
Estrogen Receptor β4 Regulates Chemotherapy Resistance and Induces Cancer Stem Cells in Triple Negative Breast Cancer
 ,
, 
Abstract
:1. Introduction
2. Results
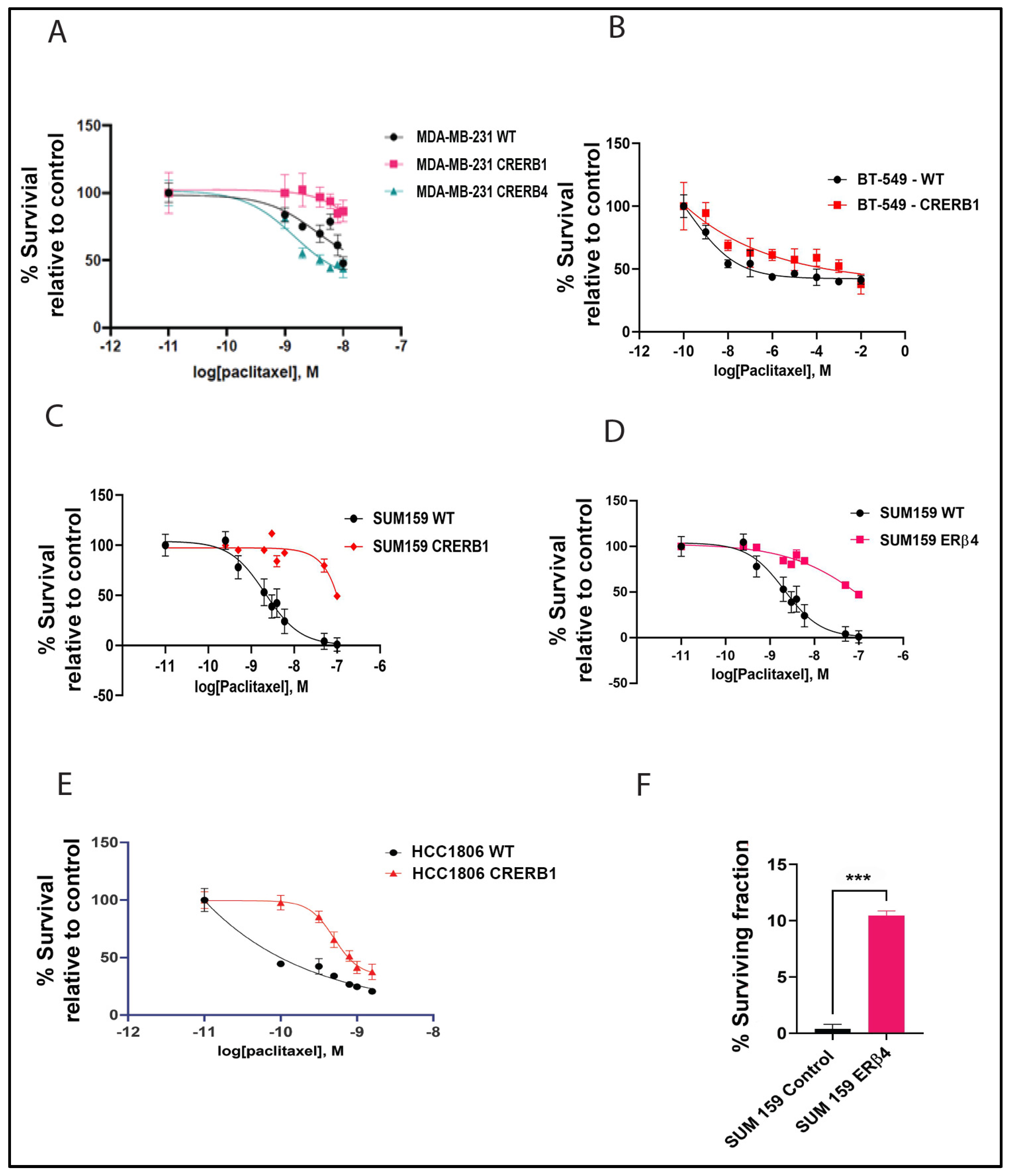
2.1. ERβ Isoforms Endogenously Expressed in TNBC Cell Lines Affect Response to Chemotherapy
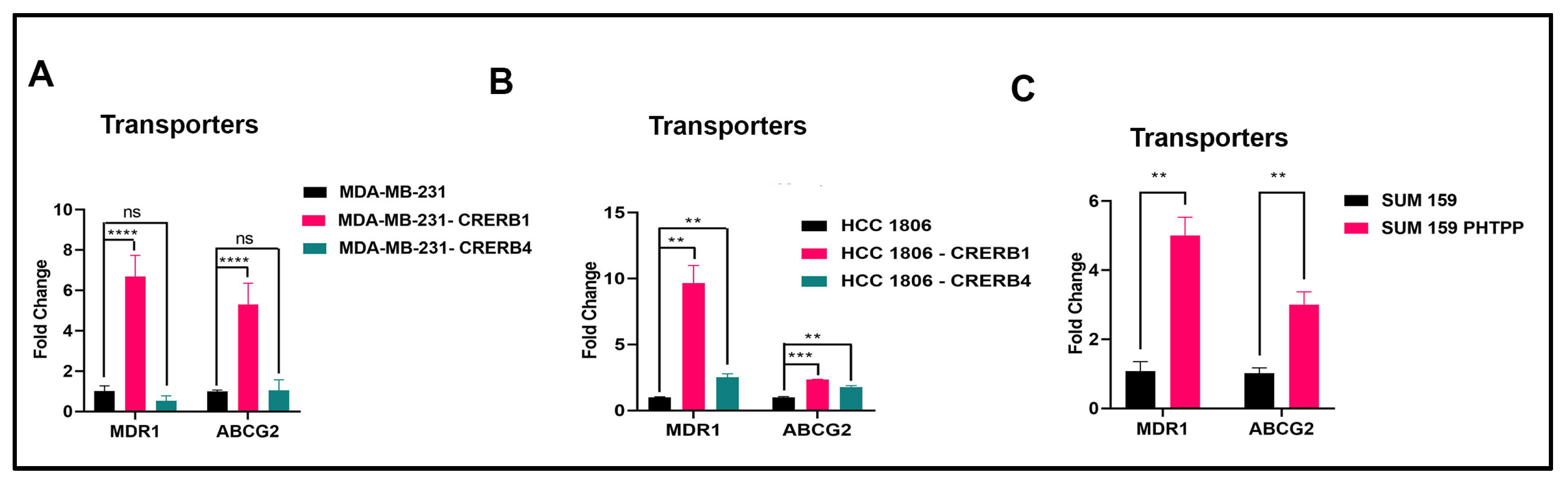
2.2. Specific Truncation of Endogenous ERβ1 LBD or Treatment with the ERβ1 Antagonist PHTPP Increase Expression of ABC Transporters
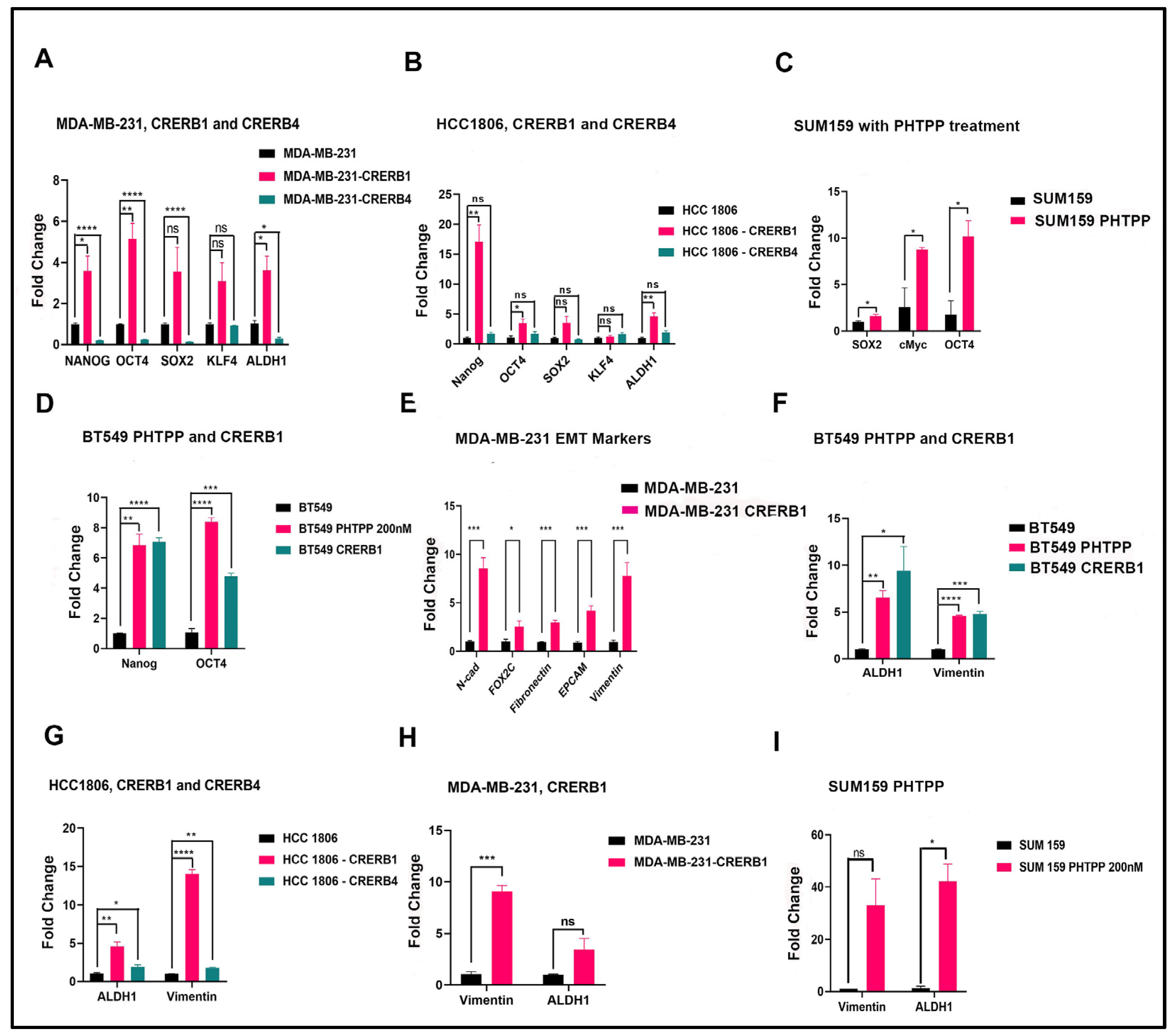
2.3. Truncation of Endogenous ERβ1 LBD or Treatment with the ERβ1 Antagonist PHTPP Induces Expression of Pluripotency Factors in MDA-MB-231, HCC1806, BT549, and SUM159 Cells
2.4. Increased Cancer Stem Cell Population in Cell Lines Treated with ERβ1 Antagonist
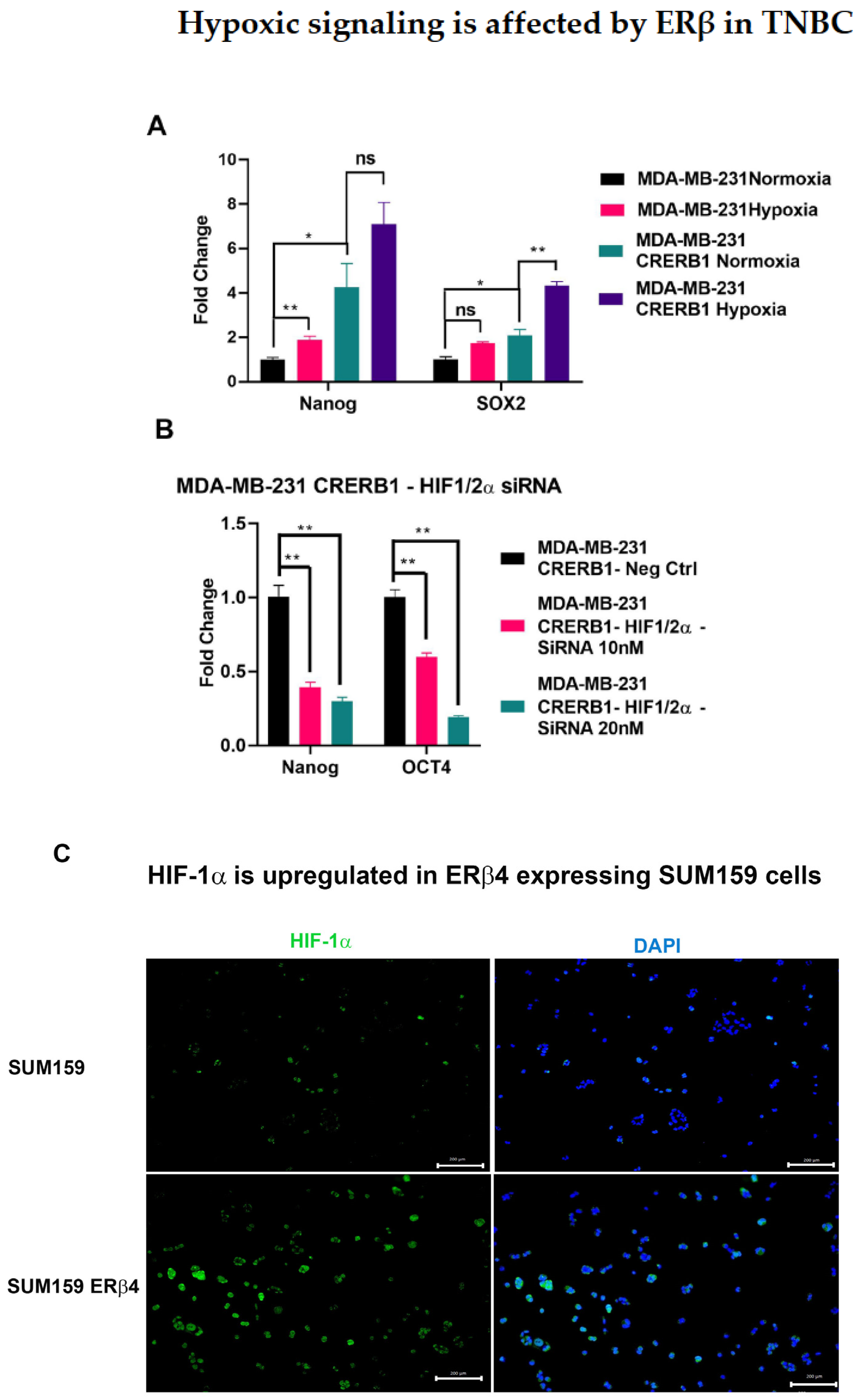
2.5. Hypoxia Inducible Factors HIF-1α and HIF-2α Are Involved in Mediating the Regulation of Chemo Resistance and Cancer Stem Cell Population in TNBC
3. Discussion
3.1. ERβ1 and ERβ4 Modulate the Chemotherapy Sensitivity via the Drug Efflux Transporters
3.2. ERβ1 and ERβ4 Regulate Stemness of Cancer Cells and Promote Epithelial to Mesenchymal Transition
3.3. Increased Frequency of BCSC Population with ERβ1 Antagonist Treatment
3.4. HIFs Mediate the Cancer Cell Stemness
4. Materials and Methods
4.1. Cell Lines and Materials
4.2. Preparation of RNA and qPCR
| 36B4 | F, 5′—GTGTTCGACAATGGCAGCAT—3′ |
| R, 5′—GACACCCTCCAGGAAGCGA—3′ (reference gene) | |
| ALDH1A | F, 5′—GCTGGCGACAATGGAGTCAA—3′ |
| R, 5′—ACGGCCCTGGATCTTGTCAG—3′ | |
| OCT4 | F, 5′—GGATGCTGTGAGCCAAGG—3′ |
| R, 5′—GAACAATGATGAGTGACAGACAG—3′ | |
| CAIX | F, 5′—AGATGAGAAGGCAGCACAGAA—3′ |
| R, 5′—GAAGTGGCATAATGAGCAGGA—3′ | |
| c-Myc | F, 5′—CTGGTGCTCCATGAGGAGAC—3′ |
| R, 5′—CTTTTCCACAGAAACAACATC—3′ | |
| SOX2 | F, 5′—TTGCTGCCTCTTTAAGACTAGGA—3′ |
| R, 5′—CTGGGGCTCAAACTTCTCTC—3′ | |
| Nanog | F, 5′—ACCCAGCTGTGTGTACTCAA—3′ |
| R, 5′—GGAAGAGTAAAGGCTGGGGT—3′ | |
| KLF4 | F, 5′—CGGACATCAACGACGTGAG—3′ |
| R, 5′—GACGCCTTCAGCACGAACT—3′ | |
| Vimentin | F, 5′—TCGTTTCGAGGTTTTCGCGTT—3′ |
| R, 5′—CGACTAAAACTCGACCGACTC—3′ | |
| MDR-1 | F, 5′—AGGAAGCCAATGCCTATGACTTTA—3′ |
| R, 5′—CAACTGGGCCCCTCTCTCTC—3′ | |
| ABCG2 | F, 5′—TTTCCAAGCGTTCATTCAAAAA—3′ |
| R, 5′—TACGACTGTGACAATGATCTGAGC—3′ | |
| ERβ4 | F, 5′—ACTTGCTGAACGCCGTGACC—3′ |
| R, 5′—TTTTCTCCCCATCTCGCATGC—3′ | |
| Fibronectin | F, 5′—AAACTTGCATCTGGAGGCAAACCC—3′ |
| R, 5′—AGCTCTGATCAGCATGGACCACTT | |
| N-Cadherin | F, 5′—GGTGGAGGAGAAGAAGACCAG—3′ |
| R,5′—GGCATCAGGCTCCACAGT—3′ | |
| EpCAM | F, 5′—CTGCCAAATGTTTGGTGATG—3′ |
| R, 5′—ACGCGTTGTGATCTCCTTCT—3′ | |
| FOXC2 | F, 5′—CCGTCTCGGAAGCAGCAT—3′ |
| R, 5′—TGAGCGCGATGTAGCTGTAG—3′ |
4.3. CRISPR/Cas9 Manipulation of Cell Lines to Cause LBD Truncation of Endogenous ERβ1 and Mutation of ERβ4 5′ of ERβ4 Unique Exon Splice Site
| ERβ LBD truncation | F, 5—CACCGCAACATGAAGTGCAAAAATG—3′ |
| R, 5—AAACCATTTTTGCACTTCATGTTGC—3′ | |
| ERβ4 knock down | F, 5′—CACCGCTTTTCTCCCCATCTGTAA—3′ |
| R, 5′—AAACTTACAGATGGGGAGAAAAGC—3′ | |
| Control | F, 5′—CACCGTATTACTGATATTGGTGGG—3′ |
| R, 5′—AAACCCCACCAATATCAGTAATAC—3′ |
4.4. DsiRNA for ERβ1
4.5. ERβ4 Ectopic Expression
4.6. Cytotoxicity MTS Assay
4.7. Clonogenic Survival Assay
4.8. Immuno Cyto Chemistry
4.9. FACS Analysis
4.10. Aldefluor Assay
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; Fluge, O.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertucci, F.; Finetti, P.; Cervera, N.; Esterni, B.; Hermitte, F.; Viens, P.; Birnbaum, D. How basal are triple-negative breast cancers? Int. J. Cancer 2008, 123, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Almansour, N.M. Triple-Negative Breast Cancer: A Brief Review About Epidemiology, Risk Factors, Signaling Pathways, Treatment and Role of Artificial Intelligence. Front. Mol. Biosci. 2022, 9, 836417. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.-Y.; Chang, C.-J.; Cheng, J.-S. Survival, treatment regimens and medical costs of women newly diagnosed with metastatic triple-negative breast cancer. Sci. Rep. 2022, 12, 729. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Strietz, J.; Bleilevens, A.; Stickeler, E.; Maurer, J. Chemotherapeutic Stress Influences Epithelial–Mesenchymal Transition and Stemness in Cancer Stem Cells of Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2020, 21, 404. [Google Scholar] [CrossRef] [Green Version]
- Bos, R.; van der Groep, P.; Greijer, A.E.; Shvarts, A.; Meijer, S.; Pinedo, H.M.; Semenza, G.L.; van Diest, P.; van der Wall, E. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer 2003, 97, 1573–1581. [Google Scholar] [CrossRef]
- Mathieu, J.; Zhang, Z.; Zhou, W.; Wang, A.J.; Heddleston, J.M.; Pinna, C.M.A.; Hubaud, A.; Stadler, B.; Choi, M.; Bar, M.; et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011, 71, 4640–4652. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wei, Q.; Li, Q.; Zhang, B.; Xiao, Q. Down-regulating HIF-1alpha by lentivirus-mediated shRNA for therapy of triple negative breast cancer. Cancer Biol. Ther. 2015, 16, 866–875. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Gilkes, D.M.; Xiang, L.; Chaturvedi, P.; Semenza, G.L. Enrichment of Breast Cancer Stem Cells following Cytotoxic Chemotherapy is Mediated by Hypoxia-Inducible Factors. bioRxiv 2022. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [Green Version]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.A.; Simon, M.C.; Keith, B. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef] [Green Version]
- Ezashi, T.; Das, P.; Roberts, R.M. Low O2 tensions and the prevention of differentiation of hES cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4783–4788. [Google Scholar] [CrossRef] [Green Version]
- Takubo, K.; Suda, T. Roles of the hypoxia response system in hematopoietic and leukemic stem cells. Int. J. Hematol. 2012, 95, 478–483. [Google Scholar] [CrossRef] [Green Version]
- Yong, L.; Tang, S.; Yu, H.; Zhang, H.; Zhang, Y.; Wan, Y.; Cai, F. The role of hypoxia-inducible factor-1 alpha in multidrug-resistant breast cancer. Front. Oncol. 2022, 12, 964934. [Google Scholar] [CrossRef]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.-A. Differential expression of estrogen receptor alpha, beta1, and beta2 in lobular and ductal breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef] [Green Version]
- Skliris, G.P.; Carder, P.J.; Lansdown, M.R.; Speirs, V. Immunohistochemical detection of ERbeta in breast cancer: Towards more detailed receptor profiling? Br. J. Cancer 2001, 84, 1095–1098. [Google Scholar] [CrossRef] [Green Version]
- Inic, Z.; Zegarac, M.; Inic, M.; Maekovic, I.; Kozomara, Z.; Djurisic, I.; Inic, I.; Pupic, G.; Jancic, S. Difference between Luminal A and Luminal B Subtypes According to Ki-67, Tumor Size, and Progesterone Receptor Negativity Providing Prognostic Information. Clin. Med. Insights Oncol. 2014, 8, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Moore, J.T.; McKee, D.D.; Slentz-Kesler, K.; Moore, L.B.; Jones, S.A.; Horne, E.L.; Su, J.L.; Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Cloning and characterization of human estrogen receptor beta isoforms. Biochem. Biophys. Res. Commun. 1998, 247, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Girault, I.; Andrieu, C.; Tozlu, S.; Spyratos, F.; Bièche, I.; Lidereau, R. Altered expression pattern of alternatively spliced estrogen receptor β transcripts in breast carcinoma. Cancer Lett. 2004, 215, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Poola, I.; Abraham, J.; Baldwin, K.; Saunders, A.; Bhatnagar, R. Estrogen receptors beta4 and beta5 are full length functionally distinct ERβ isoforms. Endocrine 2005, 27, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The dynamic structure of the estrogen receptor. J. Amino Acids 2011, 2011, 812540. [Google Scholar] [CrossRef] [Green Version]
- Leung, Y.K.; Mak, P.; Hassan, S.; Ho, S.M. Estrogen receptor (ER)-beta isoforms: A key to understanding ER-beta signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13162–13167. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Lam, E.W.; Sunters, A.; Enmark, E.; De Bella, M.T.; Charles Coombes, R.; Gustafsson, J.-A.; Dahlman-Wright, K. Expression of estrogen receptor beta isoforms in normal breast epithelial cells and breast cancer: Regulation by methylation. Oncogene 2003, 22, 7600–7606. [Google Scholar] [CrossRef] [Green Version]
- Božović, A.; Markićević, M.; Dimitrijević, B.; Ćupić, S.J.; Krajnović, M.; Lukić, S.; Mandušić, V. Potential clinical significance of ERβ ON promoter methylation in sporadic breast cancer. Med. Oncol. 2013, 30, 642. [Google Scholar] [CrossRef]
- Zhu, X.; Leav, I.; Leung, Y.K.; Wu, M.; Liu, Q.; Gao, Y.; McNeal, J.E.; Ho, S.-M. Dynamic regulation of estrogen receptor-beta expression by DNA methylation during prostate cancer development and metastasis. Am. J. Pathol. 2004, 164, 2003–2012. [Google Scholar] [CrossRef]
- Zhang, X.; Leung, Y.K.; Ho, S.M. AP-2 regulates the transcription of estrogen receptor (ER)-beta by acting through a methylation hotspot of the 0N promoter in prostate cancer cells. Oncogene 2007, 26, 7346–7354. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Dey, P.; Ziegler, Y.; Jiao, X.; Kim, S.H.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Contrasting activities of estrogen receptor beta isoforms in triple negative breast cancer. Breast Cancer Res. Treat. 2021, 185, 281–292. [Google Scholar] [CrossRef]
- Dey, P.; Jonsson, P.; Hartman, J.; Williams, C.; Strom, A.; Gustafsson, J.A. Estrogen receptors beta1 and beta2 have opposing roles in regulating proliferation and bone metastasis genes in the prostate cancer cell line PC3. Mol. Endocrinol. 2012, 26, 1991–2003. [Google Scholar] [CrossRef]
- Faria, M.; Karami, S.; Granados-Principal, S.; Dey, P.; Verma, A.; Choi, D.S.; Elemento, O.; Bawa-Khalfe, T.; Chang, J.C.; Strom, A.M.; et al. The ERbeta4 variant induces transformation of the normal breast mammary epithelial cell line MCF-10A; the ERbeta variants ERbeta2 and ERbeta5 increase aggressiveness of TNBC by regulation of hypoxic signaling. Oncotarget 2018, 9, 12201–12211. [Google Scholar] [CrossRef] [Green Version]
- Mal, R.; Magner, A.; David, J.; Datta, J.; Vallabhaneni, M.; Kassem, M.; Manouchehri, J.; Willingham, N.; Stover, D.; Vandeusen, J.; et al. Estrogen Receptor Beta (ERbeta): A Ligand Activated Tumor Suppressor. Front. Oncol. 2020, 10, 587386. [Google Scholar] [CrossRef]
- Dey, P.; Velazquez-Villegas, L.A.; Faria, M.; Turner, A.; Jonsson, P.; Webb, P.; Williams, C.; Gustafsson, J.-A.; Ström, A.M. Estrogen Receptor beta2 Induces Hypoxia Signature of Gene Expression by Stabilizing HIF-1alpha in Prostate Cancer. PLoS ONE 2015, 10, e0128239. [Google Scholar]
- Compton, D.R.; Sheng, S.; Carlson, K.E.; Rebacz, N.A.; Lee, I.Y.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Pyrazolo[1,5-a]pyrimidines: Estrogen receptor ligands possessing estrogen receptor beta antagonist activity. J. Med. Chem. 2004, 47, 5872–5893. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.H.; Wientjes, M.G.; Au, J.L. Kinetics of P-glycoprotein-mediated efflux of paclitaxel. J. Pharmacol. Exp. Ther. 2001, 298, 1236–1242. [Google Scholar] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Duan, J.-J.; Bian, X.-W.; Yu, S.-C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Lemesle, M.; Geoffroy, M.; Alpy, F.; Tomasetto, C.L.; Kuntz, S.; Grillier-Vuissoz, I. CLDN1 Sensitizes Triple-Negative Breast Cancer Cells to Chemotherapy. Cancers 2022, 14, 5026. [Google Scholar] [CrossRef]
- Spratlin, J.; Sawyer, M.B. Pharmacogenetics of paclitaxel metabolism. Crit. Rev. Oncol. Hematol. 2007, 61, 222–229. [Google Scholar] [CrossRef]
- Mukhopadhyay, U.K.; Oturkar, C.C.; Adams, C.; Wickramasekera, N.; Bansal, S.; Medisetty, R.; Miller, A.; Swetzig, W.M.; Silwal-Pandit, L.; Børresen-Dale, A.-L.; et al. TP53 Status as a Determinant of Pro- vs Anti-Tumorigenic Effects of Estrogen Receptor-Beta in Breast Cancer. JNCI J. Natl. Cancer Inst. 2019, 111, 1202–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, M.; Shepherd, P.; Pan, Y.; Chatterjee, S.S.; Navone, N.; Gustafsson, J.-A.; Storm, A. The estrogen receptor variants beta2 and beta5 induce stem cell characteristics and chemotherapy resistance in prostate cancer through activation of hypoxic signaling. Oncotarget 2018, 9, 36273–36288. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Hao, Q.; Lu, H. Mutant p53 in cancer therapy-the barrier or the path. J. Mol. Cell Biol. 2019, 11, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Conde, I.; Ribeiro, A.S.; Paredes, J. Breast Cancer Stem Cell Membrane Biomarkers: Therapy Targeting and Clinical Implications. Cells 2022, 11, 934. [Google Scholar] [CrossRef]
- Mohan, A.; Raj, R.R.; Mohan, G.; Padmaja, K.P.; Maliekal, T.T. Reporters of Cancer Stem Cells as a Tool for Drug Discovery. Front Oncol. 2021, 11, 669250. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liu, Y.; Deng, L.; Wang, D.; He, X.; Zhou, L.; Wicha, M.S.; Bai, F.; Liu, S. Transcriptional profiles of different states of cancer stem cells in triple-negative breast cancer. Mol. Cancer 2018, 17, 65. [Google Scholar] [CrossRef] [PubMed]
- Kuburich, N.A.; den Hollander, P.; Pietz, J.T.; Mani, S.A. Vimentin and cytokeratin: Good alone, bad together. Semin. Cancer Biol. 2021, 86, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018, 16, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alison, M.R.; Lin, W.-R.; Lim, S.M.L.; Nicholson, L.J. Cancer stem cells: In the line of fire. Cancer Treat. Rev. 2012, 38, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Wang, A.; Ziegler, Y.; Kumar, S.; Yan, S.; Kim, S.H.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Estrogen Receptor Beta 1: A Potential Therapeutic Target for Female Triple Negative Breast Cancer. Endocrinology 2022, 163, 172. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; MH, H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Wicha, M.S. Cancer stem cells and metastasis: Lethal seeds. Clin. Cancer Res. 2006, 12, 5606–5607. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, G.G.; Deshmukh, A.P.; den Hollander, P.; Luo, M.; Soundararajan, R.; Jia, D.; Levine, H.; Mani, S.A.; Wicha, M.S. Breast cancer dormancy: Need for clinically relevant models to address current gaps in knowledge. NPJ Breast Cancer 2021, 7, 66. [Google Scholar] [CrossRef]
- Hadjimichael, C.; Chanoumidou, K.; Papadopoulou, N.; Arampatzi, P.; Papamatheakis, J.; Kretsovali, A. Common stemness regulators of embryonic and cancer stem cells. World J. Stem Cells 2015, 7, 1150–1184. [Google Scholar]
- Mohan, A.; Raj Rajan, R.; Mohan, G.; Kollenchery Puthenveettil, P.; Maliekal, T.T. Markers and Reporters to Reveal the Hierarchy in Heterogeneous Cancer Stem Cells. Front. Cell Dev. Biol. 2021, 9, 668851. [Google Scholar] [CrossRef]
- Pádua, D.; Barros, R.; Amaral, A.L.; Mesquita, P.; Freire, A.F.; Sousa, M.; Maia, A.F.; Caiado, I.; Fernandes, H.; Pombinho, A.; et al. A SOX2 Reporter System Identifies Gastric Cancer Stem-Like Cells Sensitive to Monensin. Cancers 2020, 12, 495. [Google Scholar] [CrossRef] [Green Version]
- Tang, B.; Raviv, A.; Esposito, D.; Flanders, K.C.; Daniel, C.; Nghiem, B.T.; Garfield, S.; Lim, L.; Mannan, P.; Robles, A.I.; et al. A flexible reporter system for direct observation and isolation of cancer stem cells. Stem Cell Rep. 2015, 4, 155–169. [Google Scholar] [CrossRef] [Green Version]
- Wiechert, A.; Saygin, C.; Thiagarajan, P.S.; Rao, V.S.; Hale, J.S.; Gupta, N.; Hitomi, M.; Nagaraj, A.B.; DiFeo, A.; Lathia, J.D.; et al. Cisplatin induces stemness in ovarian cancer. Oncotarget 2016, 7, 30511–30522. [Google Scholar] [CrossRef] [Green Version]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 95, S2–S7. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Sarmiento-Castro, A.; Caamaño-Gutiérrez, E.; Sims, A.H.; Hull, N.J.; James, M.I.; Santiago-Gómez, A.; Eyre, R.; Clark, C.; Brown, M.E.; Brooks, M.D.; et al. Increased Expression of Interleukin-1 Receptor Characterizes Anti-estrogen-Resistant ALDH(+) Breast Cancer Stem Cells. Stem Cell Rep. 2020, 15, 307–316. [Google Scholar] [CrossRef]
- Chen, J.; Ding, Z.; Peng, Y.; Pan, F.; Li, J.; Zou, L.; Zhang, Y.; Liang, H. HIF-1alpha inhibition reverses multidrug resistance in colon cancer cells via downregulation of MDR1/P-glycoprotein. PLoS ONE 2014, 9, e98882. [Google Scholar]
- Yoshida, Y.; Takahashi, K.; Okita, K.; Ichisaka, T.; Yamanaka, S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell 2009, 5, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Hartman, J.; Lindberg, K.; Morani, A.; Inzunza, J.; Strom, A.; Gustafsson, J.A. Estrogen receptor beta inhibits angiogenesis and growth of T47D breast cancer xenografts. Cancer Res. 2006, 66, 11207–11213. [Google Scholar] [CrossRef] [Green Version]
- Mak, P.; Chang, C.; Pursell, B.; Mercurio, A.M. Estrogen receptor beta sustains epithelial differentiation by regulating prolyl hydroxylase 2 transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 4708–4713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, P.; Leav, I.; Pursell, B.; Bae, D.; Yang, X.; Taglienti, C.A.; Gouvin, L.M.; Sharma, V.M.; Mercurio, A.M. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: Implications for Gleason grading. Cancer Cell 2010, 17, 319–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Lathrop, K.I.; Kaklamani, V.G.; Brenner, A.J.; Li, R.; Nazarullah, A.; Hackman, S.; Thomas, C.; Gelfond, J.; Rodriguez, M.; Elledge, R. Novel estrogen receptor beta agonist S-equol decreases tumor proliferation in patients with triple negative breast cancer (TNBC). J. Clin. Oncol. 2020, 38, 560. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lehmann TNBC Subtype [41] | Genetic Abnormalities [41] | Cell Line [35] | CLDN-1 Status | Gene Mutations [35] |
|---|---|---|---|---|
| Basal-like 2 | Growth factor-signaling pathways (EGFR, MET, NGF, Wnt/β-catenin, IGF-1R), Glycolysis, gluconeogenesis, Expression of myoepithelial markers | HCC1806 | High | TP53; CDKN2A; UTX |
| Mesenchymal-like | Cell motility, Cell differentiation Growth factor signaling EMT | BT-549 | Low | TP53; PTEN; RB1 |
| Mesenchymal stem-like | Similar to M above + Low proliferation, Angiogenesis genes | SUM159PT | Low | TP53; PIK3CA |
| MDA-MB-231 | Low | TP53; BRAF; CDKN2A; KRAS; NF2; PDGFRA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bano, A.; Stevens, J.H.; Modi, P.S.; Gustafsson, J.-Å.; Strom, A.M. Estrogen Receptor β4 Regulates Chemotherapy Resistance and Induces Cancer Stem Cells in Triple Negative Breast Cancer. Int. J. Mol. Sci. 2023, 24, 5867. https://doi.org/10.3390/ijms24065867
Bano A, Stevens JH, Modi PS, Gustafsson J-Å, Strom AM. Estrogen Receptor β4 Regulates Chemotherapy Resistance and Induces Cancer Stem Cells in Triple Negative Breast Cancer. International Journal of Molecular Sciences. 2023; 24(6):5867. https://doi.org/10.3390/ijms24065867
Chicago/Turabian StyleBano, Ayesha, Jessica H. Stevens, Paulomi S. Modi, Jan-Åke Gustafsson, and Anders M. Strom. 2023. "Estrogen Receptor β4 Regulates Chemotherapy Resistance and Induces Cancer Stem Cells in Triple Negative Breast Cancer" International Journal of Molecular Sciences 24, no. 6: 5867. https://doi.org/10.3390/ijms24065867