


Systematic Selection Signature Analysis of Chinese Gamecocks Based on Genomic and Transcriptomic Data

Abstract
:1. Introduction
2. Results
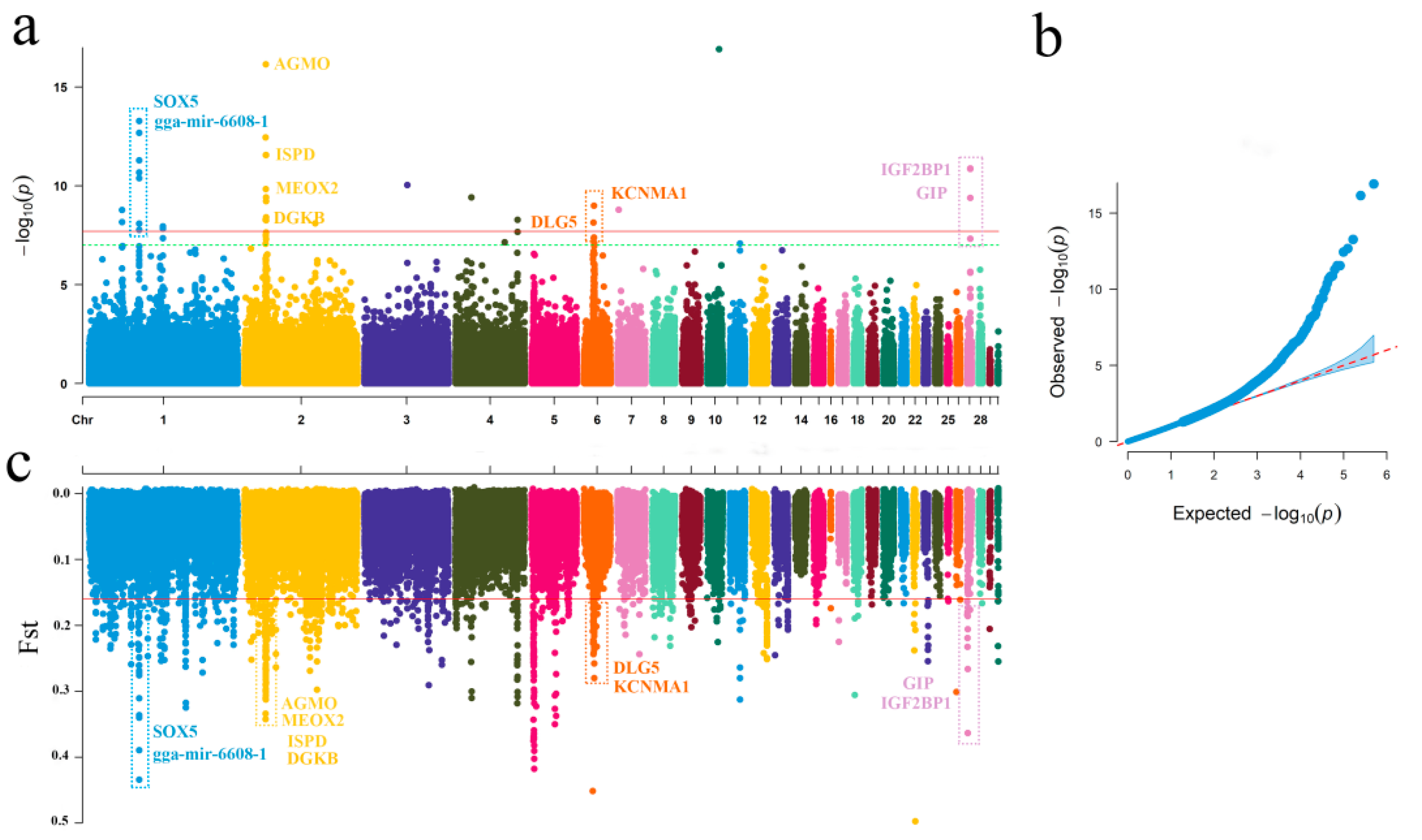
2.1. Genome-Wide Putatively Selective Signatures in Gamecocks
2.2. Overview of the RNA Sequencing Data
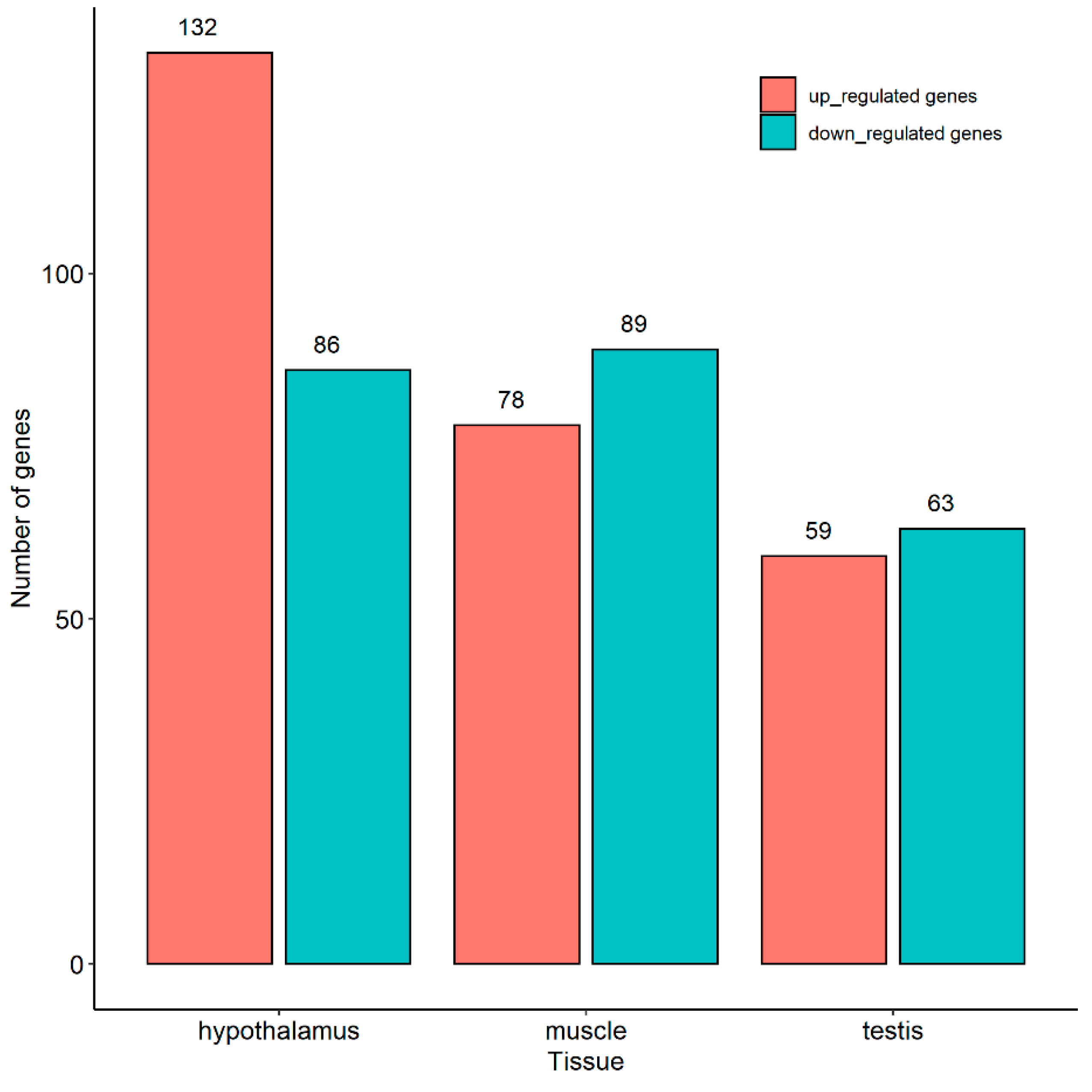
2.3. Identification of Differentially Expressed Genes
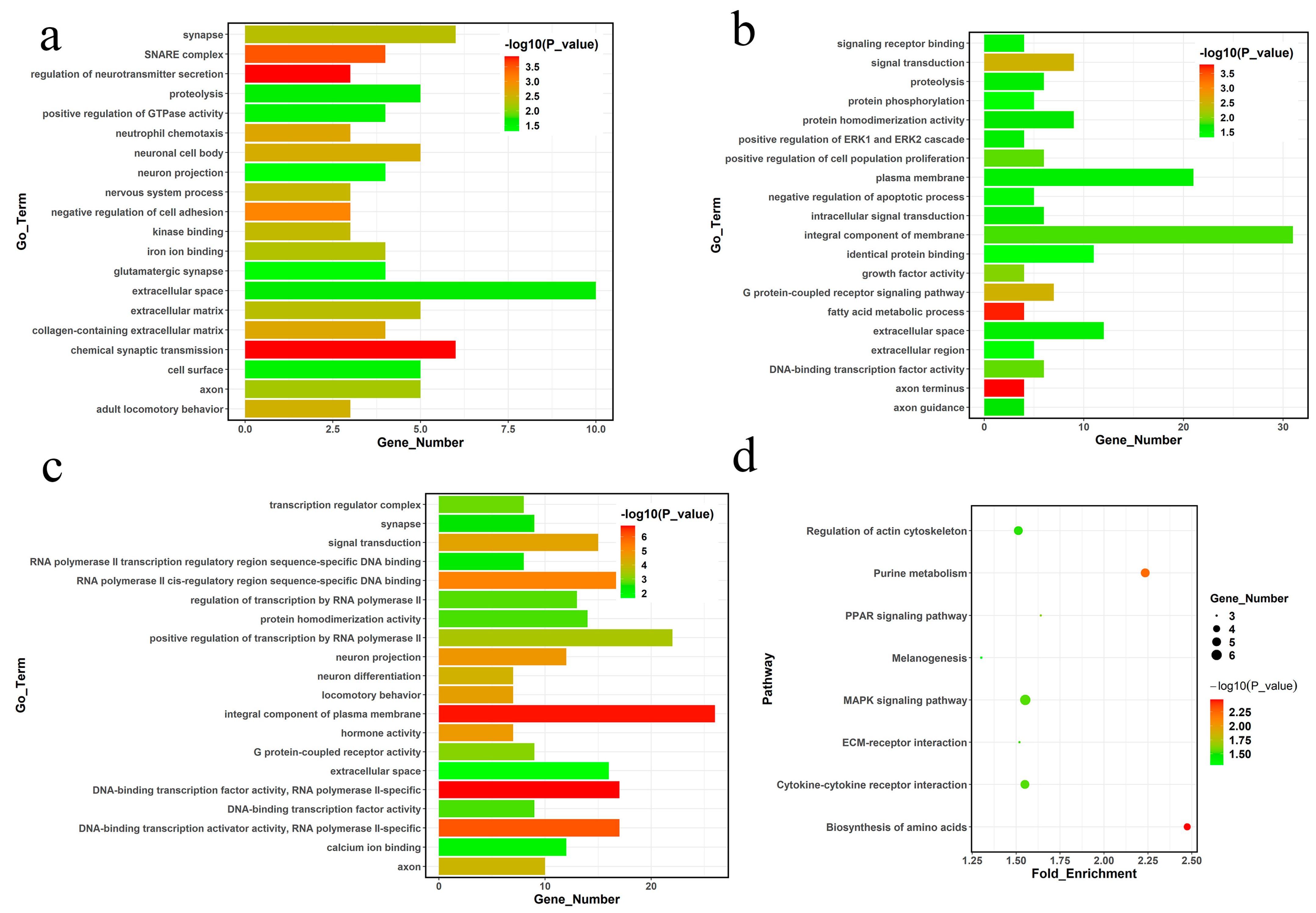
2.4. Functional Annotation of Differentially Expressed Genes
3. Discussion
4. Materials and Methods
4.1. Sample Collection and SNP Genotyping
4.2. Genome-Wide Association Studies
4.3. Genome-Wide Selective Sweep Analysis
4.4. RNA Sequencing
4.5. Analysis of Differentially Expressed Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qanbari, S.; Seidel, M.; Strom, T.M.; Mayer, K.F.; Preisinger, R.; Simianer, H. Parallel Selection Revealed by Population Sequencing in Chicken. Genome Biol. Evol. 2015, 7, 3299–3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rostamzadeh Mahdabi, E.; Esmailizadeh, A.; Ayatollahi Mehrgardi, A.; Asadi Fozi, M. A genome-wide scan to identify signatures of selection in two Iranian indigenous chicken ecotypes. Genet. Sel. Evol. 2021, 53, 72. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R. Molecular signatures of natural selection. Annu. Rev. Genet. 2005, 39, 197–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qanbari, S.; Simianer, H. Mapping signatures of positive selection in the genome of livestock. Livest. Sci. 2014, 166, 133–143. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Zhu, Q.; Yao, Y.-G. Genetic relationship of Chinese and Japanese gamecocks revealed by mtDNA sequence variation. Biochem. Genet. 2006, 44, 19–29. [Google Scholar] [CrossRef]
- Endo, H.; Mori, K.; Hosojima, M.; Mekwichai, W.; Ogawa, H.; Tsunekawa, N.; Yamasaki, T.; Hayashi, Y.; Akishinonomiya, F. Functional-morphological characteristics in the musculoskeletal system of standing-type cocks including some game breeds. Jpn. J. Zoo Wildl. Med. 2012, 17, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Amos, W.; Driscoll, E.; Hoffman, J.I. Candidate genes versus genome-wide associations: Which are better for detecting genetic susceptibility to infectious disease? Proc. Biol. Sci. 2011, 278, 1183–1188. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Zou, L.; Sun, T.; Xu, W.; Zeng, L.; Jia, Y.; Jiang, J.; Deng, J.; Yang, X. Genome-Wide Association Study Using Whole-Genome Sequencing Identifies a Genomic Region on Chromosome 6 Associated With Comb Traits in Nandan-Yao Chicken. Front. Genet. 2021, 12, 682501. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Luo, C.; Wang, M.; Guo, L.; Chen, X.; Li, Z.; Zheng, M.; Folaniyi, B.S.; Luo, W.; Shu, D.; et al. Genome diversity of Chinese indigenous chicken and the selective signatures in Chinese gamecock chicken. Sci. Rep. 2020, 10, 14532. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tu, Y.; Zhang, M.; Ji, G.; Wang, K.; Shan, Y.; Ju, X.; Zhang, D.; Shu, J.; Zou, J. Identification of molecular pathways and candidate genes associated with cocks’ comb size trait by genome-wide transcriptome analysis. Sci. Rep. 2018, 8, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Liu, A.; Wang, Q.; Wang, H.; Dong, D.; Liu, L. Transcriptome analysis of embryonic muscle development in Chengkou Mountain Chicken. BMC Genom. 2021, 22, 431. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Wang, Y.; Shu, D.; Qu, H.; Luo, C.; Hu, X. Food intake-related genes in chicken determined through combinatorial genome-wide association study and transcriptome analysis. Anim. Genet. 2020, 51, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, J.; Ma, C.; Wang, Z.; Chen, H.; Su, H.; Wan, Y.; Jiang, R. Genome-wide re-sequencing and transcriptome analysis reveal candidate genes associated with the pendulous comb phenotype in domestic chickens. Anim. Sci. J. 2020, 91, e13308. [Google Scholar] [CrossRef]
- Liu, X.; Wu, Z.; Li, J.; Bao, H.; Wu, C. Genome-Wide Association Study and Transcriptome Differential Expression Analysis of the Feather Rate in Shouguang Chickens. Front. Genet. 2020, 11, 613078. [Google Scholar] [CrossRef]
- Guo, X.; Fang, Q.; Ma, C.; Zhou, B.; Wan, Y.; Jiang, R. Whole-genome resequencing of Xishuangbanna fighting chicken to identify signatures of selection. Genet. Sel. Evol. 2016, 48, 62. [Google Scholar] [CrossRef] [Green Version]
- Rietveld, C.A.; Medland, S.E.; Derringer, J.; Yang, J.; Esko, T.; Martin, N.W.; Westra, H.J.; Shakhbazov, K.; Abdellaoui, A.; Agrawal, A.; et al. GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. Science 2013, 340, 1467–1471. [Google Scholar] [CrossRef] [Green Version]
- Hormozdiari, F.; van de Bunt, M.; Segrè, A.V.; Li, X.; Joo, J.W.J.; Bilow, M.; Sul, J.H.; Sankararaman, S.; Pasaniuc, B.; Eskin, E. Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet. 2016, 99, 1245–1260. [Google Scholar] [CrossRef] [Green Version]
- Ghoreishifar, S.M.; Eriksson, S. Signatures of selection reveal candidate genes involved in economic traits and cold acclimation in five Swedish cattle breeds. Genet Sel Evol. 2020, 521, 52. [Google Scholar] [CrossRef]
- González-Rodríguez, A.; Munilla, S.; Mouresan, E.F.; Cañas-Álvarez, J.J.; Díaz, C.; Piedrafita, J. On the performance of tests for the detection of signatures of selection: A case study with the Spanish autochthonous beef cattle populations. Genet. Sel. Evol. 2016, 481, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundu, S.; Nandhu, M.S.; Longo, S.L.; Longo, J.A.; Rai, S.; Chin, L.S.; Richardson, T.E.; Viapiano, M.S. The scaffolding protein DLG5 promotes glioblastoma growth by controlling Sonic Hedgehog signaling in tumor stem cells. Neuro. Oncol. 2022, 24, 1230–1242. [Google Scholar] [CrossRef]
- Wright, D.; Boije, H.; Meadows, J.R.S.; Bed’hom, B.; Gourichon, D.; Vieaud, A.; Tixier-Boichard, M.; Rubin, C.-J.; Imsland, F.; Hallböök, F.; et al. Copy number variation in intron 1 of SOX5 causes the Pea-comb phenotype in chickens. PLoS Genet. 2009, 5, e1000512. [Google Scholar] [CrossRef] [PubMed]
- Pallotta, B.S.; Magleby, K.L.; Barrett, J.N. Single channel recordings of Ca2+-activated K+ currents in rat muscle cell culture. Nature 1981, 293, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Squire, L.G.; Petersen, O.H. Modulation of Ca2+- and voltage-activated K+ channels by internal Mg2+ in salivary acinar cells. Biochim. Biophys Acta 1987, 899, 171–175. [Google Scholar] [CrossRef]
- Schubert, R.; Krien, U.; Gagov, H. Protons inhibit the BK(Ca) channel of rat small artery smooth muscle cells. J. Vasc. Res. 2001, 38, 30–38. [Google Scholar] [CrossRef]
- Reijntjes, S.; Stricker, S.; Mankoo, B.S. A comparative analysis of Meox1 and Meox2 in the developing somites and limbs of the chick embryo. Int. J. Dev. Biol. 2007, 51, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, L.; Zhu, Y.-H.; Zhang, Y.-E.; Zheng, F.; Yang, J.-Y.; Guo, L.-Y.; Li, X.-Y.; Wang, L.; Tang, J.-M.; et al. Mesoderm/mesenchyme homeobox gene l promotes vascular smooth muscle cell phenotypic modulation and vascular remodeling. Int. J. Cardiol. 2018, 251, 82–89. [Google Scholar] [CrossRef]
- Cirak, S.; Foley, A.R.; Herrmann, R.; Willer, T.; Yau, S.; Stevens, E.; Torelli, S.; Brodd, L.; Kamynina, A.; Vondracek, P.; et al. ISPD gene mutations are a common cause of congenital and limb-girdle muscular dystrophies. Brain 2013, 136, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Hu, H.; Tian, Y.; Li, J.; Scheben, A.; Zhang, C.; Li, Y.; Wu, J.; Yang, L.; Fan, X.; et al. The Chicken Pan-Genome Reveals Gene Content Variation and a Promoter Region Deletion in IGF2BP1 Affecting Body Size. Mol. Biol. Evol. 2021, 38, 5066–5081. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, M.; Cheng, H.; Fan, W.; Yuan, Z.; Gao, Q.; Xu, Y.; Guo, Z.; Zhang, Y.; Hu, J.; et al. An intercross population study reveals genes associated with body size and plumage color in ducks. Nat. Commun. 2018, 9, 2648. [Google Scholar] [CrossRef] [Green Version]
- Keren, A.; Tamir, Y.; Bengal, E. The p38 MAPK signaling pathway: A major regulator of skeletal muscle development. Mol. Cell. Endocrinol. 2006, 252, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Watschinger, K.; Keller, M.A.; McNeill, E.; Alam, M.T.; Lai, S.; Sailer, S.; Rauch, V.; Patel, J.; Hermetter, A.; Golderer, G.; et al. Tetrahydrobiopterin and alkylglycerol monooxygenase substantially alter the murine macrophage lipidome. Proc. Natl. Acad. Sci. USA 2015, 112, 2431–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Zhang, R.; Wang, C.; Wang, J.; Ma, X.; Hou, X.; Lu, J.; Yu, W.; Jiang, F.; Bao, Y.; et al. Variants from GIPR, TCF7L2, DGKB, MADD, CRY2, GLIS3, PROX1, SLC30A8 and IGF1 are associated with glucose metabolism in the Chinese. PLoS ONE 2010, 5, e15542. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Pfeiffer, A.F.H. The evolving story of incretins (GIP and GLP-1) in metabolic and cardiovascular disease: A pathophysiological update. Diabetes Obes. Metab. 2021, 23 (Suppl. S3), 5–29. [Google Scholar] [CrossRef]
- Cheng, L.; Yang, J.; Rao, Q.; Liu, Z.; Song, W.; Guan, S.; Zhao, Z.; Song, W. Toxic effects of Decabromodiphenyl ether (BDE-209) on thyroid of broiler chicks by transcriptome profile analysis. Ecotoxicol. Environ. Saf. 2021, 219, 112305. [Google Scholar] [CrossRef]
- Adkins, D.E.; Khachane, A.N.; McClay, J.L.; Aberg, K.; Bukszár, J.; Sullivan, P.F.; van den Oord, E.J. SNP-based analysis of neuroactive ligand-receptor interaction pathways implicates PGE2 as a novel mediator of antipsychotic treatment response: Data from the CATIE study. Schizophr. Res. 2012, 135, 200–201. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Liang, X.; Liu, L.; Zhang, D.; Wan, C.; Gan, Z.; Yuan, L. High Throughput Sequencing Identifies MicroRNAs Mediating α-Synuclein Toxicity by Targeting Neuroactive-Ligand Receptor Interaction Pathway in Early Stage of Drosophila Parkinson’s Disease Model. PLoS ONE 2015, 10, e0137432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Liu, J.; Liang, S.; Sun, M.; Duan, J. Low-Dose Exposure of Silica Nanoparticles Induces Neurotoxicity via Neuroactive Ligand-Receptor Interaction Signaling Pathway in Zebrafish Embryos. Int. J. Nanomed. 2020, 15, 4407–4415. [Google Scholar] [CrossRef]
- Brand, A.; Leibfritz, D.; Hamprecht, B.; Dringen, R. Metabolism of cysteine in astroglial cells: Synthesis of hypotaurine and taurine. J. Neurochem. 1998, 71, 827–832. [Google Scholar] [CrossRef]
- Lombardini, J.B. Effects of ATP and taurine on calcium uptake by membrane preparations of the rat retina. J. Neurochem. 1983, 40, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Ripps, H.; Shen, W. Review: Taurine: A “very essential” amino acid. Mol. Vis. 2012, 18, 2673–2686. [Google Scholar]
- Beauchet, O. Testosterone and cognitive function: Current clinical evidence of a relationship. Eur. J. Endocrinol. 2006, 155, 773–781. [Google Scholar] [CrossRef] [Green Version]
- Storer, T.W.; Basaria, S.; Traustadottir, T.; Harman, S.M.; Pencina, K.; Li, Z.; Travison, T.G.; Miciek, R.; Tsitouras, P.; Hally, K.; et al. Effects of Testosterone Supplementation for 3 Years on Muscle Performance and Physical Function in Older Men. J. Clin. Endocrinol. Metab. 2017, 102, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHenry, J.; Carrier, N.; Hull, E.; Kabbaj, M. Sex differences in anxiety and depression: Role of testosterone. Front. Neuroendocrinol. 2014, 35, 42–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 2014, 11, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [Green Version]
- Pearson, T.A.; Manolio, T.A. How to interpret a genome-wide association study. JAMA 2008, 299, 1335–1344. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Population/Breed | Geographic Origin | Classification | Number |
|---|---|---|---|
| Henan Game | Henan Province, China | Fight breed | 13 F |
| Luxi Game | Shandong Province, China | Fight breed | 2 M/8 F |
| Turpan Game | Xinjiang Province, China | Fight breed | 6 M/5 F |
| Xishuangbanna Game | Yunnan Province, China | Fight breed | 10 |
| Zhangzhou Game | Fujian Province, China | Fight breed | 10 |
| Big Bone | Liaoning Province, China | Indigenous breed | 5 M/5 F |
| Beijing You | Beijing, China | Indigenous breed | 50 |
| Chahua | Yunnan Province, China | Indigenous breed | 5 M/6 F |
| Hongshan | Hubei Province, China | Indigenous breed | 24 M/24 F |
| Piaoji | Yunnan Province, China | Indigenous breed | 9F |
| Shouguang | Shandong Province, China | Indigenous breed | 50 |
| Taihe Silky | Jiangxi Province, China | Indigenous breed | 50 |
| Tibetan | Tibet, China | Indigenous breed | 40 |
| Wenchang | Hainan Province China | Indigenous breed | 11 |
| RIR | Rhode Island, America | Commercial breed | 50 |
| WhiteLeghorn | Tuscany, Italy | Commercial breed | 40 |
| Dwarf Layer | Guizhou Province China | Cultivated breed | 50 |
| Dwarf Yellow Broiler | Guizhou Province China | Cultivated breed | 50 |
| Dongtao | Vietnam | Foreign breed | 20 M/10 F |
| Houdan | France | Foreign breed | 50 |
| Total | 603 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, X.; Guan, Z.; Zhao, X.; Zhang, X.; Wen, J.; Cheng, H.; Zhang, Y.; Cheng, X.; Liu, Y.; Ning, Z.; et al. Systematic Selection Signature Analysis of Chinese Gamecocks Based on Genomic and Transcriptomic Data. Int. J. Mol. Sci. 2023, 24, 5868. https://doi.org/10.3390/ijms24065868
Ren X, Guan Z, Zhao X, Zhang X, Wen J, Cheng H, Zhang Y, Cheng X, Liu Y, Ning Z, et al. Systematic Selection Signature Analysis of Chinese Gamecocks Based on Genomic and Transcriptomic Data. International Journal of Molecular Sciences. 2023; 24(6):5868. https://doi.org/10.3390/ijms24065868
Chicago/Turabian StyleRen, Xufang, Zi Guan, Xiurong Zhao, Xinye Zhang, Junhui Wen, Huan Cheng, Yalan Zhang, Xue Cheng, Yuchen Liu, Zhonghua Ning, and et al. 2023. "Systematic Selection Signature Analysis of Chinese Gamecocks Based on Genomic and Transcriptomic Data" International Journal of Molecular Sciences 24, no. 6: 5868. https://doi.org/10.3390/ijms24065868
APA StyleRen, X., Guan, Z., Zhao, X., Zhang, X., Wen, J., Cheng, H., Zhang, Y., Cheng, X., Liu, Y., Ning, Z., & Qu, L. (2023). Systematic Selection Signature Analysis of Chinese Gamecocks Based on Genomic and Transcriptomic Data. International Journal of Molecular Sciences, 24(6), 5868. https://doi.org/10.3390/ijms24065868