17620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoletti, E.; De Nicola, L.; Gabbai, F.B.; Chiodini, P.; Ravera, M.; Pieracci, L.; Marre, S.; Cassottana, P.; Lucà, S.; Vettoretti, S.; et al. Associations of Left Ventricular Hypertrophy and Geometry with Adverse Outcomes in Patients with CKD and Hypertension. Clin. J. Am. Soc. Nephrol. 2016, 11, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.D.; Ambler, S.K.; Mitchell, M.D.; Long, C.S. The cardiac fibroblast: Therapeutic target in myocardial remodeling and failure. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 657–687. [Google Scholar] [CrossRef] [PubMed]
- van Rooij, E.; Olson, E.N. MicroRNA therapeutics for cardiovascular disease: Opportunities and obstacles. Nat. Rev. Drug Discov. 2012, 11, 860–872. [Google Scholar] [CrossRef]
- Bao, J.; Lu, Y.; She, Q.; Dou, W.; Tang, R.; Xu, X.; Zhang, M.; Zhu, L.; Zhou, Q.; Li, H.; et al. MicroRNA-30 regulates left ventricular hypertrophy in chronic kidney disease. JCI Insight 2021, 6, e138027. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Lu, J.R.; Antos, C.L.; Markham, B.; Richardson, J.; Robbins, J.; Grant, S.R.; Olson, E.N. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 1998, 93, 215–228. [Google Scholar] [CrossRef] [Green Version]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Osada-Oka, M.; Shiota, M.; Izumi, Y.; Nishiyama, M.; Tanaka, M.; Yamaguchi, T.; Sakurai, E.; Miura, K.; Iwao, H. Macrophage-derived exosomes induce inflammatory factors in endothelial cells under hypertensive conditions. Hypertens. Res. 2016, 40, 353–360. [Google Scholar] [CrossRef]
- Jiao, Y.; Li, Z.; Loughran, P.A.; Fan, E.K.; Scott, M.J.; Li, Y.; Billiar, T.R.; Wilson, M.A.; Shi, X.; Fan, J. Frontline Science: Macrophage-derived exosomes promote neutrophil necroptosis following hemorrhagic shock. J. Leukoc. Biol. 2018, 103, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Heymans, S.; Corsten, M.F.; Verhesen, W.; Carai, P.; van Leeuwen, R.E.; Custers, K.; Peters, T.; Hazebroek, M.; Stöger, L.; Wijnands, E.; et al. Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation 2013, 128, 1420–1432. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wang, Z.M.; Ji, J.L.; Gan, W.; Zhang, A.; Shi, H.J.; Wang, H.; Lv, L.; Li, Z.; Tang, T.; et al. Macrophage-Derived Exosomal Mir-155 Regulating Cardiomyocyte Pyroptosis and Hypertrophy in Uremic Cardiomyopathy. JACC Basic Transl. Sci. 2020, 5, 148–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, A.; Wang, H.; Klein, J.D.; Tan, L.; Wang, Z.M.; Du, J.; Naqvi, N.; Liu, B.-C.; Wang, X.H. miR-26a Limits Muscle Wasting and Cardiac Fibrosis through Exosome-Mediated microRNA Transfer in Chronic Kidney Disease. Theranostics 2019, 9, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1540–H1559. [Google Scholar] [CrossRef] [Green Version]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Sarkozy, M.; Gaspar, R.; Zvara, A.; Siska, A.; Kovari, B.; Szucs, G.; Márványkövi, F.; Kovács, M.G.; Diószegi, P.; Bodai, L.; et al. Chronic kidney disease induces left ventricular overexpression of the pro-hypertrophic microRNA-212. Sci. Rep. 2019, 9, 1302. [Google Scholar] [CrossRef] [Green Version]
- Chuppa, S.; Liang, M.; Liu, P.; Liu, Y.; Casati, M.C.; Cowley, A.W.; Patullo, L.; Kriegel, A.J. MicroRNA-21 regulates peroxisome proliferator-activated receptor alpha, a molecular mechanism of cardiac pathology in Cardiorenal Syndrome Type 4. Kidney Int. 2018, 93, 375–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssef, J.; Badr, M. Role of Peroxisome Proliferator-Activated Receptors in Inflammation Control. J. Biomed. Biotechnol. 2004, 2004, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Wen, G.; Li, H. Role of peroxisome proliferator-activated receptor α in atherosclerosis. Mol. Med. Rep. 2014, 9, 1755–1760. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wang, K.C.; Wu, W.; Subramaniam, S.; Shyy, J.Y.; Chiu, J.J.; Li, J.Y.; Chien, S. MicroRNA-21 targets peroxisome proliferators-activated receptor-alpha in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proc. Natl. Acad. Sci. USA 2011, 108, 10355–10360. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Huang, X.; Xie, D.; Shen, M.; Lin, H.; Zhu, Y.; Ma, S.; Zheng, C.; Chen, L.; Liu, Y.; et al. RNA interactions in right ventricular dysfunction induced type II cardiorenal syndrome. Aging 2021, 13, 4215–4241. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; Lennartz, S.; Leonardis, D.; D’Arrigo, G.; Tripepi, R.; Emrich, I.E.; Ma, S.; Zheng, C.; Chen, L.; Liu, Y.; et al. High estimated pulmonary artery systolic pressure predicts adverse cardiovascular outcomes in stage 2-4 chronic kidney disease. Kidney Int. 2015, 88, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolignano, D.; Rastelli, S.; Agarwal, R.; Fliser, D.; Massy, Z.; Ortiz, A.; Wiecek, A.; Martinez-Castelao, A.; Covic, A.; Goldsmith, D.; et al. Pulmonary hypertension in CKD. Am. J. Kidney Dis. 2013, 61, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; De Rosa, S.; Greco, M.; Presta, P.; Patella, G.; Crugliano, G.; Sabatino, J.; Strangio, A.; Romano, L.R.; Comi, A.; et al. Marinobufagenin, left ventricular geometry and cardiac dysfunction in end-stage kidney disease patients. Int. Urol. Nephrol. 2022, 54, 2581–2589. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; Greco, M.; Presta, P.; Crugliano, G.; Sabatino, J.; Carullo, N.; Arena, R.; Leo, I.; Comi, A.; Andreucci, M.; et al. Altered circulating marinobufagenin levels and recurrent intradialytic hypotensive episodes in chronic hemodialysis patients: A pilot, prospective study. Rev. Cardiovasc. Med. 2021, 22, 1577–1587. [Google Scholar] [CrossRef]
- Tian, J.; Cai , T.; Yuan , Z.; Wang , H.; Liu , L.; Haas , M.; Maksimova , E.; Huang , X.Y.; Xie , Z.J. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z. AANK-AaastEJBM-dj-. Na(+)/K(+)-ATPase as a signal transducer. Eur. J. Biochem. 2002. [Google Scholar]
- Qin, W.; Chung, A.C.K.; Huang, X.R.; Meng, X.M.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. TGF-β/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef] [Green Version]
- Drummond, C.A.; Fan, X.; Haller, S.T.; Kennedy, D.J.; Liu, J.; Tian, J. Na/K-ATPase signaling mediates miR-29b-3p regulation and cardiac fibrosis formation in mice with chronic kidney disease. PLoS ONE 2018, 13, e0197688. [Google Scholar] [CrossRef] [Green Version]
- Drummond, C.A.; Hill, M.C.; Shi, H.; Fan, X.; Xie, J.X.; Haller, S.T.; Kennedy, D.J.; Liu, J.; Garrett, M.R.; Xie, Z.; et al. Na/K-ATPase signaling regulates collagen synthesis through microRNA-29b-3p in cardiac fibroblasts. Physiol. Genom. 2016, 48, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Castoldi, G.; Di Gioia, C.R.; Bombardi, C.; Catalucci, D.; Corradi, B.; Gualazzi, M.G.; Leopizzi, M.; Mancini, M.; Zerbini, G.; Condorelli, G.; et al. MiR-133a regulates collagen 1A1: Potential role of miR-133a in myocardial fibrosis in angiotensin II-dependent hypertension. J. Cell. Physiol. 2012, 227, 850–856. [Google Scholar] [CrossRef]
- Simeoni, M.; Nicotera, R.; Colao, M.; Citraro, M.L.; Pelagi, E.; Cerantonio, A.; Comi, N.; Coppolino, G.; Fuiano, G. Direct inhibition of plasmatic renin activity with aliskiren: A promising but under-investigated therapeutic option for non-diabetic glomerulonephritis. Int. Urol. Nephrol. 2016, 48, 229–237. [Google Scholar] [CrossRef]
- Pei, Z.; Meng, R.; Li, G.; Yan, G.; Xu, C.; Zhuang, Z.; Ren, J.; Wu, Z. Angiotensin-(1-7) ameliorates myocardial remodeling and interstitial fibrosis in spontaneous hypertension: Role of MMPs/TIMPs. Toxicol. Lett. 2010, 199, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhong, Y.; Cheng, C.; Liu, B.; Wang, L.; Li, A.; Xiong, L.; Liu, S. MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS ONE 2013, 8, e53950. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Duan, L.; Gao, Y.; Zhou, S.; Liu, Y.; Wei, S.; An, S.; Liu, J.; Tian, L.; Wang, S. Angiotensin II receptor blocker valsartan ameliorates cardiac fibrosis partly by inhibiting miR-21 expression in diabetic nephropathy mice. Mol. Cell. Endocrinol. 2018, 472, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhang, C. MicroRNA-21 in cardiovascular disease. J. Cardiovasc. Transl. Res. 2010, 3, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Katz, D.H.; Burns, J.A.; Aguilar, F.G.; Beussink, L.; Shah, S.J. Albuminuria is independently associated with cardiac remodeling, abnormal right and left ventricular function, and worse outcomes in heart failure with preserved ejection fraction. JACC Heart Fail. 2014, 2, 586–596. [Google Scholar] [CrossRef]
- Rana, I.; Velkoska, E.; Patel, S.K.; Burrell, L.M.; Charchar, F.J. MicroRNAs mediate the cardioprotective effect of angiotensin-converting enzyme inhibition in acute kidney injury. Am. J. Physiol. Ren. Physiol. 2015, 309, F943–F954. [Google Scholar] [CrossRef] [Green Version]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef] [Green Version]
- He, B.; Xiao, J.; Ren, A.J.; Zhang, Y.F.; Zhang, H.; Chen, M.; Xie, B.; Gao, X.G.; Wang, Y.W. Role of miR-1 and miR-133a in myocardial ischemic postconditioning. J. Biomed. Sci. 2011, 18, 22. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Song, G.; Liu, M.; Li, X.; Tang, H. miRNA-1 targets fibronectin1 and suppresses the migration and invasion of the HEp2 laryngeal squamous carcinoma cell line. FEBS Lett. 2011, 585, 3263–3269. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Zheng, J.; Sun, Y.; Wu, Z.; Liu, Z.; Huang, G. MicroRNA-1 regulates cardiomyocyte apoptosis by targeting Bcl-2. Int. Heart J. 2009, 50, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panizo, S.; Barrio-Vázquez, S.; Naves-Díaz, M.; Carrillo-López, N.; Rodríguez, I.; Fernández-Vázquez, A.; Valdivielso, J.M.; Thadhani, R.; Cannata-Andía, J.B. Vitamin D receptor activation, left ventricular hypertrophy and myocardial fibrosis. Nephrol. Dial. Transplant. 2013, 28, 2735–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolignano, D.; Greco, M.; Arcidiacono, V.; Tripolino, O.; Vita, C.; Provenzano, M.; Donato, C.; Chiarella, S.; Fuiano, G.; De Sarro, G.; et al. Cathepsin-K is a potential cardiovascular risk biomarker in prevalent hemodialysis patients. Int. Urol. Nephrol. 2021, 53, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, C.; Nie, L.; Zhao, X.; Gu, J.; Guan, X.; Wang, S.; Xiao, T.; Xu, X.; He, T.; et al. Klotho Protects Against Indoxyl Sulphate-Induced Myocardial Hypertrophy. J. Am. Soc. Nephrol. 2015, 26, 2434–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meems, L.M.; Cannon, M.V.; Mahmud, H.; Voors, A.A.; van Gilst, W.H.; Silljé, H.H.; Ruifrok, W.P.; de Boer, R.A. The vitamin D receptor activator paricalcitol prevents fibrosis and diastolic dysfunction in a murine model of pressure overload. J. Steroid Biochem. Mol. Biol 2012, 132, 282–289. [Google Scholar] [CrossRef]
- Mizobuchi, M.; Nakamura, H.; Tokumoto, M.; Finch, J.; Morrissey, J.; Liapis, H.; Slatopolsky, E. Myocardial effects of VDR activators in renal failure. J. Steroid Biochem. Mol. Biol. 2010, 121, 188–192. [Google Scholar] [CrossRef] [Green Version]
- Panizo, S.; Carrillo-López, N.; Naves-Díaz, M.; Solache-Berrocal, G.; Martínez-Arias, L.; Rodrigues-Díez, R.R.; Fernández-Vázquez, A.; Martínez-Salgado, C.; Ruiz-Ortega, M.; Dusso, A.; et al. Regulation of miR-29b and miR-30c by vitamin D receptor activators contributes to attenuate uraemia-induced cardiac fibrosis. Nephrol. Dial. Transplant. 2017, 32, 1831–1840. [Google Scholar] [CrossRef] [Green Version]
- Lim, V.S. Thyroid function in patients with chronic renal failure. Am. J. Kidney Dis. 2001, 38, S80–S84. [Google Scholar] [CrossRef]
- Williams, A.J.; O’Shea, P.J.; Williams, G.R. Complex interactions between thyroid hormone and fibroblast growth factor signalling. Curr. Opin. Endocrinol. Diabetes Obes. 2007, 14, 410–415. [Google Scholar] [CrossRef]
- Lim, V.S.; Zavala, D.C.; Flanigan, M.J.; Freeman, R.M. Blunted peripheral tissue responsiveness to thyroid hormone in uremic patients. Kidney Int. 1987, 31, 808–814. [Google Scholar] [CrossRef] [Green Version]
- van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, L.E.; Sigel, A.V.; Douglas, J.A.; Eghbali-Webb, M. Upregulation of collagen type I gene expression in the ventricular myocardium of thyroidectomized male and female rats. J. Mol. Cell Cardiol. 1996, 28, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Ghose Roy, S.; Mishra, S.; Ghosh, G.; Bandyopadhyay, A. Thyroid hormone induces myocardial matrix degradation by activating matrix metalloproteinase-1. Matrix Biol. 2007, 26, 269–279. [Google Scholar] [CrossRef]
- Huang, X.H.; Li, J.L.; Li, X.Y.; Wang, S.X.; Jiao, Z.H.; Li, S.Q.; Liu, J.; Ding, J. miR-208a in Cardiac Hypertrophy and Remodeling. Front. Cardiovasc. Med. 2021, 8, 773314. [Google Scholar] [CrossRef] [PubMed]
- McDermott, F.T. The effect of 10 percent human uremic serum upon human fibroblastic cell cultures. J. Surg. Res. 1971, 11, 119–123. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Hullinger, T.G.; Semus, H.M.; Dickinson, B.A.; Seto, A.G.; Lynch, J.M.; Stack, C.; Latimer, P.A.; Olson, E.N.; Van Rooij, E. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation 2011, 124, 1537–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prado-Uribe, M.D.; Soto-Abraham, M.V.; Mora-Villalpando, C.J.; Gallardo, J.M.; Bonilla, E.; Avila, M.; Tena, E.; Paniagua, R. Role of thyroid hormones and mir-208 in myocardial remodeling in 5/6 nephrectomized rats. Arch. Med. Res. 2013, 44, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Neugarten, J.; Acharya, A.; Silbiger, S.R. Effect of gender on the progression of nondiabetic renal disease: A meta-analysis. J. Am. Soc. Nephrol 2000, 11, 319–329. [Google Scholar] [CrossRef]
- Carrero, J.J.; Hecking, M.; Chesnaye, N.C.; Jager, K.J. Sex and gender disparities in the epidemiology and outcomes of chronic kidney disease. Nat. Rev. Nephrol. 2018, 14, 151–164. [Google Scholar] [CrossRef]
- Dubey, R.K.; Jackson, E.K. Estrogen-induced cardiorenal protection: Potential cellular, biochemical, and molecular mechanisms. Am. J. Physiol. Ren. Physiol. 2001, 280, F365–F388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, M.R.; Geurts, A.M.; Kriegel, A.J. miR-146b-5p has a sex-specific role in renal and cardiac pathology in a rat model of chronic kidney disease. Kidney Int. 2019, 96, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.; Song, D.; Ye, H.; Wu, X.; Jiang, L.; Tang, B.; Zhou, Y.; Fang, L.; Cao, H.; He, W.; et al. Circulating MiR-133a as a biomarker predicts cardiac hypertrophy in chronic hemodialysis patients. PLoS ONE 2014, 9, e103079. [Google Scholar] [CrossRef] [Green Version]
- Stopic, B.; Dragicevic, S.; Medic-Brkic, B.; Nikolic, A.; Stojanovic, M.; Budisavljevic, S.; Dimkovic, N. Biomarkers of Uremic Cardiotoxicity. Toxins 2021, 13, 639. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; Greco, M.; Presta, P.; Duni, A.; Vita, C.; Pappas, E.; Mirabelli, M.; Lakkas, L.; Naka, K.K.; Brunetti, A.; et al. A small circulating miRNAs signature predicts mortality and adverse cardiovascular outcomes in chronic hemodialysis patients. Clin. Kidney J. 2023, in press. [Google Scholar] [CrossRef]
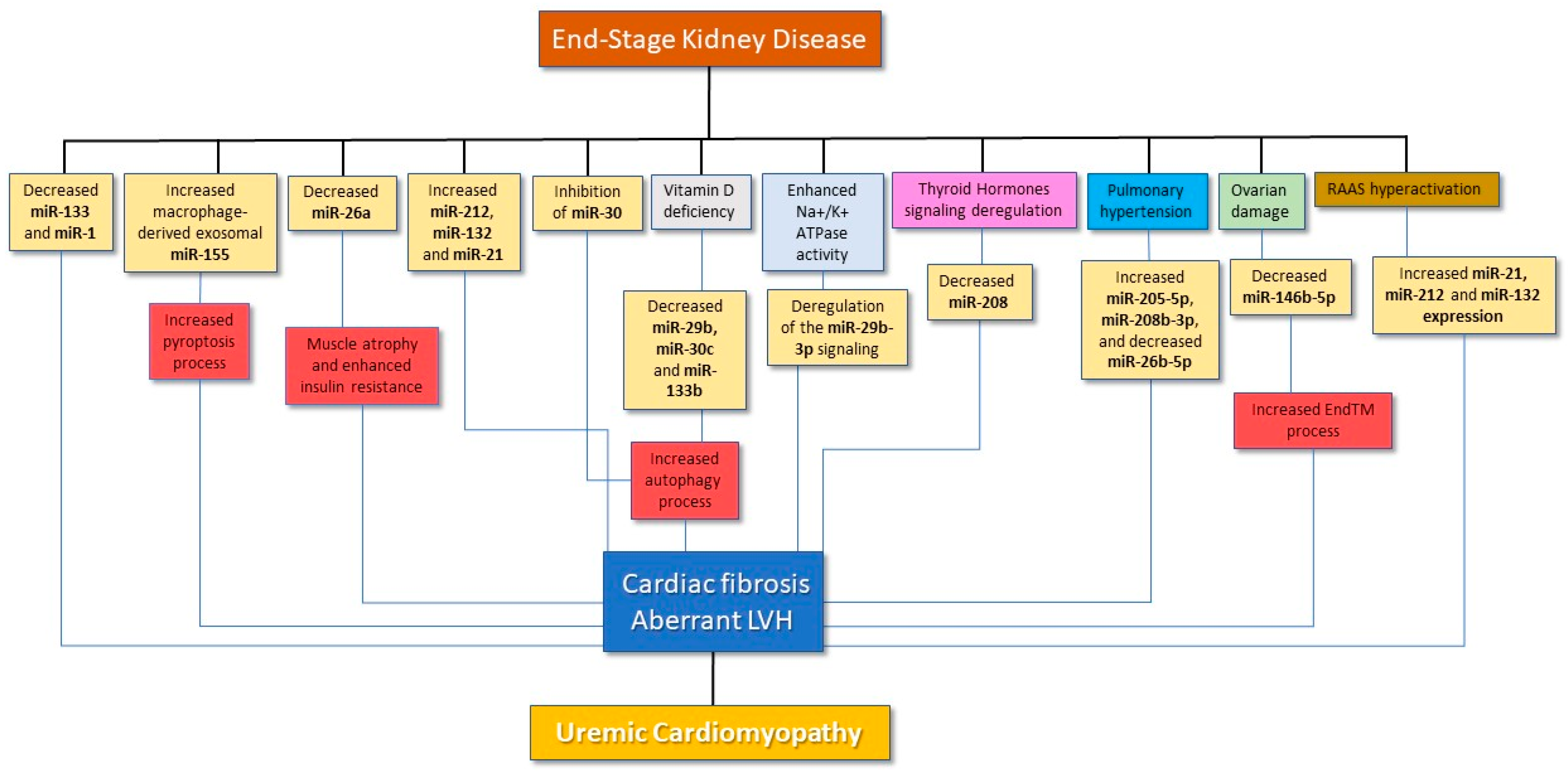

{kind=link}
| Study | Methods and Model | miRNAs | Key-Findings |
|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Study | Design | Population/Methods | miRNAs | Results |
|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Agostino, M.; Mauro, D.; Zicarelli, M.; Carullo, N.; Greco, M.; Andreucci, M.; Coppolino, G.; Bolignano, D. miRNAs in Uremic Cardiomyopathy: A Comprehensive Review. Int. J. Mol. Sci. 2023, 24, 5425. https://doi.org/10.3390/ijms24065425
D’Agostino M, Mauro D, Zicarelli M, Carullo N, Greco M, Andreucci M, Coppolino G, Bolignano D. miRNAs in Uremic Cardiomyopathy: A Comprehensive Review. International Journal of Molecular Sciences. 2023; 24(6):5425. https://doi.org/10.3390/ijms24065425
Chicago/Turabian StyleD’Agostino, Mario, Davide Mauro, Mariateresa Zicarelli, Nazareno Carullo, Marta Greco, Michele Andreucci, Giuseppe Coppolino, and Davide Bolignano. 2023. "miRNAs in Uremic Cardiomyopathy: A Comprehensive Review" International Journal of Molecular Sciences 24, no. 6: 5425. https://doi.org/10.3390/ijms24065425