
Molecular Analysis of SARS-CoV-2 Spike Protein-Induced Endothelial Cell Permeability and vWF Secretion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
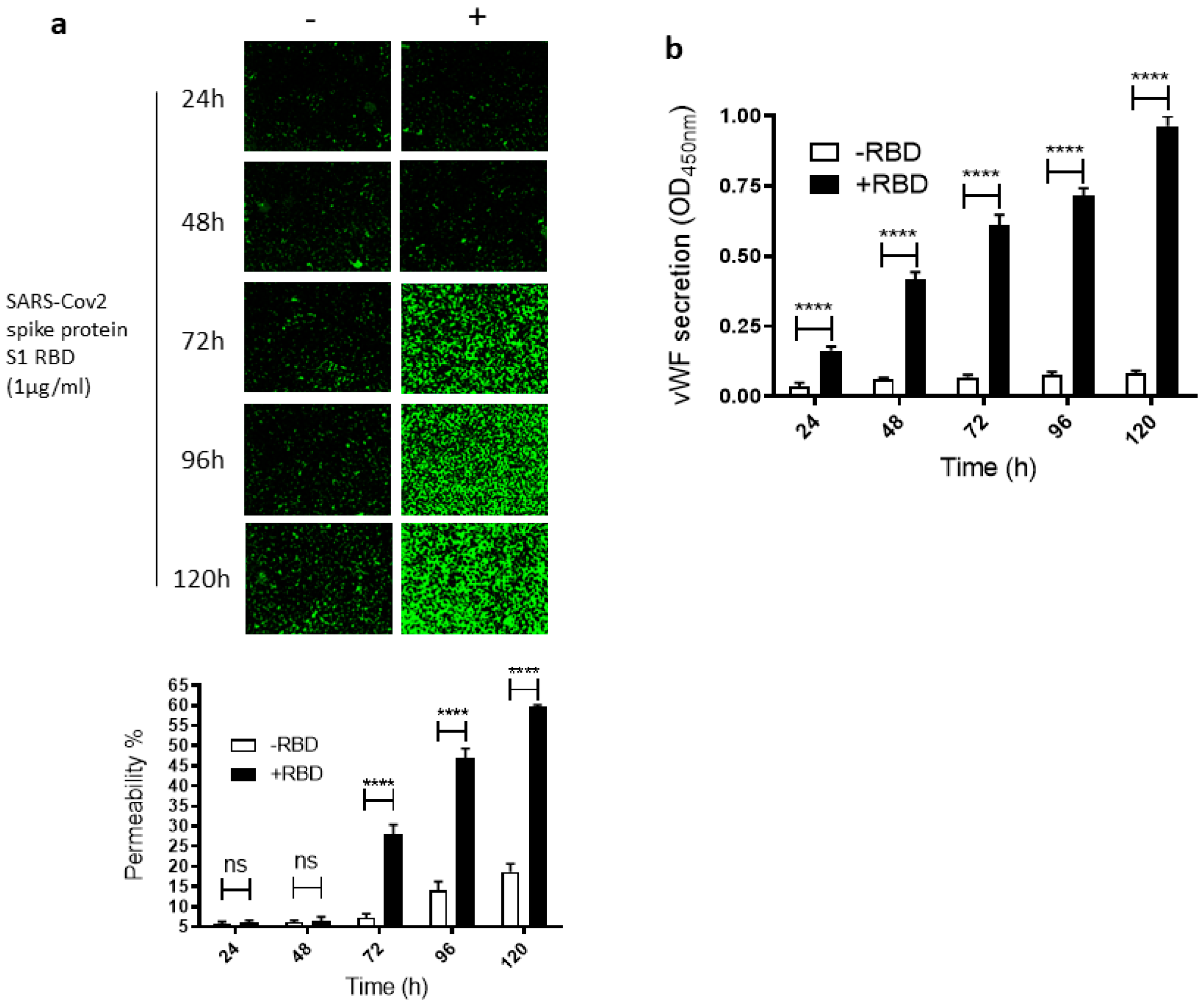
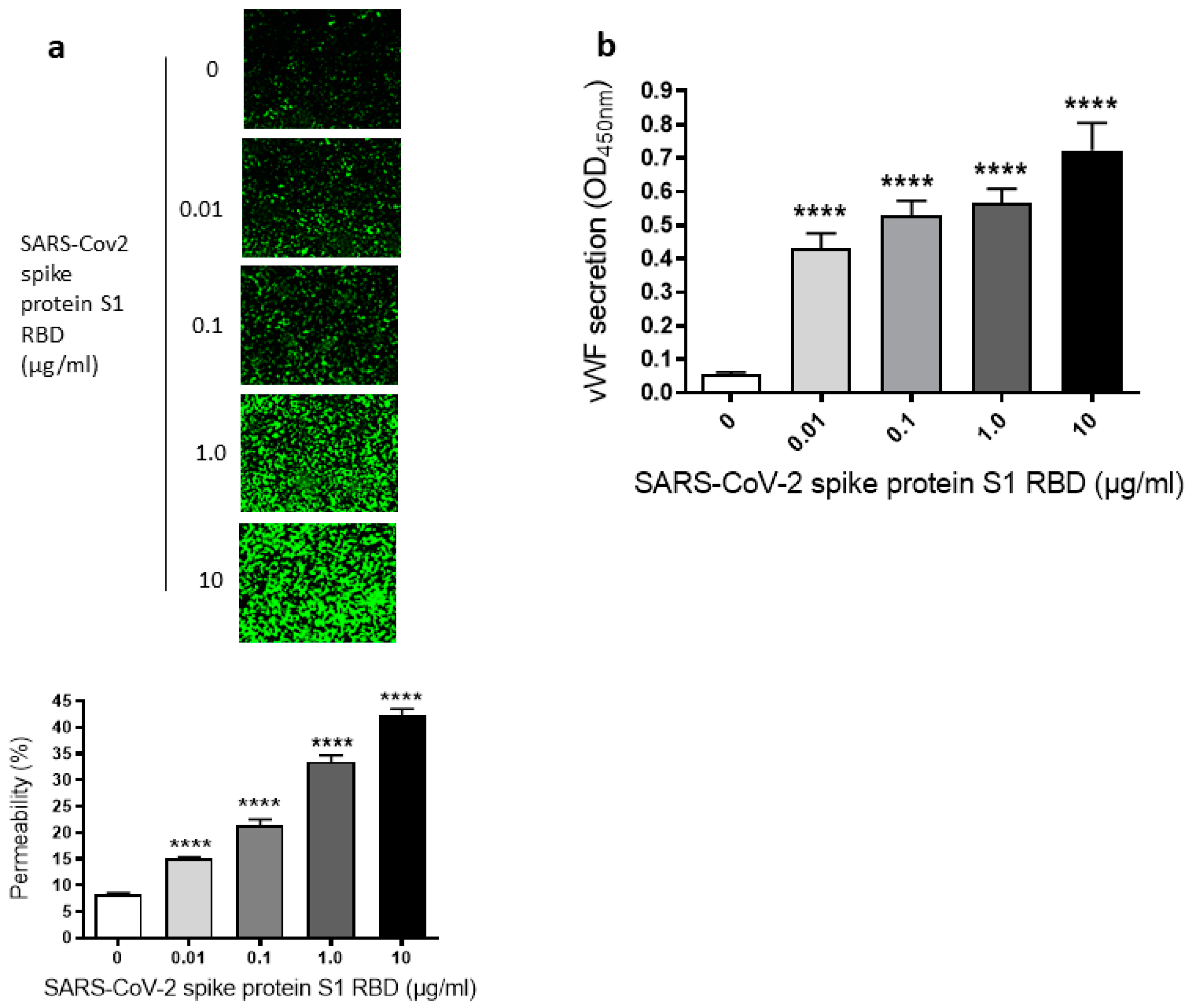
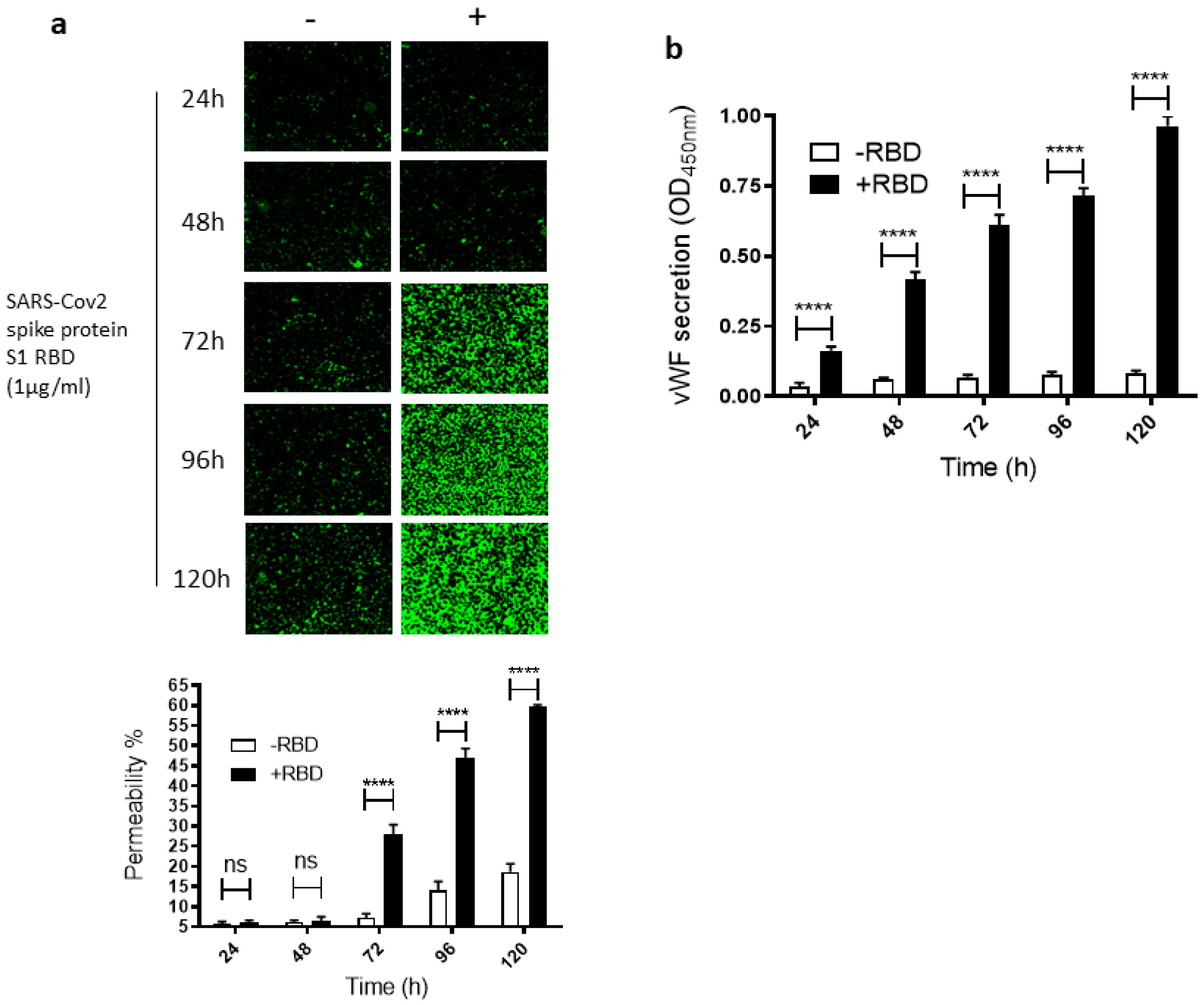
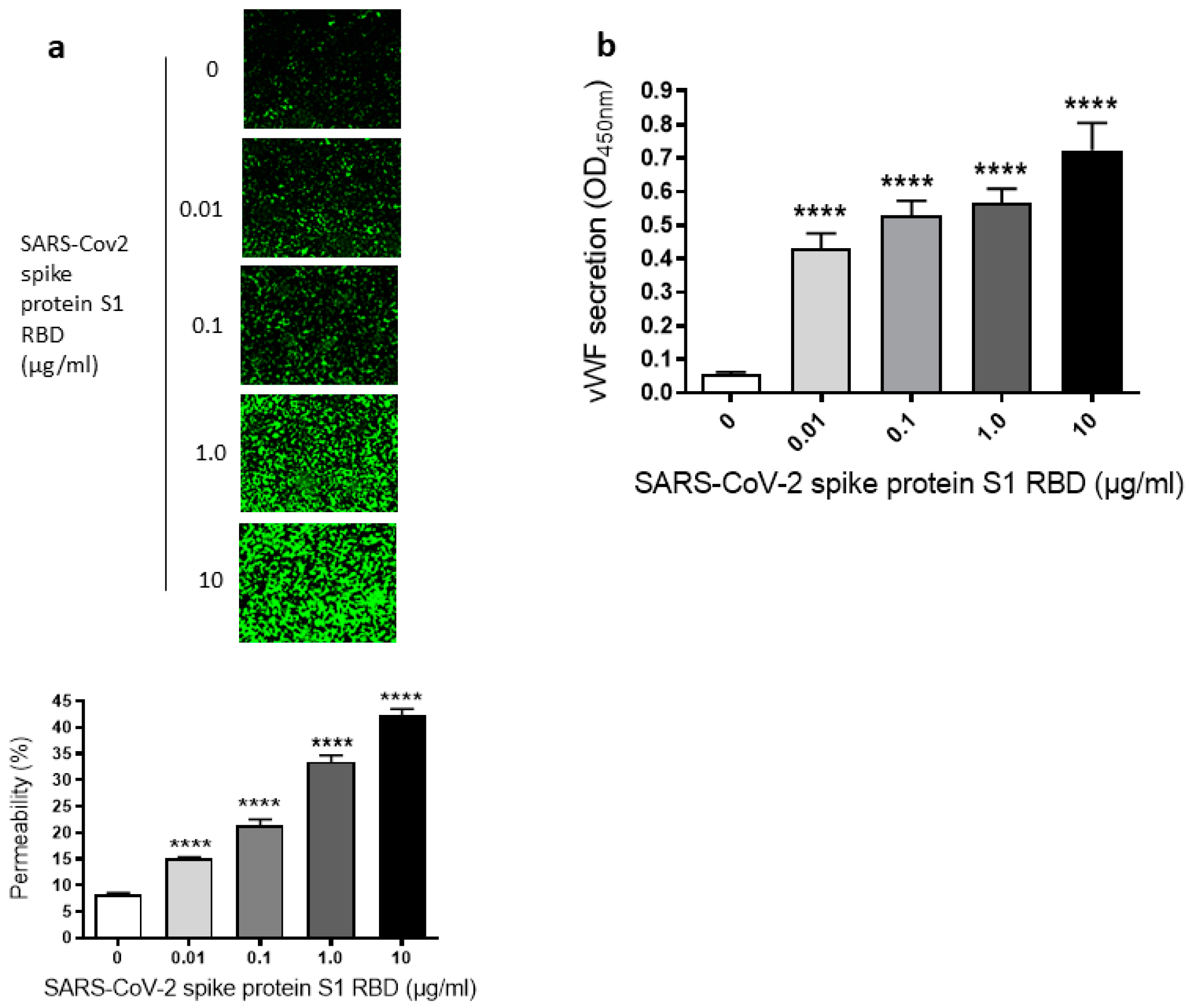
2.1. SARS-CoV-2 Spike Protein S1 RBD Induces EC Permeability and vWF Secretion
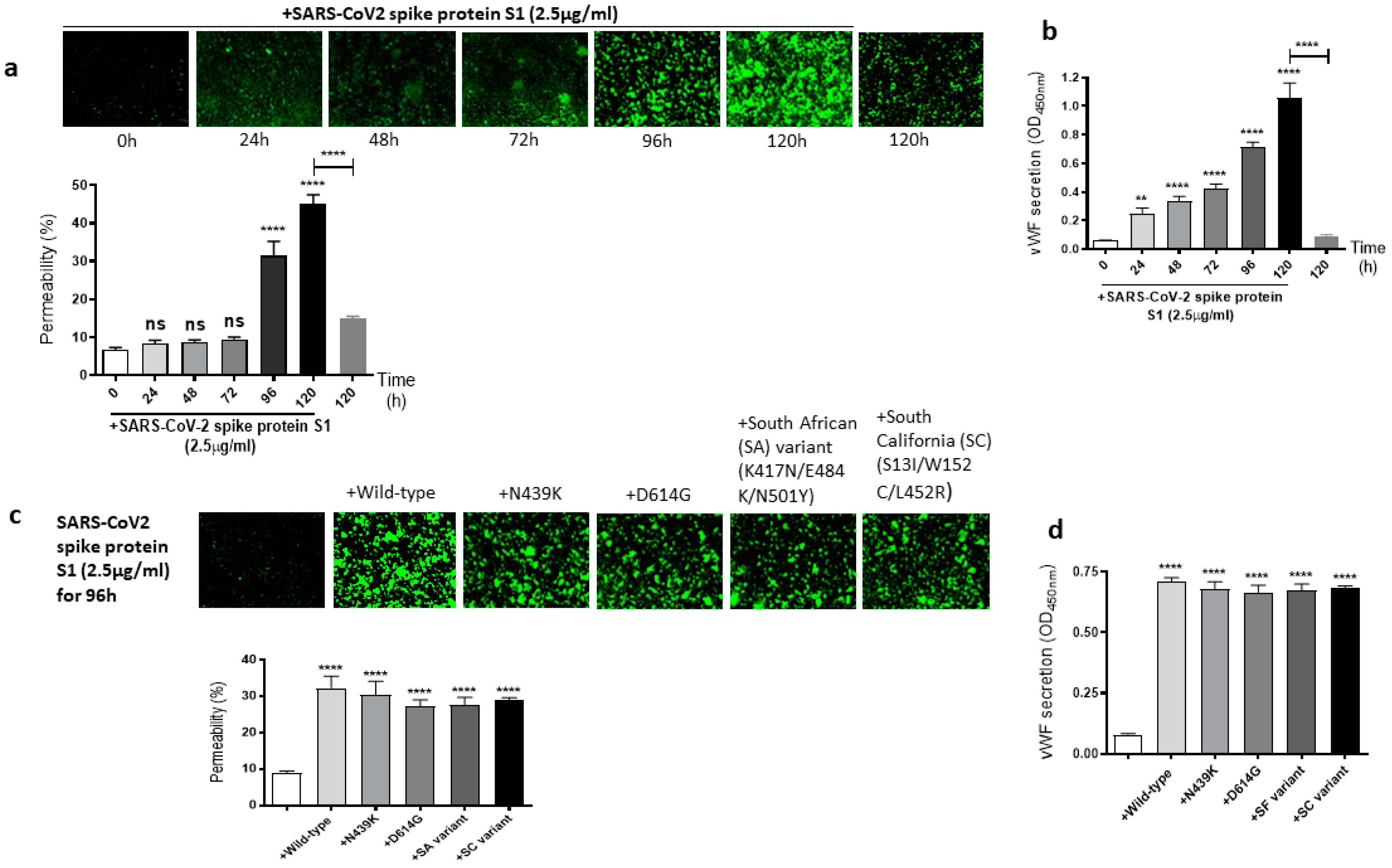
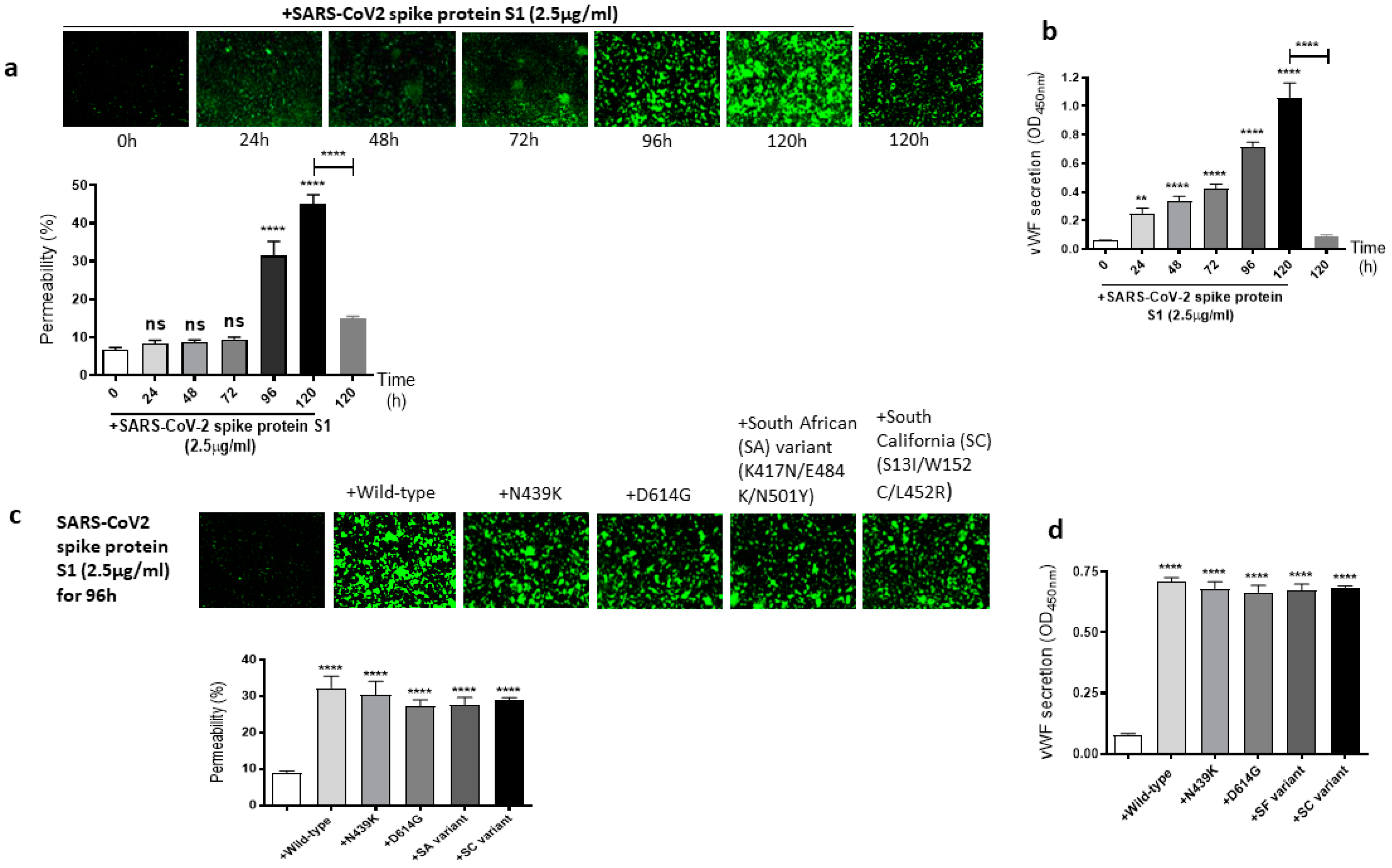
2.2. The Mutations of SARS-CoV-2 Spike Protein S1 Unaffected Its Induced EC Permeability and vWF Secretion
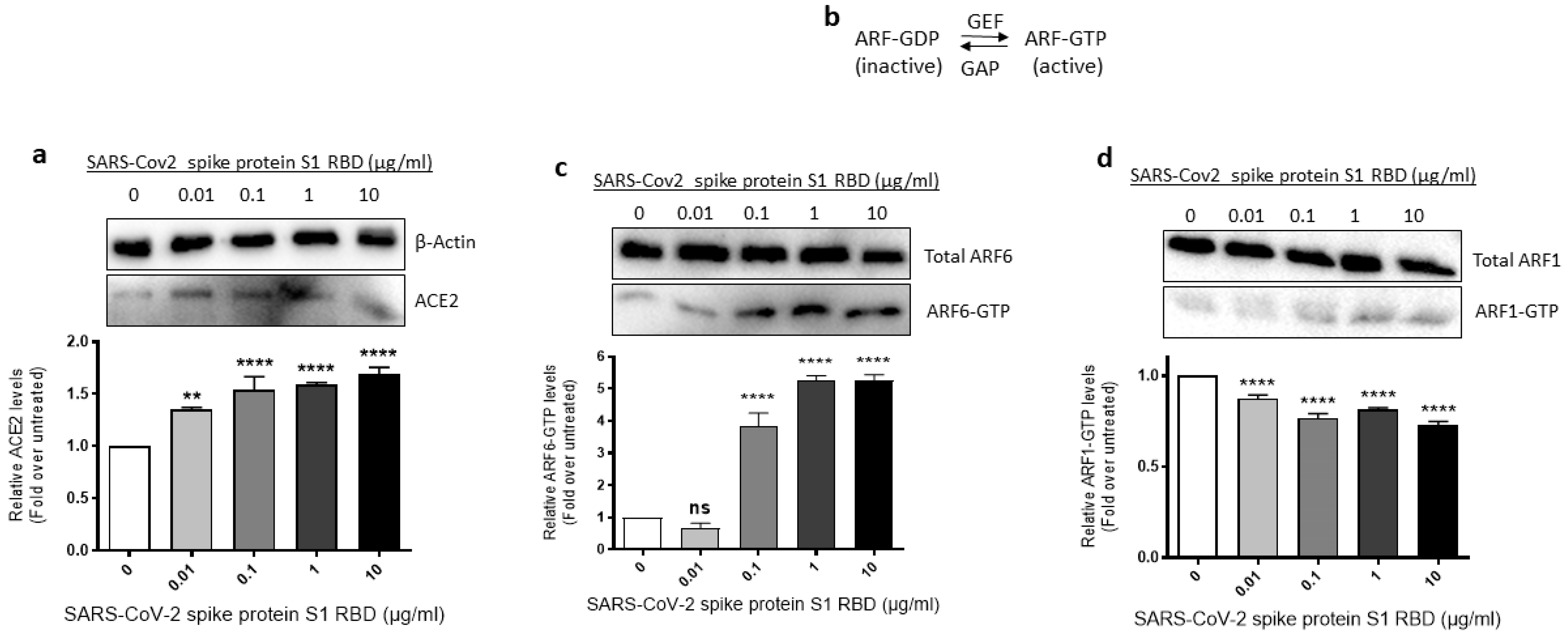
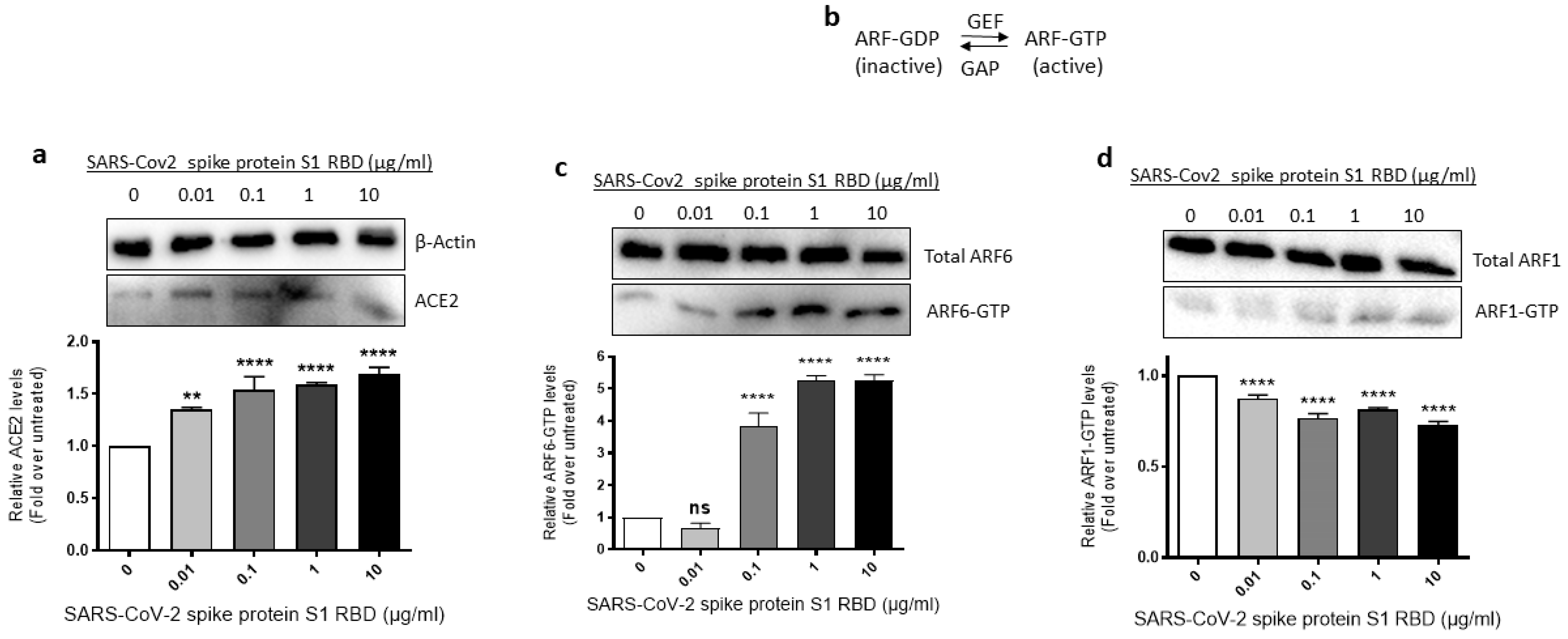
2.3. ACE2 Expression and ARF6 Activation Are Increased in EA.hy 296 Cells Treated with SARS-CoV-2 Spike Protein S1 RBD
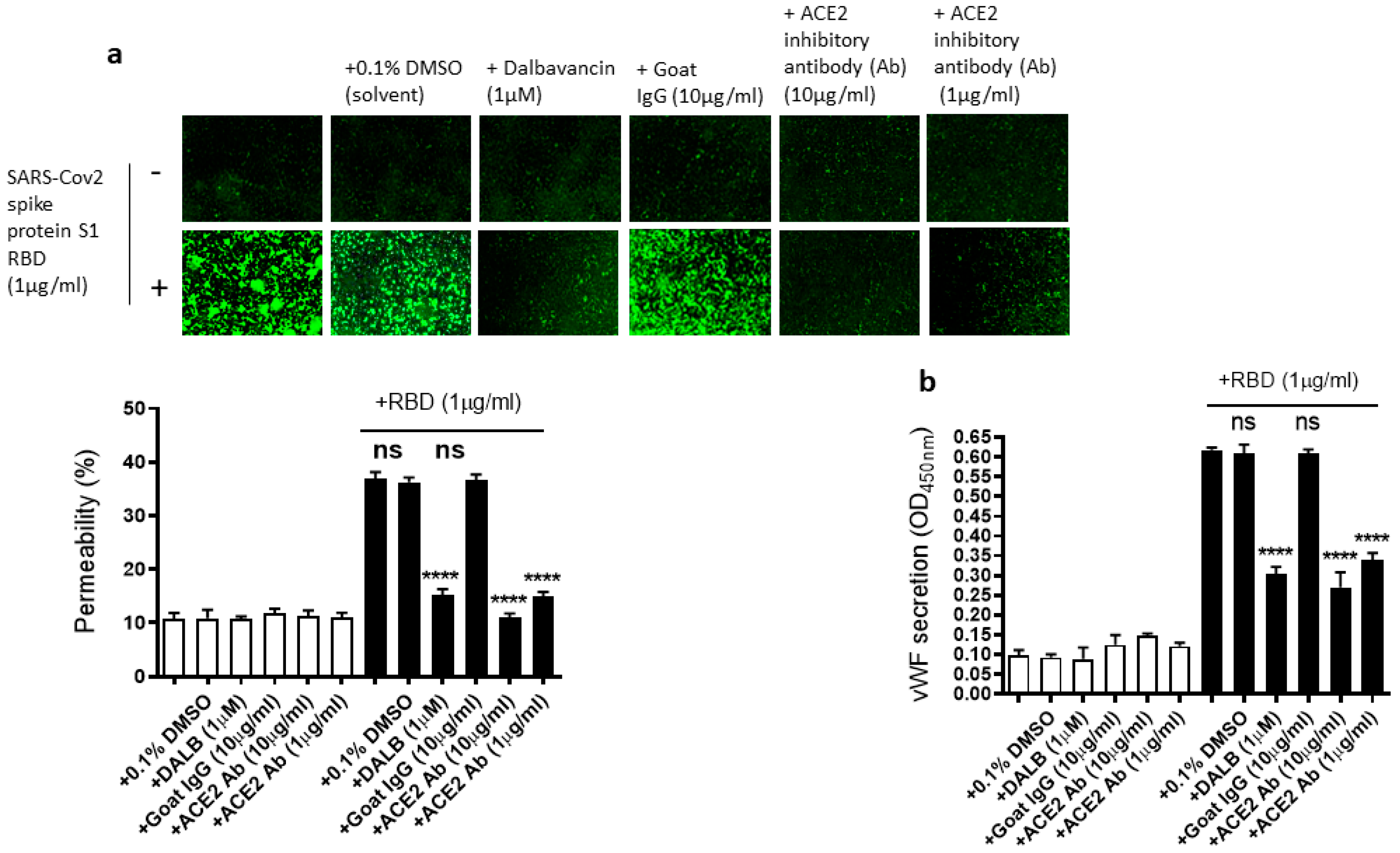
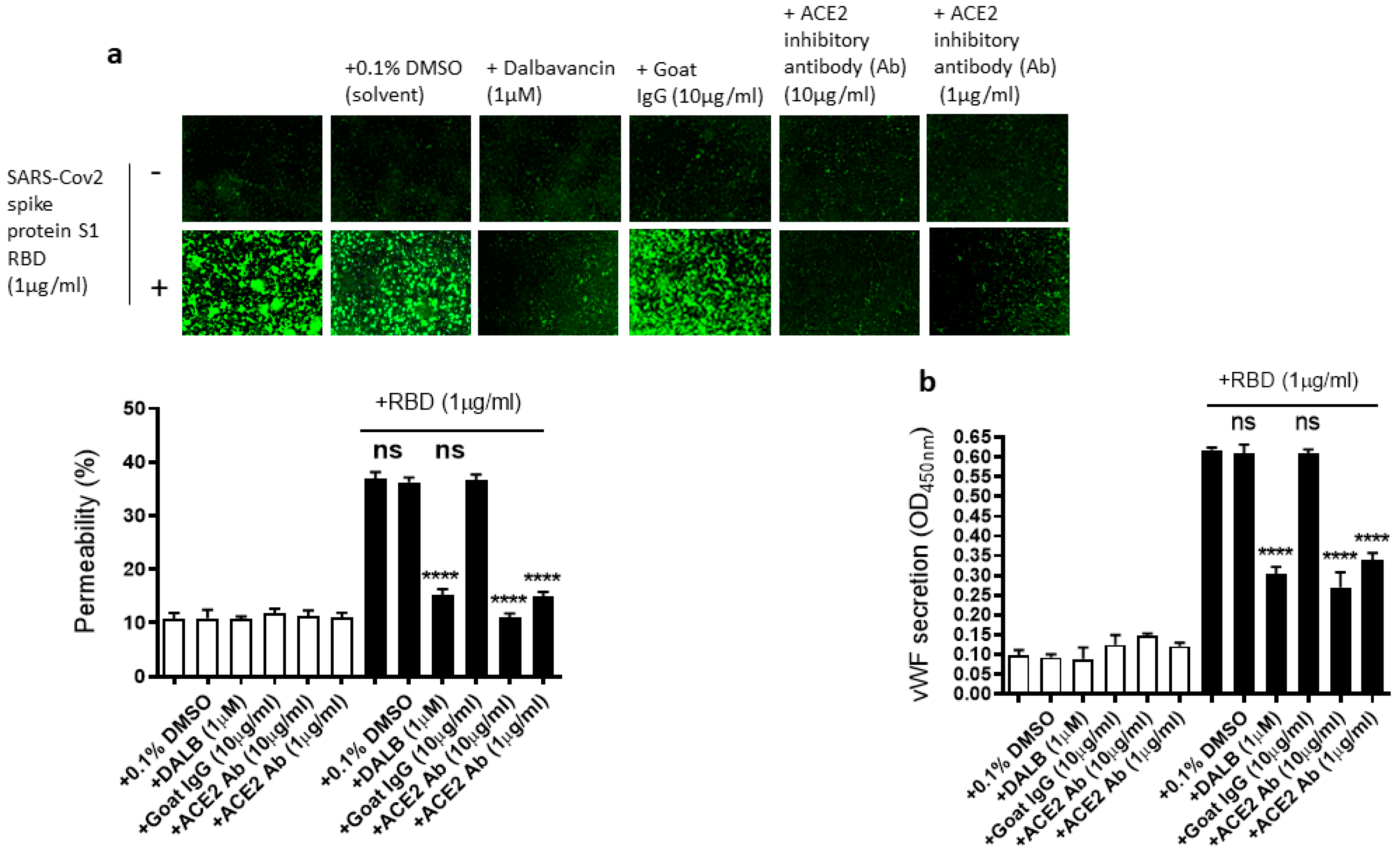
2.4. The EC Permeability and vWF Secretion in SARS-CoV-2 Spike Protein S1 RBD-Stimulated EA.hy 296 Cells Is Mediated by ACE2
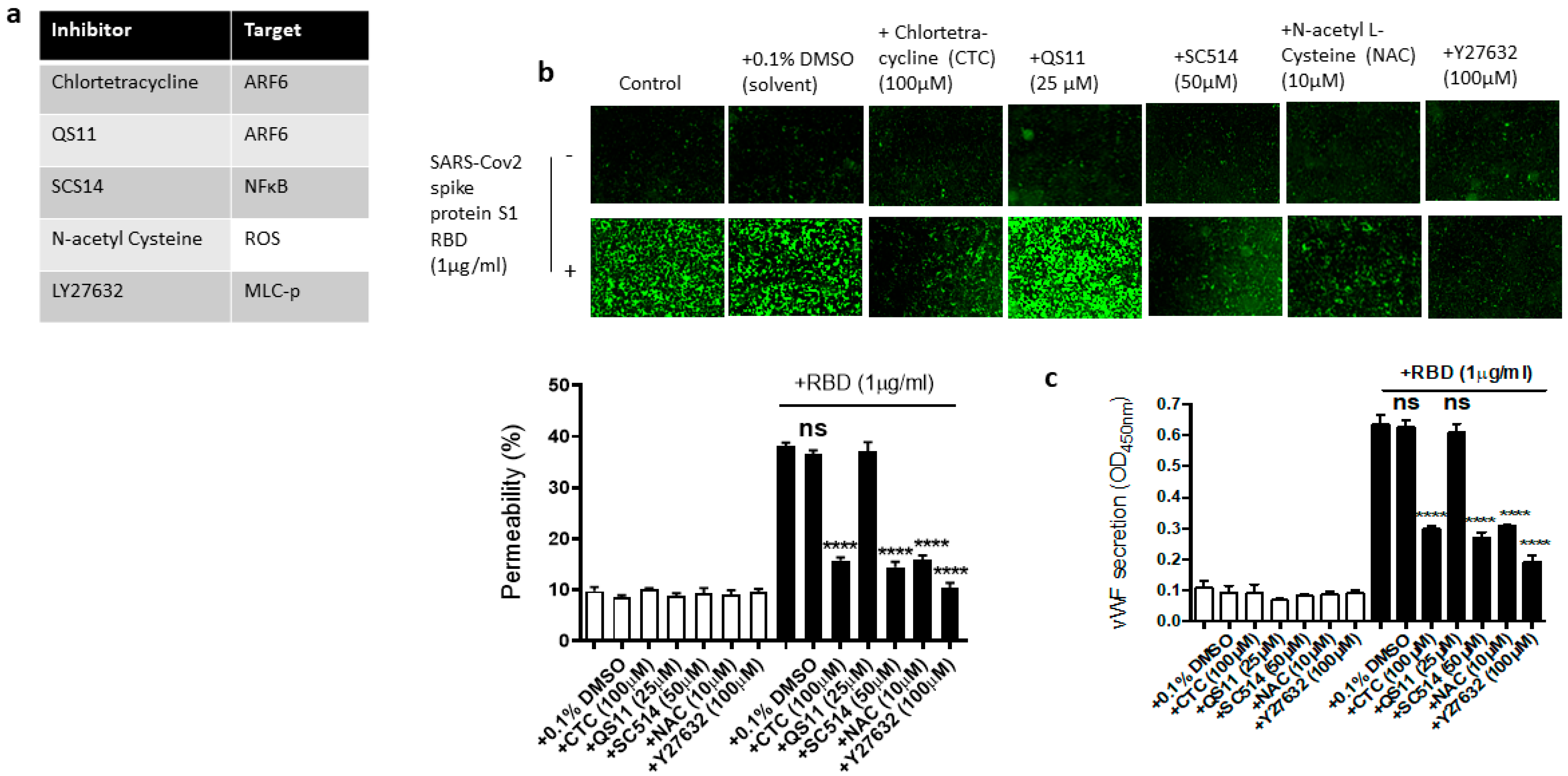
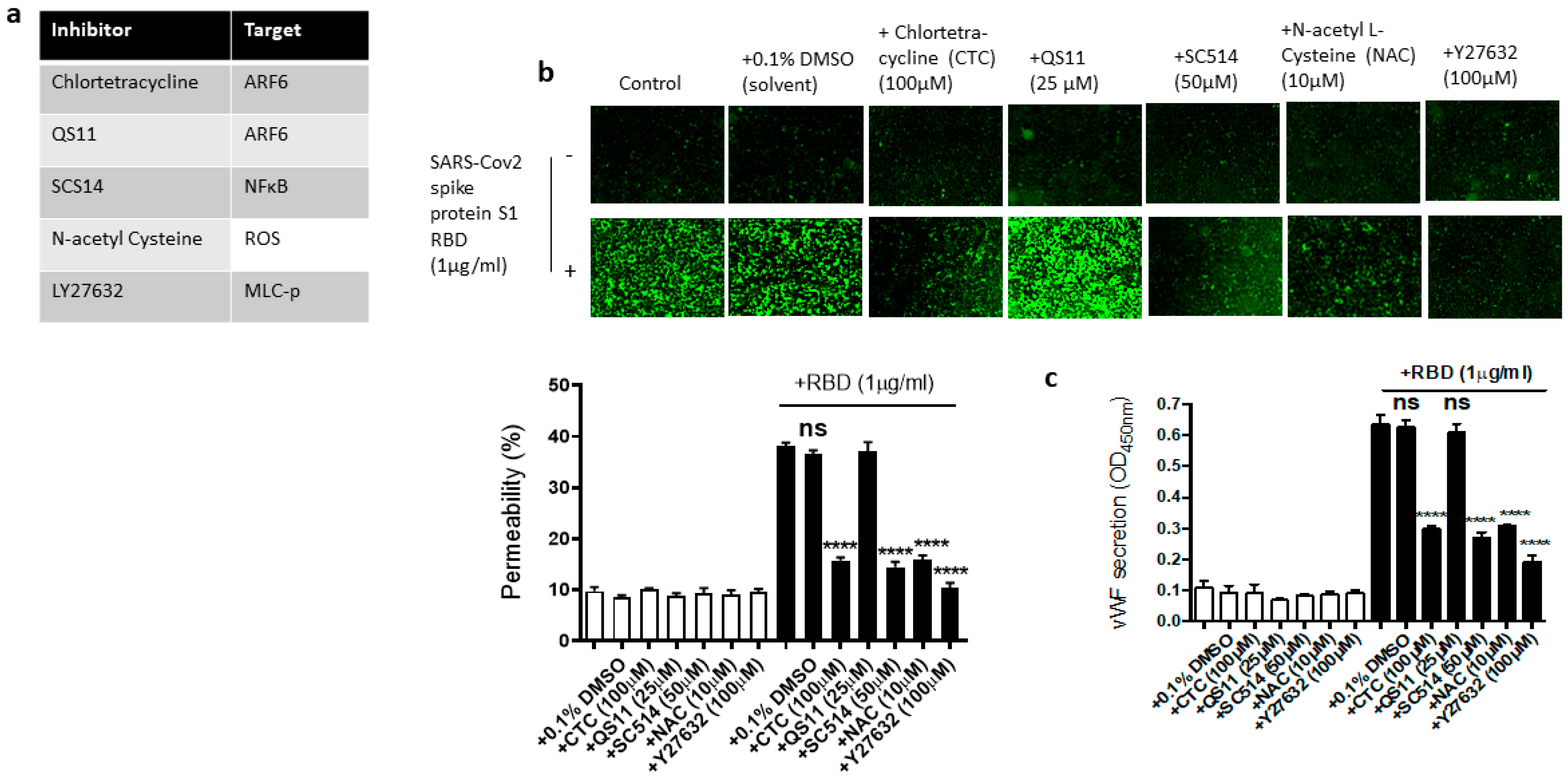
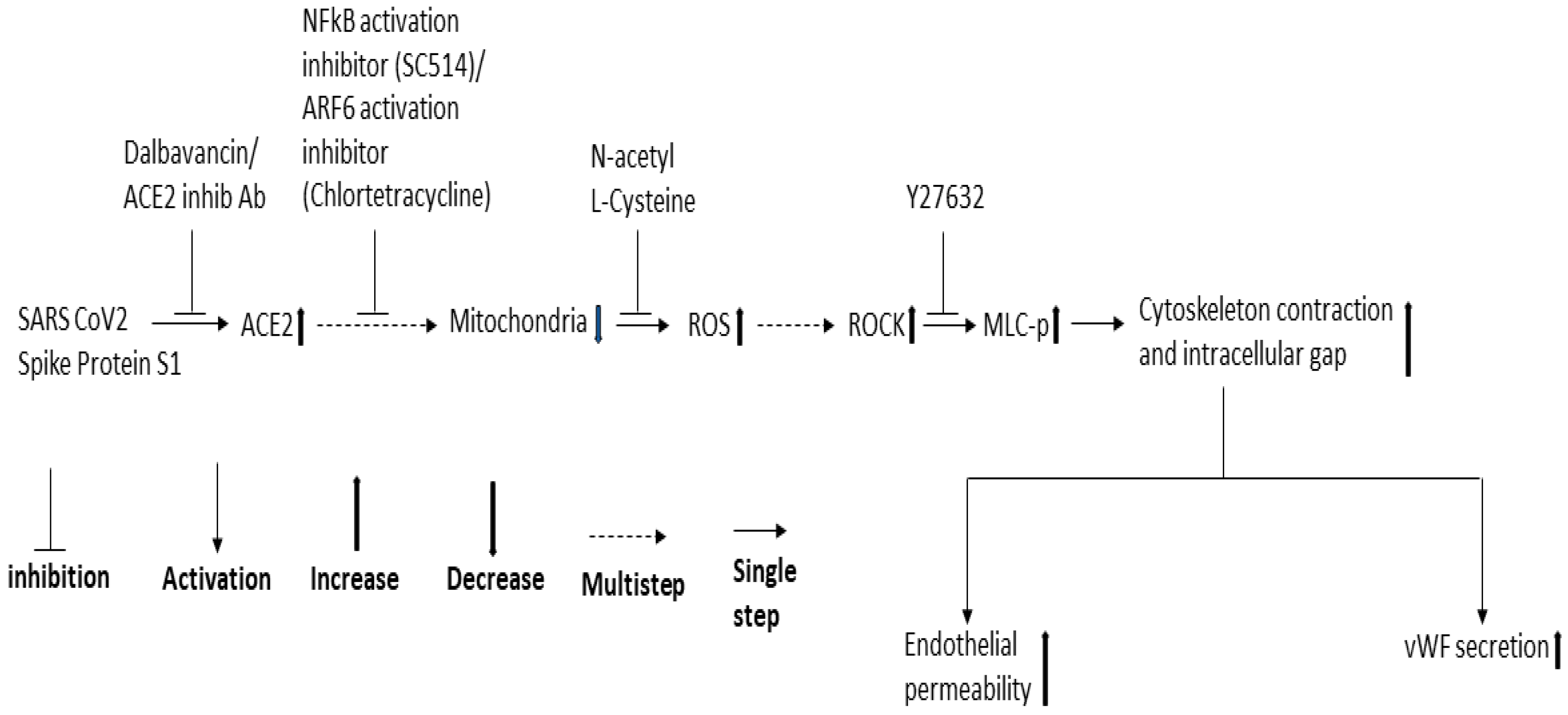
2.5. Dissecting the Signaling Pathway for EC Permeability and vWF Secretion in SARS-CoV-2 Spike Protein S1 RBD-Stimulated EA.hy 296 Cells
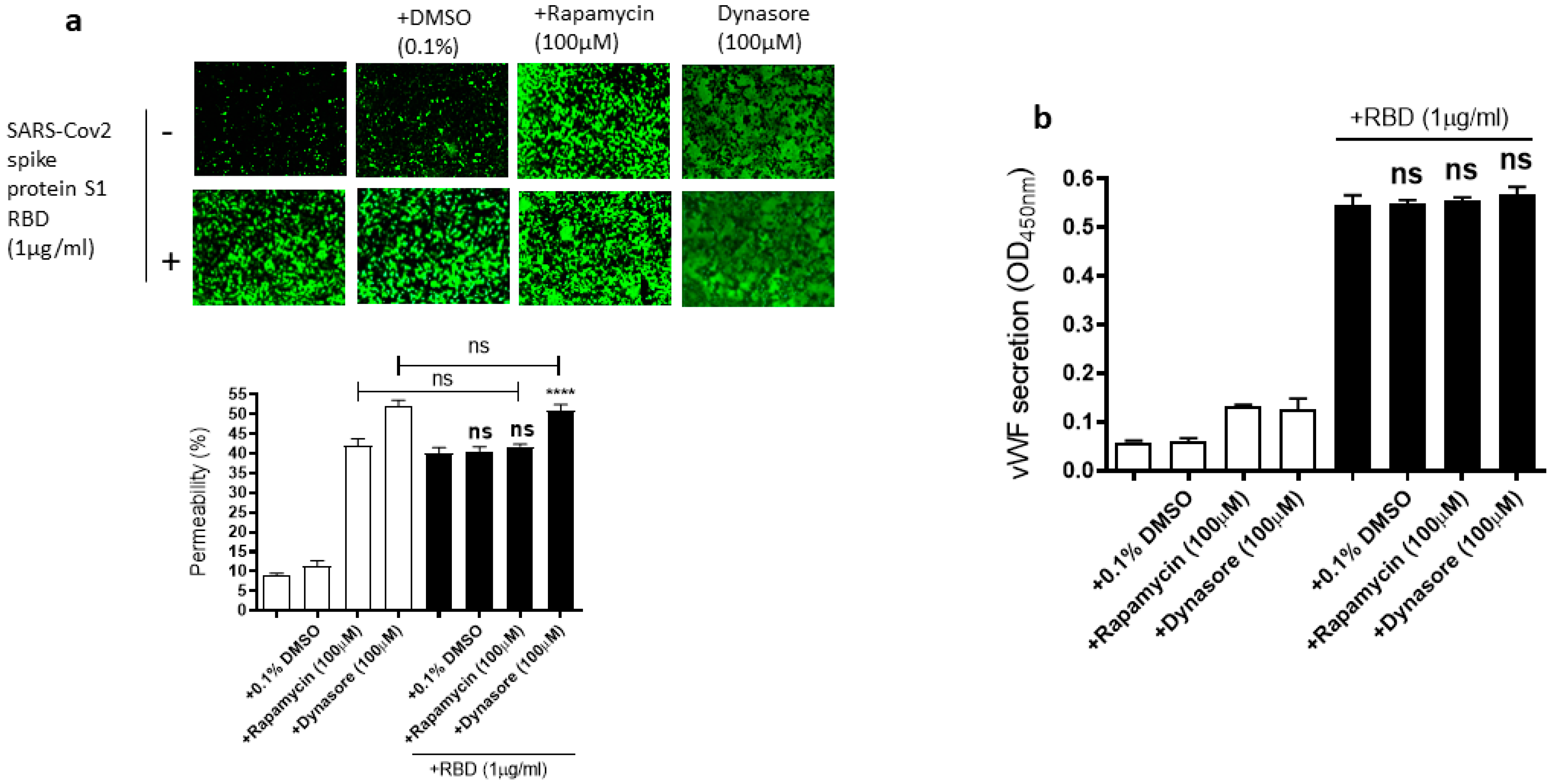
2.6. Rapamycin and Dynasore Do Not Alter the RBD-Induced EC Permeability and vWF Secretion
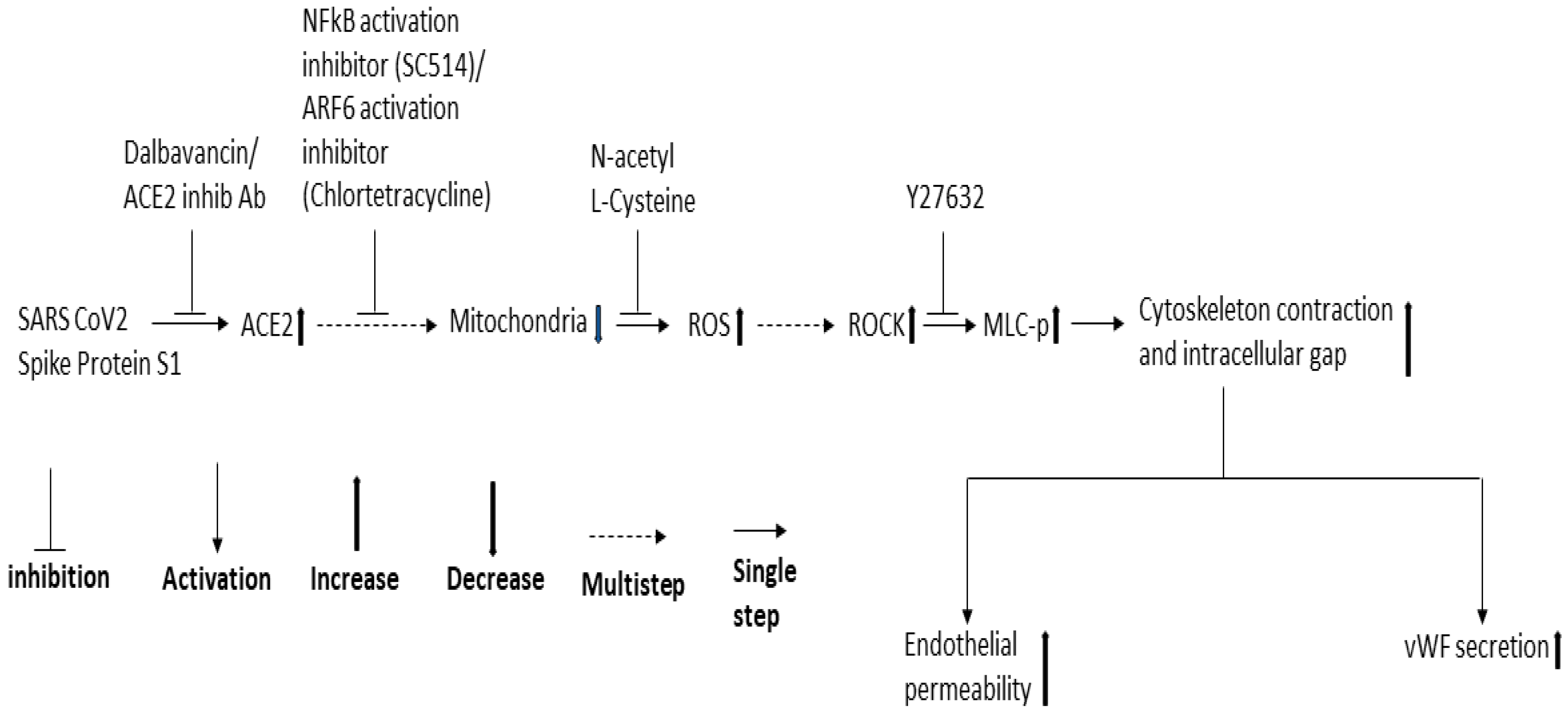
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. 96-Well Trans-Monolayer Endothelial Permeability Assay
4.4. ARF Activation Assay
4.5. Immunoblotting
4.6. Quantification of Secreted vWF and Cytokines by ELISA
4.7. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kamel Boulos, M.N.; Geraghty, E.M. Geographical tracking and mapping of coronavirus disease COVID-19/severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) epidemic and associated events around the world: How 21st century GIS technologies are supporting the global fight against outbreaks and epidemics. Int. J. Health Geogr. 2020, 19, 1–12. [Google Scholar]
- Chen, J. Pathogenicity and transmissibility of 2019-nCoV—A quick overview and comparison with other emerging viruses. Microbes Infect. 2020, 22, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Geng, X.; Tan, Y.; Li, Q.; Xu, C.; Xu, J.; Hao, L.; Zeng, Z.; Luo, X.; Liu, F. New understanding of the damage of SARS-CoV-2 infection outside the respiratory system. Biomed. Pharmacother. 2020, 127, 110195. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Ahmad, I.; Pawara, R.; Surana, S.; Patel, H. The repurposed ACE2 inhibitors: SARS-CoV-2 entry blockers of COVID-19. Top. Curr. Chem. 2021, 379, 40. [Google Scholar] [CrossRef]
- Chen, L.-F.; Greene, W.C. Shaping the nuclear action of NF-κB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef]
- Su, C.M.; Wang, L.; Yoo, D. Activation of NF-kappaB and induction of proinflammatory cytokine expressions mediated by ORF7a protein of SARS-CoV-2. Sci. Rep. 2021, 11, 13464. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Kanan, T.; Kanan, D.; Al Shardoub, E.J.; Durdagi, S. Transcription factor NF-κB as target for SARS-CoV-2 drug discovery efforts using inflammation-based QSAR screening model. J. Mol. Graph. Model. 2021, 108, 107968. [Google Scholar] [CrossRef] [PubMed]
- Kanamarlapudi, V.; Tamaddon-Jahromi, S.; Murphy, K. ADP-ribosylation factor 6 expression increase in oesophageal adenocarcinoma suggests a potential biomarker role for it. PLoS ONE 2022, 17, e0263845. [Google Scholar] [CrossRef]
- Zhou, Y.-Q.; Wang, K.; Wang, X.-Y.; Cui, H.-Y.; Zhao, Y.; Zhu, P.; Chen, Z.-N. SARS-CoV-2 pseudovirus enters the host cells through spike protein-CD147 in an Arf6-dependent manner. Emerg. Microbes Infect. 2022, 11, 1135–1144. [Google Scholar] [CrossRef]
- Olwal, C.O.; Nganyewo, N.N.; Tapela, K.; Djomkam Zune, A.L.; Owoicho, O.; Bediako, Y.; Duodu, S. Parallels in sepsis and COVID-19 conditions: Implications for managing severe COVID-19. Front. Immunol. 2021, 12, 602848. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-H.; Perl, D.P.; Nair, G.; Li, W.; Maric, D.; Murray, H.; Dodd, S.J.; Koretsky, A.P.; Watts, J.A.; Cheung, V. Microvascular injury in the brains of patients with Covid-19. N. Engl. J. Med. 2021, 384, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Aid, M.; Busman-Sahay, K.; Vidal, S.J.; Maliga, Z.; Bondoc, S.; Starke, C.; Terry, M.; Jacobson, C.A.; Wrijil, L.; Ducat, S. Vascular disease and thrombosis in SARS-CoV-2-infected rhesus macaques. Cell 2020, 183, 1354–1366.e13. [Google Scholar] [CrossRef]
- Barbosa, L.C.; Goncalves, T.L.; de Araujo, L.P.; de Oliveira Rosario, L.V.; Ferrer, V.P. Endothelial cells and SARS-CoV-2: An intimate relationship. Vasc. Pharmacol. 2021, 137, 106829. [Google Scholar] [CrossRef]
- Lin, T.; Luo, W.; Li, Z.; Zhang, L.; Zheng, X.; Mai, L.; Yang, W.; Guan, G.; Su, Z.; Liu, P. Rhamnocitrin extracted from Nervilia fordii inhibited vascular endothelial activation via miR-185/STIM-1/SOCE/NFATc3. Phytomedicine 2020, 79, 153350. [Google Scholar] [CrossRef]
- Matarese, A.; Gambardella, J.; Sardu, C.; Santulli, G. miR-98 regulates TMPRSS2 expression in human endothelial cells: Key implications for COVID-19. Biomedicines 2020, 8, 462. [Google Scholar] [CrossRef] [PubMed]
- Potus, F.; Mai, V.; Lebret, M.; Malenfant, S.; Breton-Gagnon, E.; Lajoie, A.C.; Boucherat, O.; Bonnet, S.; Provencher, S. Novel insights on the pulmonary vascular consequences of COVID-19. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 319, L277–L288. [Google Scholar] [CrossRef] [PubMed]
- Pum, A.; Ennemoser, M.; Adage, T.; Kungl, A.J. Cytokines and chemokines in SARS-CoV-2 infections—Therapeutic strategies targeting cytokine storm. Biomolecules 2021, 11, 91. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, H.K.; Libby, P.; Ridker, P.M. COVID-19-A vascular disease. Trends. Cardiovasc. Med. 2021, 31, 1–5. [Google Scholar] [CrossRef]
- Teuwen, L.-A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Wazny, V.; Siau, A.; Wu, K.X.; Cheung, C. Vascular underpinning of COVID-19. Open Biol. 2020, 10, 200208. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, B.; Su, Z.; Liu, Y.; Shelite, T.R.; Chang, Q.; Wang, P.; Bukreyev, A.; Soong, L.; Jin, Y. EPAC regulates von Willebrand factor secretion from endothelial cells in a PI3K/eNOS-dependent manner during inflammation. bioRxiv 2020, 2020, 282806. [Google Scholar]
- Choudhary, S.; Sharma, K.; Singh, P.K. Von Willebrand factor: A key glycoprotein involved in thrombo-inflammatory complications of COVID-19. Chem.-Biol. Interact. 2021, 348, 109657. [Google Scholar] [CrossRef]
- Chen, W.; Pan, J.Y. Anatomical and Pathological Observation and Analysis of SARS and COVID-19: Microthrombosis Is the Main Cause of Death. Biol. Proced. Online 2021, 23, 4. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L. SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Kenchappa, D.B.; Leo, M.D. SARS-CoV-2 Spike Protein Induces Degradation of Junctional Proteins That Maintain Endothelial Barrier Integrity. Front. Cardiovasc. Med. 2021, 8, 687783. [Google Scholar] [CrossRef]
- Rauti, R.; Shahoha, M.; Leichtmann-Bardoogo, Y.; Nasser, R.; Paz, E.; Tamir, R.; Miller, V.; Babich, T.; Shaked, K.; Ehrlich, A. Effect of SARS-CoV-2 proteins on vascular permeability. eLife 2021, 10, e69314. [Google Scholar] [CrossRef] [PubMed]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef]
- Flaumenhaft, R.; Enjyoji, K.; Schmaier, A.A. Vasculopathy in COVID-19. Blood 2022, 140, 222–235. [Google Scholar] [CrossRef]
- Karki, P.; Birukova, A.A. Substrate stiffness-dependent exacerbation of endothelial permeability and inflammation: Mechanisms and potential implications in ALI and PH (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045894018773044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McVerry, B.J.; Garcia, J.G. Endothelial cell barrier regulation by sphingosine 1-phosphate. J. Cell. Biochem. 2004, 92, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Kümper, S.; Mardakheh, F.K.; McCarthy, A.; Yeo, M.; Stamp, G.W.; Paul, A.; Worboys, J.; Sadok, A.; Jørgensen, C.; Guichard, S. Rho-associated kinase (ROCK) function is essential for cell cycle progression, senescence and tumorigenesis. eLife 2016, 5, e12203. [Google Scholar] [CrossRef]
- Kim, S.; Kim, S.A.; Han, J.; Kim, I.-S. Rho-kinase as a target for cancer therapy and its immunotherapeutic potential. Int. J. Mol. Sci. 2021, 22, 12916. [Google Scholar] [CrossRef]
- Fernandez, P.; Bordenave, L.; Celerier, C.; Bareille, R.; Brouillaud, B.; Basse-Cathalinat, B. A novel potential application for 99mTc-HMPAO: Endothelial cell labeling for in vitro investigation of cell-biomaterial interactions. J. Nucl. Med. 1999, 40, 1756–1763. [Google Scholar] [PubMed]
- Maestro, A.; Terdoslavich, M.; Vanzo, A.; Kuku, A.; Tramer, F.; Nicolin, V.; Micali, F.; Decorti, G.; Passamonti, S. Expression of bilitranslocase in the vascular endothelium and its function as a flavonoid transporter. Cardiovasc. Res. 2010, 85, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Dubrovskyi, O.; Birukova, A.A.; Birukov, K.G. Measurement of local permeability at subcellular level in cell models of agonist-and ventilator-induced lung injury. Lab. Investig. 2013, 93, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Nightingale, T.; Cutler, D. The secretion of von W illebrand factor from endothelial cells; an increasingly complicated story. J. Thromb. Haemost. 2013, 11, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Estrada, E. Fractional diffusion on the human proteome as an alternative to the multi-organ damage of SARS-CoV-2 <A3B2 show [editpick]>. Chaos: Interdiscip. J. Nonlinear Sci. 2020, 30, 081104. [Google Scholar]
- Mirabelli, C.; Sherman, E.J.; Wotring, J.W.; El Saghir, J.; Cunha, J.B.; Harder, J.; Sexton, J.Z.; Emmer, B.T.; Wobus, C.E. ARF6 is an important host factor for SARS-CoV-2 infection in vitro. bioRxiv 2022, 2022, 495482. [Google Scholar]
- Cohen, L.A.; Donaldson, J.G. Analysis of Arf GTP-binding protein function in cells. Curr. Protoc. Cell Biol. 2010, 48, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Kanamarlapudi, V.; Thompson, A.; Kelly, E.; Bernal, A.L. ARF6 activated by the LHCG receptor through the cytohesin family of guanine nucleotide exchange factors mediates the receptor internalization and signaling. J. Biol. Chem. 2012, 287, 20443–20455. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Jin, Y.; Pöhlmann, S. Dalbavancin: Novel candidate for COVID-19 treatment. Cell Res. 2021, 31, 243–244. [Google Scholar] [CrossRef]
- Wang, G.; Yang, M.-L.; Duan, Z.-L.; Liu, F.-L.; Jin, L.; Long, C.-B.; Zhang, M.; Tang, X.-P.; Xu, L.; Li, Y.-C. Dalbavancin binds ACE2 to block its interaction with SARS-CoV-2 spike protein and is effective in inhibiting SARS-CoV-2 infection in animal models. Cell Res. 2021, 31, 17–24. [Google Scholar] [CrossRef]
- Chaouat, A.E.; Achdout, H.; Kol, I.; Berhani, O.; Roi, G.; Vitner, E.B.; Melamed, S.; Politi, B.; Zahavy, E.; Brizic, I. SARS-CoV-2 receptor binding domain fusion protein efficiently neutralizes virus infection. PLoS Pathog. 2021, 17, e1010175. [Google Scholar] [CrossRef]
- Svilenov, H.L.; Sacherl, J.; Reiter, A.; Wolff, L.S.; Cheng, C.C.; Stern, M.; Grass, V.; Feuerherd, M.; Wachs, F.P.; Simonavicius, N.; et al. Picomolar inhibition of SARS-CoV-2 variants of concern by an engineered ACE2-IgG4-Fc fusion protein. Antiviral. Res. 2021, 196, 105197. [Google Scholar] [CrossRef]
- Macia, E.; Vazquez-Rojas, M.; Robiolo, A.; Fayad, R.; Abélanet, S.; Mus-Veteau, I.; Fontaine-Vive, F.; Mehiri, M.; Luton, F.; Franco, M. Chlortetracycline, a novel arf inhibitor that decreases the Arf6-dependent invasive properties of breast cancer cells. Molecules 2021, 26, 969. [Google Scholar] [CrossRef]
- Zhang, Q.; Major, M.B.; Takanashi, S.; Camp, N.D.; Nishiya, N.; Peters, E.C.; Ginsberg, M.H.; Jian, X.; Randazzo, P.A.; Schultz, P.G. Small-molecule synergist of the Wnt/β-catenin signaling pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 7444–7448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2020, 9, 1703244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karam, B.S.; Morris, R.S.; Bramante, C.T.; Puskarich, M.; Zolfaghari, E.J.; Lotfi-Emran, S.; Ingraham, N.E.; Charles, A.; Odde, D.J.; Tignanelli, C.J. mTOR inhibition in COVID-19: A commentary and review of efficacy in RNA viruses. J. Med. Virol. 2021, 93, 1843–1846. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.; Byrareddy, S.N. Rapamycin as a potential repurpose drug candidate for the treatment of COVID-19. Chem.-Biol. Interact. 2020, 331, 109282. [Google Scholar] [CrossRef] [PubMed]
- Karthika, T.; Joseph, J.; Das, V.A.; Nair, N.; Charulekha, P.; Roji, M.D.; Raj, V.S. SARS-CoV-2 cellular entry is independent of the ace2 cytoplasmic domain signaling. Cells 2021, 10, 1814. [Google Scholar] [CrossRef]
- Iba, T.; Connors, J.M.; Levy, J.H. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm. Res. 2020, 69, 1181–1189. [Google Scholar] [CrossRef]
- Ladikou, E.E.; Sivaloganathan, H.; Milne, K.M.; Arter, W.E.; Ramasamy, R.; Saad, R.; Stoneham, S.M.; Philips, B.; Eziefula, A.C.; Chevassut, T. Von Willebrand factor (vWF): Marker of endothelial damage and thrombotic risk in COVID-19? Clin. Med. 2020, 20, e178. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, S.; Gou, J.; Wen, Y.; Fan, L.; Zhou, J.; Zhou, G.; Xu, G.; Zhang, Z. Spike-mediated ACE2 down-regulation was involved in the pathogenesis of SARS-CoV-2 infection. J. Infect. 2022, 85, 418–427. [Google Scholar] [CrossRef]
- Wang, D.; Chen, Z.; Wai Kan Yeung, A.; Atanasov, A.G. Differences between common endothelial cell models (primary human aortic endothelial cells and EA.hy926 cells) revealed through transcriptomics, bioinformatics, and functional analysis. Curr. Res. Biotechnol. 2021, 3, 135–145. [Google Scholar] [CrossRef]
- Davis, C.T.; Zhu, W.; Gibson, C.C.; Bowman-Kirigin, J.A.; Sorensen, L.; Ling, J.; Sun, H.; Navankasattusas, S.; Li, D.Y. ARF6 inhibition stabilizes the vasculature and enhances survival during endotoxic shock. J. Immunol. 2014, 192, 6045–6052. [Google Scholar] [CrossRef] [Green Version]
- Ahamed, J.; Laurence, J. Long COVID endotheliopathy: Hypothesized mechanisms and potential therapeutic approaches. J. Clin. Investig. 2022, 132, e161167. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.W.; Ilyas, I.; Weng, J.P. Endothelial dysfunction in COVID-19: An overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kanamarlapudi, V. Exchange factor EFA6R requires C-terminal targeting to the plasma membrane to promote cytoskeletal rearrangement through the activation of ADP-ribosylation factor 6 (ARF6). J. Biol. Chem. 2014, 289, 33378–33390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenchik, N.I.; Desiderio, D.M.; Gerling, I.C. Two-dimensional gel electrophoresis characterization of the mouse leukocyte proteome, using a tri-reagent for protein extraction. Proteomics 2005, 5, 2202–2209. [Google Scholar] [CrossRef]
- Thompson, A.; Kanamarlapudi, V. The regions within the N-terminus critical for human glucagon like peptide-1 receptor (hGLP-1R) cell surface expression. Sci. Rep. 2014, 4, 7410. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Kanamarlapudi, V. Molecular Analysis of SARS-CoV-2 Spike Protein-Induced Endothelial Cell Permeability and vWF Secretion. Int. J. Mol. Sci. 2023, 24, 5664. https://doi.org/10.3390/ijms24065664
Guo Y, Kanamarlapudi V. Molecular Analysis of SARS-CoV-2 Spike Protein-Induced Endothelial Cell Permeability and vWF Secretion. International Journal of Molecular Sciences. 2023; 24(6):5664. https://doi.org/10.3390/ijms24065664
Chicago/Turabian StyleGuo, Yuexi, and Venkateswarlu Kanamarlapudi. 2023. "Molecular Analysis of SARS-CoV-2 Spike Protein-Induced Endothelial Cell Permeability and vWF Secretion" International Journal of Molecular Sciences 24, no. 6: 5664. https://doi.org/10.3390/ijms24065664
APA StyleGuo, Y., & Kanamarlapudi, V. (2023). Molecular Analysis of SARS-CoV-2 Spike Protein-Induced Endothelial Cell Permeability and vWF Secretion. International Journal of Molecular Sciences, 24(6), 5664. https://doi.org/10.3390/ijms24065664