
Quercetin and Its Fermented Extract as a Potential Inhibitor of Bisphenol A-Exposed HT-29 Colon Cancer Cells’ Viability
 , , and
, , and
Abstract
:1. Introduction
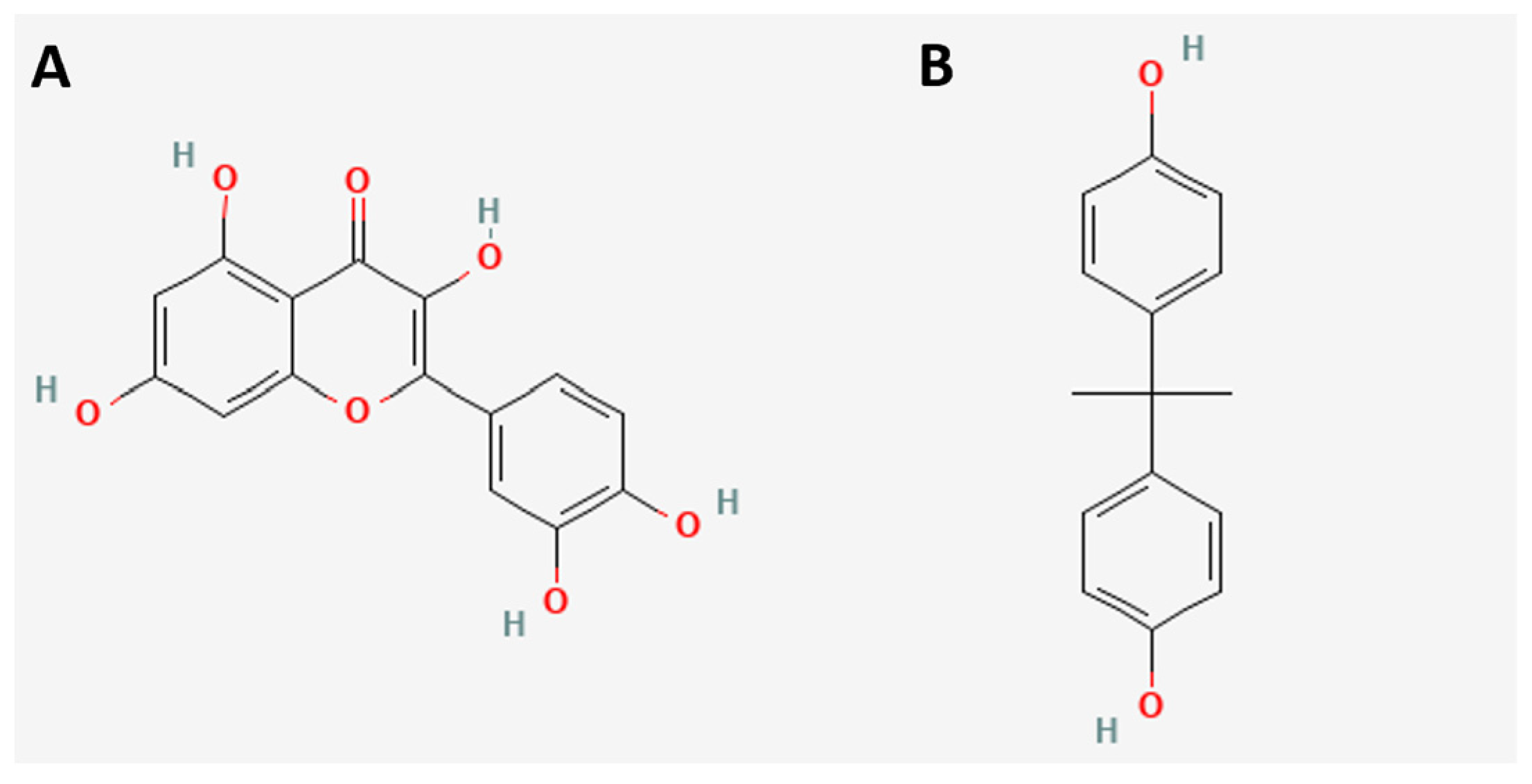
2. Results and Discussions
3. Materials and Methods
3.1. Gastrointestinal Digestion and Colonic Fermentation In Vitro
3.2. Analysis of Polyphenols by HPLC-DAD
3.3. Quantification of Antioxidant Capacity by DPPH and ORAC
3.4. Cell Viability and IC50 Determination
3.5. Treatments
3.6. Cytotoxicity Analysis
3.7. DNA Quantification in the Phases of the Cell Cycle
3.8. Gene Expression
3.9. Assessment of Gene Expression of the p53 Signaling Pathway
3.10. Molecular In Silico Docking Analysis
3.11. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gen | Sequence | Alignment Temperature |
|---|---|---|
| ESR1 | FWD: TGCTGGCTACATCATCTCGG | 60 °C |
| REV: CAGGAACTTATCCCTCATATAG | ||
| ESR2 | FWD: TCCCACTTCGTAACACTTCCG | 64 °C |
| REV: ACATTCTATAGCCCTGCTGTGA | ||
| Actin | FWD: ACGGGGTCACCCACACTGTGC | 62 °C |
| REV: CTAGAAGCATTTGCGGTGGACGATG | ||
| GPR30 | FWD: AGTCGGATGTGAGGTTCAG | 60 °C |
| REV: TCGTGTTGAGGGAGTGCAAG |
References
- Pombo Arias, M.; Castro Feijóo, L.; Barreiro Conde, J.; Cabanas Rodríguez, P. Una revisión sobre los disruptores endocrinos y su posible impacto sobre la salud de los humanos. Rev. Esp. Endocrinol. Pediatr. 2020, 11, 2. [Google Scholar] [CrossRef]
- Geens, T.; Goeyens, L.; Covaci, A. Are potential sources for human exposure to bisphenol-A overlooked? Int. J. Hyg. Environ. Health 2011, 214, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef]
- Bolli, A.; Galluzzo, P.; Ascenzi, P.; Del Pozzo, G.; Manco, I.; Vietri, M.T.; Mita, L.; Altucci, L.; Mita, D.G.; Marino, M. Laccase treatment impairs bisphenol A-induced cancer cell proliferation affecting estrogen receptor alpha-dependent rapid signals. IUBMB Life 2008, 60, 843–852. [Google Scholar] [CrossRef]
- Lorber, M.; Schecter, A.; Paepke, O.; Shropshire, W.; Christensen, K.; Birnbaum, L. Exposure assessment of adult intake of bisphenol A (BPA) with emphasis on canned food dietary exposures. Environ. Int. 2015, 77, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.S.; Hwang, K.A.; Hyun, S.H.; Nam, K.H.; Lee, C.K.; Choi, K.C. Bisphenol A and nonylphenol have the potential to stimulate the migration of ovarian cancer cells by inducing epithelial-mesenchymal transition via an estrogen receptor dependent pathway. Chem. Res. Toxicol. 2015, 28, 662–671. [Google Scholar] [CrossRef]
- Chen, Z.J.; Yang, X.L.; Liu, H.; Wei, W.; Zhang, K.S.; Huang, H.B.; Giesy, J.P.; Liu, H.L.; Du, J.; Wang, H.S. Bisphenol A modulates colorectal cancer protein profile and promotes the metastasis via induction of epithelial to mesenchymal transitions. Arch. Toxicol. 2015, 89, 1371–1381. [Google Scholar] [CrossRef]
- Buer, C.S.; Imin, N.; Djordjevic, M.A. Flavonoids: New Roles for Old Molecules. J. Integr. Plant Biol. 2010, 52, 98–111. [Google Scholar] [CrossRef]
- Nikolić, I.; Savić-Gajić, I.; Tačić, A.; Savić, I. Classification and biological activity of phytoestrogens: A review. Adv. Technol. 2017, 6, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Serra, A.; Macià, A.; Romero, M.P.; Reguant, J.; Ortega, N.; Motilva, M.J. Metabolic pathways of the colonic metabolism of flavonoids (flavonols, flavones and flavanones) and phenolic acids. Food Chem. 2012, 130, 383–393. [Google Scholar] [CrossRef]
- Lamson, D.W.; Brignall, M.S. Antioxidants and cancer, part 3: Quercetin. Altern. Med. Rev. 2000, 5, 196–208. [Google Scholar] [PubMed]
- Watson, W.; Cai, J.; Jones, D. Diet and apoptosis. Annu. Rev. Nutr. 2000, 20, 485–505. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.T.; Lee, S.H.; Kim, J.I.; Kim, Y.M. Quercetin regulates the sestrin 2-AMPK-p38 MAPK signaling pathway and induces apoptosis by increasing the generation of intracellular ROS in a p53-independent manner. Int. J. Mol. Med. 2014, 33, 863–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.W.; Hou, W.C.; Shen, S.C.; Juan, S.H.; Ko, C.H.; Wang, L.M.; Chen, Y.C. Quercetin inhibition of tumor invasion via suppressing PKC /ERK/AP-1-dependent matrix metalloproteinase-9 activation in breast carcinoma cells. Carcinogenesis 2008, 29, 1807–1815. [Google Scholar] [CrossRef] [Green Version]
- Rosa, L.S.; Jordão, N.A.; da Costa Pereira Soares, N.; deMesquita, J.F.; Monteiro, M.; Teodoro, A.J. Pharmacokinetic, antiproliferative and apoptotic effects of phenolic acids in human colon adenocarcinoma cells using in vitro and in silico approaches. Molecules 2018, 23, 2569. [Google Scholar] [CrossRef] [Green Version]
- Bulzomi, P.; Galluzzo, P.; Bolli, A.; Leone, S.; Acconcia, F.; Marino, M. The pro-apoptotic effect of quercetin in cancer cell lines requires ERβ-dependent signals. J. Cell. Physiol. 2012, 227, 1891–1898. [Google Scholar] [CrossRef]
- Peng, X.; Zhang, Z.; Zhang, N.; Liu, L.; Li, S.; Wei, H. In vitro catabolism of quercetin by human fecal bacteria and the antioxidant capacity of its catabolites. Food Nutr. Res. 2014, 58, 10. [Google Scholar] [CrossRef] [Green Version]
- Chebotarev, A.N.; Snigur, D.V. Study of the acid-base properties of quercetin in aqueous solutions by color measurements. J. Anal. Chem. 2015, 70, 55–59. [Google Scholar] [CrossRef]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2015, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Ou, B.; Hampsch-Woodill, R.L. Development and validation of an improved oxygen radical absorbance capacity assay using fluorescein as the fluorescent probe. J. Agric. Food Chem. 2001, 49, 4619–4626. [Google Scholar] [CrossRef]
- Sirivibulkovit, K.; Nouanthavong, S.; Sameenoi, Y. Paper-based DPPH Assay for Antioxidant Activity Analysis. Anal. Sci. 2018, 34, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brett, A.M.O.; Ghica, M.E. Electrochemical Oxidation of Quercetin. Electroanalysis 2003, 15, 1745. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Maqueda, D.; Miralles, B.; Recio, I. HT29 Cell Line. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Springer: Berlin/Heidelberg, Germany, 2015; Chapter 11. Available online: https://www.ncbi.nlm.nih.gov/books/NBK500137 (accessed on 6 March 2023).
- Catalán, M.; Ferreira, J.; Carrasco-Pozo, C. The Microbiota-Derived Metabolite of Quercetin, 3,4-Dihydroxyphenylacetic Acid Prevents Malignant Transformation and Mitochondrial Dysfunction Induced by Hemin in Colon Cancer and Normal Colon Epithelia Cell Lines. Molecules 2020, 25, 4138. [Google Scholar] [CrossRef] [PubMed]
- Gao, K.; Xu, A.; Krul, C.; Venema, K.; Liu, Y.; Niu, Y.; Lu, J.; Bensoussan, L.; Seeram, N.P.; Heber, D. Of the major phenolic acids formed during human microbial fermentation of tea, citrus, and soy flavonoid supplements, only 3,4-dihydroxyphenylacetic acid has antiproliferative activity. J. Nutr. 2006, 136, 52–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrbek, S.; Rufer, C.E.; Marko, D.; Esselen, M. Quercetin and its microbial degradation product 3,4-dihydroxyphenylacetic acid generate hydrogen peroxide modulating their stability under in vitro conditions. J. Food Nutr. Res. 2009, 48, 129–140. [Google Scholar]
- Qu, W.; Zhao, Z.; Chen, S.; Zhang, L.; Wu, D.; Chen, Z. Bisphenol A suppresses proliferation and induces apoptosis in colonic epithelial cells through mitochondrial and MAPK/AKT pathways. Life Sci. 2018, 208, 167–174. [Google Scholar] [CrossRef]
- Hwang, K.A.; Kang, N.H.; Yi, B.R.; Lee, H.R.; Park, M.A.; Choi, K.C. Genistein, a soy phytoestrogen, prevents the growth of BG-1 ovarian cancer cells induced by 17β-estradiol or bisphenol A via the inhibition of cell cycle progression. Int. J. Oncol. 2013, 42, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Liu, Y.; Wang, M.; Qian, Y.; Dong, X.; Gu, H.; Wang, H.; Guo, S.; Hisamitsu, T. Quercetin-induced apoptosis of HT-29 colon cancer cells via inhibition of the Akt-CSN6-Myc signaling axis. Mol. Med. Rep. 2016, 14, 4559–4566. [Google Scholar] [CrossRef] [Green Version]
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, S.K.; Kim, B.S.; Lee, S.H.; Park, Y.S.; Park, B.K.; Jung, J.Y. Apoptotic Effect of Quercetin on HT-29 Colon Cancer Cells via the AMPK Signaling Pathway. J. Agric. Food Chem. 2010, 58, 8643–8650. [Google Scholar] [CrossRef]
- Martineti, V.; Picariello, L.; Tognarini, I.; Sala, S.C.; Gozzini, A.; Azzari, C.; Brandi, M.L. ERβ is a potent inhibitor of cell proliferation in the HCT8 human colon cancer cell line through regulation of cell cycle components. Endocr.-Relat. Cancer 2005, 12, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.; Scavo, M.P.; Papagni, S.; Piscitelli, D.; Guido, R.; Di Lena, M.; Leo, A. ERβ expression in normal, adenomatous and carcinomatous tissues of patients with familial adenomatous polyposis. Scand. J. Gastroenterol. 2010, 45, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Pampaloni, B.; Palmini, G.; Mavilia, C.; Zonefrati, R.; Tanini, A.; Brandi, M.L. In vitro effects of polyphenols on colorectal cancer cells. World J. Gastrointest. Oncol. 2014, 6, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Principi, M. Ulcerative colitis: From inflammation to cancer. Do estrogen receptors have a role? World J. Gastroenterol. 2014, 20, 11496. [Google Scholar] [CrossRef]
- Maier, T.; Güell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [Green Version]
- Sonneveld, S.; Verhagen, B.M.P.; Tanenbaum, M.E. Heterogeneity in mRNA Translation. Trends Cell Biol. 2020, 30, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Choi, H.G.; Lee, H.M.; Lee, G.A.; Hwang, K.A.; Choi, K.C. Effects of bisphenol compounds on the growth and epithelial mesenchymal transition of MCF-7 CV human breast cancer cells. J. Biomed. Res. 2017, 31, 358–369. [Google Scholar] [CrossRef]
- Zhang, Y.; Bhavnani, B.G. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 2006, 7, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Gortat, A.; Sancho, M.; Mondragón, L.; Messeguer, A.; Pérez-Payá, E.; Orzáez, M. Apaf1 inhibition promotes cell recovery from apoptosis. Protein Cell 2015, 6, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Yu, X.; Asara, J.M.; Heuser, J.E.; Ludtke, S.J.; Akey, C.W. The holo-apoptosome: Activation of procaspase-9 and interactions with caspase-3. Structure 2011, 19, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.l.; Liu, Y.Q.; Wang, P.; Song, C.H.; Wang, K.J.; Dai, L.P.; Zhang, J.Y.; Ye, H. The effect of quercetin nanoparticle on cervical cancer progression by inducing apoptosis, autophagy and anti-proliferation via JAK2 suppression. Biomed. Pharmacother. 2016, 82, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, K.; Ghosh, S.; Mukherjee, A.; Sadhukhan, R.; Mondal, J.; Khuda-Bukhsh, A.R. Quercetin induces cytochrome-c release and ROS accumulation to promote apoptosis and arrest the cell cycle in G2/M, in cervical carcinoma: Signal cascade and drug-DNA interaction. Cell Prolif. 2013, 46, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Shang, H.S.; Lu, H.F.; Lee, C.H.; Chiang, H.S.; Chu, Y.L.; Chen, A.; Chung, J.G. Quercetin induced cell apoptosis and altered gene expression in AGS human gastric cancer cells. Environ. Toxicol. 2018, 33, 1168–1181. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Zaidi, S.; Cui, Z.; Zhou, D.; Saeed, S.; Inadera, H. Potential proapoptotic phytochemical agents for the treatment and prevention of colorectal cancer. Oncol. Lett. 2019, 18, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Dairkee, S.H.; Luciani-Torres, M.G.; Moore, D.H.; Goodson, W.H., 3rd. Bisphenol-A-induced inactivation of the p53 axis underlying deregulation of proliferation kinetics, and cell death in non-malignant human breast epithelial cells. Carcinogenesis 2013, 34, 703–712. [Google Scholar] [CrossRef]
- Al-Ghamdi, M.A.; AL-Enazy, A.; Huwait, E.A.; Albukhari, A.; Harakeh, S.; Moselhy, S.S. Enhancement of Annexin V in response to combination of epigallocatechin gallate and quercetin as a potent arrest the cell cycle of colorectal cancer. Braz. J. Biol. 2021, 83, e248746. [Google Scholar] [CrossRef]
- Wu, J.; Yi, J.; Wu, Y.; Chen, X.; Zeng, J.; Wu, J.; Peng, W. 3,30-dimethylquercetin inhibits the proliferation of human colon cancer RKO cells through Inducing G2/M cell cycle arrest and apoptosis. Anti Cancer Agents Med. Chem. 2019, 19, 402–409. [Google Scholar] [CrossRef]
- Goodson III, W.H.; Luciani, M.G.; Sayeed, S.A.; Jaffee, I.M.; Moore, D.H., 2nd; Dairkee, S.H. Activation of the mTOR pathway by low levels of xenoestrogens in breast epithelial cells from high-risk women. Carcinogenesis 2011, 32, 1724–1733. [Google Scholar] [CrossRef] [Green Version]
- Yani, S.; Opik, T.; Siti, R.; Ida, K.; Epa, P. In silico analysis of kaempferol as a competitor of estrogen on estrogen receptor alpha of endometrial cancer. J. Phys. 2019, 1402, 1742–6596. [Google Scholar] [CrossRef] [Green Version]
- Aehle, E.; Müller, U.; Eklund, P.C.; Willför, S.M.; Sippl, W.; Dräger, B. Lignans as food constituents with estrogen and antiestrogen activity. Phytochemistry 2011, 72, 2396–2405. [Google Scholar] [CrossRef]
- Campos-Vega, R.; Vázquez-Sánchez, K.; López-Barrera, D.; Loarca-Piña, G.; Mendoza-Díaz, S.; Oomah, B.D. Simulated gastrointestinal digestion and in vitro colonic fermentation of spent coffee (Coffea arabica L.): Bioaccessibility and intestinal permeability. Food Res. Int. 2015, 77, 156–161. [Google Scholar] [CrossRef]
- Ramírez-Jiménez, A.K.; Reynoso-Camacho, R.; Mendoza-Díaz, S.; Loarca-Piña, G. Functional and technological potential of dehydrated Phaseolus vulgaris L. flours. Food Chem. 2014, 161, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, L.R.; Mazza, G. Assessing antioxidant and prooxidant activities of phenolic compounds. J. Agric. Food Chem. 2000, 48, 3597–3604. [Google Scholar] [CrossRef] [PubMed]
- Correa-Basurto, J.; Flores-Sandoval, C.; Marín-Cruz, J.; Rojo-Domínguez, A.; Espinoza-Fonseca, L.M.; Trujillo-Ferrara, J.G. Docking and quantum mechanic studies on cholinesterases and their inhibitors. Eur. J. Med. Chem. 2007, 42, 10–19. [Google Scholar] [CrossRef] [PubMed]








| Sample | Quercetin | DOPAC | Antioxidant Capacity | |||
|---|---|---|---|---|---|---|
| Fermentation | (µg/µL) | RT | (µg/µL) | RT | DPPH (μmoles/µL) | ORAC (μmoles/µL) |
| 6 h | 0.058 a | 11.915 | 0.029 a | 6.112 | NE | NE |
| 12 h | 0.062 a | 11.823 | 0.017 a | 6.652 | NE | NE |
| 24 h | 0.083 b | 11.727 | 0.188 b | 6.147 | 4.316 b | 22.130 b |
| FC | 0.052 a | 11.470 | 0.006 a | 6.246 | 0.353 c | 2.021 c |
| Quercetin | 5.102 a | 21.114 a | ||||
| Symbol | Description | Q | BPA | Q + BPA | FEQ | FEQ + BPA |
|---|---|---|---|---|---|---|
| Fold Change | ||||||
| Genes Associated with Programmed Cell Death (Apoptosis) | ||||||
| APAF1 | Apoptotic peptidase activating factor 1 (pro-apoptosis) | 2.28 | −29.02 | 1.52 | 1.37 | −1.57 |
| BAX | BCL2-associated X protein (pro-apoptosis) | 1.61 | 1.48 | 1.07 | −1.14 | −1.08 |
| BBC3 | BCL2 binding component 3 (pro-apoptosis) | 1.38 | −1.15 | 1.28 | −1.42 | 1.10 |
| BCL2 | B-cell CLL/lymphoma 2 (anti-apoptosis) | 3.23 | 1.33 | 2.47 | 1.46 | 5.00 |
| BCL2A1 | BCL2-related protein A1 (anti-apoptosis) | 3.23 | 1.33 | 2.47 | 1.46 | 5.00 |
| BID | BH3 interacting domain death agonist (pro-apoptosis) | −1.12 | 1.40 | −1.65 | 1.07 | −1.04 |
| BIRC5 | Baculoviral IAP repeat containing 5 (anti-apoptosis) | −1.41 | −6.40 | 1.19 | 1.27 | 1.16 |
| CASP2 | Caspase-2, apoptosis-related cysteine peptidase (pro-apoptosis) | 1.54 | −2.80 | 1.16 | −1.07 | −1.02 |
| CASP9 | Caspase-9, apoptosis-related cysteine peptidase (pro-apoptosis) | 1.67 | −3.36 | 1.02 | 1.16 | 1.06 |
| CRADD | CASP2 and RIPK1 domain containing adaptor with death domain (pro-apoptosis) | −1.04 | 1.42 | −1.36 | −2.08 | 1.49 |
| EI24 | Etoposide induced 2.4 mRNA (pro-apoptosis) | −1.28 | −1.75 | −1.83 | −1.25 | −1.47 |
| FADD | Fas (TNFRSF6)-associated via death domain (pro-apoptosis) | −7.83 | −119.69 | −1.25 | −1.21 | −1.24 |
| FAS | Fas (TNF receptor superfamily, member 6) (pro-apoptosis) | 1.34 | −5.53 | 1.57 | 1.22 | −1.47 |
| FASLG | Fas ligand (TNF superfamily, member 6) (pro-apoptosis) | 3.23 | 1.33 | 2.47 | 1.46 | 5.00 |
| FOXO3 | Forkhead box O3 (pro-apoptosis) | −2.59 | −5.53 | −1.24 | −1.04 | −1.15 |
| HK2 | Hexokinase 2 (anti-apoptosis, growth induction) | −1.02 | 1.00 | −1.23 | −1.37 | −1.32 |
| IGF1R | Insulin-like growth factor 1 receptor (anti-apoptosis) | −1.00 | 1.31 | −1.38 | −1.57 | −1.54 |
| KRAS | V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (pro-apoptosis) | 12.43 | 40.23 | 88.50 | 70.48 | 75.71 |
| MCL1 | Myeloid cell leukemia sequence 1 (BCL2-related) (anti-apoptosis) | 1.52 | −2.11 | 1.05 | 1.04 | −1.81 |
| MDM4 | Mdm4 p53 binding protein homolog (mouse) (anti-apoptosis) | −1.14 | −1.86 | −1.27 | −1.16 | −1.14 |
| PIDD1 | P53-induced death domain protein (pro-apoptosis or survival is isoform-dependent) | 1.15 | −1.29 | 1.02 | −1.35 | −1.72 |
| PRKCA | Protein kinase C, alpha (regulation of cell proliferation, apoptosis, differentiation, migration, adhesion, angiogenesis) | 1.31 | −1.38 | −1.10 | −1.14 | −1.31 |
| SIAH1 | Seven in absentia homolog 1 (Drosophila) (pro-apoptosis) | −1.44 | −1.10 | −1.50 | −1.24 | −1.37 |
| TNF | Tumor necrosis factor (anti- and pro-apoptosis) | 3.23 | 1.33 | 2.47 | 7.80 | 5.00 |
| TNFRSF10B | Tumor necrosis factor receptor superfamily, member 10b (pro-apoptosis) | −1.02 | 1.04 | −1.19 | 1.08 | −1.06 |
| TNFRSF10D | Tumor necrosis factor receptor superfamily, member 10d, decoy with truncated death domain (anti-apoptosis) | 1.62 | −1.35 | −1.09 | −1.25 | 1.06 |
| TP53 | Tumor protein p53 (cell cycle regulation and apoptosis) | 1.12 | 1.05 | −1.36 | −1.75 | −1.73 |
| TP53AIP1 | Tumor protein p53 regulated apoptosis inducing protein 1 (pro-apoptosis) | 3.23 | 1.33 | 2.47 | 1.46 | 5.00 |
| TP53BP2 | Tumor protein p53 binding protein, 2 (pro-apoptosis) | 1.38 | 1.54 | −1.03 | −1.12 | −1.05 |
| TP63 | Tumor protein p63 (pro-apoptosis) | 3.23 | 1.33 | 2.47 | 1.46 | 5.00 |
| TRAF2 | TNF receptor-associated factor 2 (anti-apoptosis) | 1.13 | 1.07 | −1.15 | −1.20 | −1.11 |
| Genes associated with the cell cycle | ||||||
| ATM | Ataxia telangiectasia mutated (cell cycle inhibition) | 1.46 | −14.22 | 1.37 | 1.42 | −1.18 |
| ATR | Ataxia telangiectasia and Rad3 related (cell cycle inhibition) | 1.24 | −2.62 | −1.14 | 1.06 | −1.01 |
| BRCA1 | Breast cancer 1, early onset (cell cycle inhibition) | −2.67 | 1.17 | −1.66 | −1.35 | −1.21 |
| BRCA2 | Breast cancer 2, early onset (cell cycle inhibition) | −18.56 | −45.11 | −3.04 | −1.24 | −1.79 |
| BTG2 | BTG family, member 2 (cell cycle inhibition) | 1.03 | −5.55 | −1.57 | −1.44 | −1.47 |
| CCNB1 | Cyclin B1 (cell cycle regulation) | 1.54 | −1.02 | 1.05 | 1.07 | 1.23 |
| CCNE1 | Cyclin E1 (cell cycle regulation) | 1.19 | 1.72 | −1.20 | 1.30 | 1.06 |
| CCNG1 | Cyclin G1 (cell cycle regulation) | −1.08 | 1.22 | −1.18 | −1.66 | −1.51 |
| CCNH | Cyclin H (cell cycle regulation) | 1.64 | 2.26 | 1.18 | 1.25 | 1.21 |
| CDC25A | Cell division cycle 25 homolog A (S. pombe) (cell cycle regulation) | −1.24 | −2.06 | −1.80 | −1.06 | −1.36 |
| CDC25C | Cell division cycle 25 homolog C (S. pombe) (cell cycle regulation) | 1.37 | 1.22 | 1.13 | −1.13 | 1.22 |
| CDK1 | Cyclin-dependent kinase 1 (cell cycle regulation) | −1.19 | −6.22 | −1.31 | −1.32 | −1.30 |
| CDK4 | Cyclin-dependent kinase 4 (cell cycle regulation) | −1.38 | 1.26 | −1.60 | −1.70 | −1.57 |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A (p21, Cip1) (cell cycle regulation) | −2.02 | −2.58 | 1.24 | 1.84 | 2.08 |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A (melanoma, p16, inhibits CDK4) (cell cycle regulation) | 2.00 | −9.67 | 1.46 | 1.23 | −2.68 |
| CHEK1 | CHK1 checkpoint homolog (S. pombe) (cell cycle regulation) | 1.59 | −6.20 | −1.01 | 1.02 | −1.19 |
| CHEK2 | CHK2 checkpoint homolog (S. pombe) (cell cycle regulation) | 1.31 | −2.02 | −1.16 | −1.10 | −1.22 |
| E2F1 | E2F transcription factor 1 (cell cycle induction) | 1.36 | 2.16 | −1.11 | 1.08 | 1.01 |
| E2F3 | E2F transcription factor 3 (cell cycle induction) | −1.41 | 1.20 | −1.74 | −1.77 | −2.15 |
| EGR1 | Early growth response 1 (regulation of the cell cycle, proliferation, and cell death) | 2.62 | 1.99 | −1.05 | 2.63 | −1.07 |
| GADD45A | Growth arrest and DNA-damage-inducible, alpha (cell cycle inhibition) | −14.67 | 1.07 | −3.67 | −2.49 | −1.90 |
| GML | Glycosylphosphatidylinositol anchored molecule-like protein (cell cycle inhibition, pro-apoptosis) | 3.23 | 1.33 | 2.47 | 1.46 | 5.00 |
| MDM2 | Mdm2 p53 binding protein homolog (mouse) (cell cycle induction) | 1.58 | −1.65 | −1.09 | −1.23 | −1.09 |
| MLH1 | MutL homolog 1, colon cancer, nonpolyposis type 2 (E. coli) (cell cycle inhibition) | 1.60 | 1.32 | −1.02 | −1.18 | −1.35 |
| MSH2 | MutS homolog 2, colon cancer, nonpolyposis type 1 (E. coli) (cell cycle inhibition) | 1.10 | 1.51 | −1.43 | −1.03 | −1.34 |
| MYC | V-myc myelocytomatosis viral oncogene homolog (avian) (cell cycle inhibition) | 1.36 | 2.06 | 1.06 | 1.06 | 1.06 |
| MYOD1 | Myogenic differentiation 1 (cell cycle inhibition) | 3.23 | 1.33 | 2.47 | 1.46 | 5.00 |
| NF1 | Neurofibromin 1 (cell cycle inhibition, proliferation inhibition, survival inhibition) | 1.06 | 1.30 | −1.21 | −1.20 | −1.32 |
| NFKB1 | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 (cell cycle inhibition, proliferation inhibition, survival inhibition) | −1.03 | 1.06 | −1.57 | −1.69 | −2.19 |
| PPM1D | Protein phosphatase, Mg2+/Mn2+ dependent, 1D (cell cycle inhibition) | −1.37 | −4.83 | −1.38 | −1.29 | −1.42 |
| PCNA | Proliferating cell nuclear antigen (cell cycle induction) | 1.05 | 1.31 | −1.34 | 1.06 | −1.25 |
| PRC1 | Protein regulator of cytokinesis 1 (cell cycle induction) | 1.46 | −1.81 | −1.05 | −1.12 | −1.33 |
| PTEN | Phosphatase and tensin homolog (cell cycle inhibition, proliferation inhibition, survival inhibition, migration inhibition) | −2.12 | −2.32 | −1.25 | −1.27 | −1.26 |
| PTTG1 | Pituitary tumor-transforming 1 (cell cycle induction) | 1.89 | 1.11 | 1.56 | 1.52 | 1.52 |
| RB1 | Retinoblastoma 1 (cell cycle inhibition) | 1.29 | 1.41 | −1.24 | −1.66 | −1.55 |
| RPRM | Reprimo, TP53 dependent G2 arrest mediator candidate (cell cycle inhibition) | 1.02 | −1.09 | −1.79 | −1.38 | 1.13 |
| TADA3 | Transcriptional adaptor 3 (cell cycle induction, acetylation) | −11.94 | 1.16 | 1.08 | −1.28 | −1.19 |
| TP73 | Tumor protein p73 (pro-apoptosis) | 3.23 | 1.33 | 2.47 | 1.46 | 5.00 |
| Genes associated with angiogenesis, inflammation, autophagy, acetylation, methylation, and tumor suppression | ||||||
| ADGRB1 | Brain-specific angiogenesis inhibitor 1 (angiogenesis inhibition) | 2.18 | −1.53 | 1.32 | 2.19 | 2.93 |
| DNMT1 | DNA (cytosine-5-)-methyltransferase 1 (methylation) | 1.29 | 1.67 | −1.24 | −1.01 | −1.11 |
| EGFR | Epidermal growth factor receptor (proliferation induction) | 1.14 | 1.54 | −1.16 | −1.29 | −1.15 |
| ESR1 | Estrogen receptor 1 (proliferation induction) | 3.23 | 1.77 | 2.47 | 1.46 | 7.16 |
| HDAC1 | Histone deacetylase 1 (deacetylation) | 1.25 | 1.42 | −1.10 | −1.35 | −1.13 |
| IL6 | Interleukin 6 (interferon, beta 2) (pro-inflammation) | 3.23 | 1.33 | 2.47 | 1.46 | 5.00 |
| JUN | Jun proto-oncogene (growth regulation) | −1.11 | −4.72 | −1.39 | −1.34 | −1.93 |
| KAT2B | K(lysine) acetyltransferase 2B (acetylation) | −1.21 | 1.04 | −1.09 | −1.86 | −1.09 |
| RELA | V-rel reticuloendotheliosis viral oncogene homolog A (avian) (growth regulation) | 1.49 | 1.68 | 1.05 | 1.00 | 1.09 |
| SESN2 | Sestrin 2 (autophagy induction) | −1.50 | 1.09 | −1.97 | −1.58 | −1.67 |
| SIRT1 | Sirtuin 1 (anti- and pro-tumorogenic) | −1.24 | 1.67 | −1.61 | −1.17 | −1.04 |
| STAT1 | Signal transducer and activator of transcription 1, 91kDa (anti- and pro-tumorogenic) | −2.06 | 1.34 | −1.92 | −2.26 | −1.72 |
| TSC1 | Tuberous sclerosis 1 (tumor suppressor) | −1.03 | 1.19 | −1.10 | −1.04 | −1.11 |
| WT1 | Wilms tumor 1 (tumor suppressor) | 3.23 | 1.33 | 2.47 | 1.46 | 5.00 |
| XRCC5 | X-ray repair complementing defective repair in Chinese hamster cells 5 (double-strand-break rejoining) (DNA repair) | −1.72 | 1.31 | −1.03 | 1.01 | −1.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Gutiérrez, N.; Luna-Bárcenas, G.; Herrera-Hernández, G.; Campos-Vega, R.; Lozano-Herrera, S.J.; Sánchez-Tusié, A.A.; García-Solis, P.; Vergara-Castañeda, H.A. Quercetin and Its Fermented Extract as a Potential Inhibitor of Bisphenol A-Exposed HT-29 Colon Cancer Cells’ Viability. Int. J. Mol. Sci. 2023, 24, 5604. https://doi.org/10.3390/ijms24065604
García-Gutiérrez N, Luna-Bárcenas G, Herrera-Hernández G, Campos-Vega R, Lozano-Herrera SJ, Sánchez-Tusié AA, García-Solis P, Vergara-Castañeda HA. Quercetin and Its Fermented Extract as a Potential Inhibitor of Bisphenol A-Exposed HT-29 Colon Cancer Cells’ Viability. International Journal of Molecular Sciences. 2023; 24(6):5604. https://doi.org/10.3390/ijms24065604
Chicago/Turabian StyleGarcía-Gutiérrez, Nataly, Gabriel Luna-Bárcenas, Guadalupe Herrera-Hernández, Rocio Campos-Vega, Sara Julietta Lozano-Herrera, Ana Alicia Sánchez-Tusié, Pablo García-Solis, and Haydé Azeneth Vergara-Castañeda. 2023. "Quercetin and Its Fermented Extract as a Potential Inhibitor of Bisphenol A-Exposed HT-29 Colon Cancer Cells’ Viability" International Journal of Molecular Sciences 24, no. 6: 5604. https://doi.org/10.3390/ijms24065604