
CDDO-Me Abrogates Aberrant Mitochondrial Elongation in Clasmatodendritic Degeneration by Regulating NF-κB-PDI-Mediated S-Nitrosylation of DRP1
Abstract
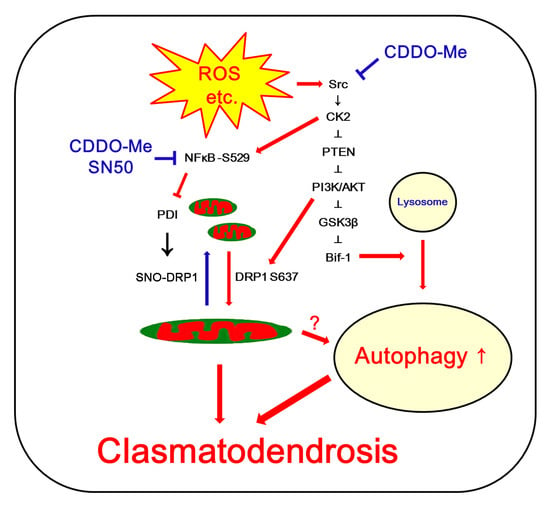
:
1. Introduction
2. Results
2.1. CDDO-Me and SN50 Ameliorates Clasmatodendritic Degeneration in CA1 Astrocytes
2.2. CDDO-Me and SN50 Inhibit NF-κB S529 Phosphorylation in CA1 Astrocytes
2.3. CDDO-Me and SN50 Ameliorate Aberrant Mitochondrial Elongation in CA1 Astrocytes
2.4. CDDO-Me and SN50 Induce Mitochondrial Fragmentation without Altering DRP1 S616 Phosphorylation
2.5. CDDO-Me and SN50 Restore the Reduced PDI Expression in CA1 Astrocytes
2.6. PDI Knockdown Leads to Mitochondrial Elongation in CA1 Astrocytes under Physiological Condition
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Chemicals
4.2. Generation of Chronic Epilepsy Rats
4.3. Surgery for CDDO-Me, SN50 or PDI siRNA Infusion
4.4. Tissue Processing
4.5. Immunohistochemistry, Cell Counts, and Mitochondrial Morphometry
4.6. Western Blot
4.7. Measurement of SNO-PDI
4.8. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eulenburg, V.; Gomeza, J. Neurotransmitter transporters expressed in glial cells as regulators of synapse function. Brain Res. Rev. 2010, 63, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Peng, H.; Kang, N.; Zhao, Z.; Lin, J.H.; Stanton, P.K.; Kang, J. Glutamate-induced exocytosis of glutamate from astrocytes. J. Biol. Chem. 2007, 282, 24185–24197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.S.; Kim, J.E.; Kwak, S.E.; Choi, K.C.; Kim, D.W.; Kwon, O.S.; Choi, S.Y.; Kang, T.C. Spatiotemporal characteristics of astroglial death in the rat hippocampo-entorhinal complex following pilocarpine-induced status epilepticus. J. Comp. Neurol. 2008, 511, 581–598. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Lewén, A.; Noshita, N.; Gasche, Y.; Chan, P.H. Effects of global ischemia duration on neuronal, astroglial, oligodendroglial, and microglial reactions in the vulnerable hippocampal CA1 subregion in rats. J. Neurotrauma 2002, 19, 85–98. [Google Scholar] [CrossRef]
- Penfield, W. Neuroglia and microglia—The interstitial tissue of the central nervous system. In Special Cytology, the Form and Function of the Cell in Health and Disease; Cowdry, E.V., Ed.; Hoeber: New York, NY, USA, 1928; pp. 1033–1068. [Google Scholar]
- Friede, R.L.; van Houten, W.H. Relations between postmortem alterations and glycolytic metabolism in the brain. Exp. Neurol. 1961, 4, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Kraig, R.P.; Chesler, M. Astrocytic acidosis in hyperglycemic and complete ischemia. J. Cereb. Blood Flow Metab. 1990, 10, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Hulse, R.E.; Winterfield, J.; Kunkler, P.E.; Kraig, R.P. Astrocytic clasmatodendrosis in hippocampal organ culture. Glia 2001, 33, 169–179. [Google Scholar] [CrossRef]
- Kim, J.E.; Ryu, H.J.; Yeo, S.I.; Kang, T.C. P2X7 receptor differentially modulates astroglial apoptosis and clasmatodendrosis in the rat brain following status epilepticus. Hippocampus 2011, 21, 1318–1333. [Google Scholar] [CrossRef]
- Kim, J.E.; Ko, A.R.; Hyun, H.W.; Min, S.J.; Kang, T.C. P2RX7-MAPK1/2-SP1 axis inhibits MTOR independent HSPB1-mediated astroglial autophagy. Cell Death Dis. 2018, 9, 546. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Kang, T.C. CDDO-Me attenuates astroglial autophagy via Nrf2-, ERK1/2-SP1- and Src-CK2-PTEN-PI3K/AKT-mediated signaling pathways in the hippocampus of chronic epilepsy rats. Antioxidants 2021, 10, 655. [Google Scholar] [CrossRef]
- Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kim, D.W.; Kwon, O.S.; Choi, S.Y.; Kang, T.C. F-actin depolymerization accelerates clasmatodendrosis via activation of lysosome-derived autophagic astroglial death. Brain Res. Bull. 2011, 85, 368–373. [Google Scholar] [CrossRef]
- Ryu, H.J.; Kim, J.E.; Yeo, S.I.; Kang, T.C. p65/RelA-Ser529 NF-κB subunit phosphorylation induces autophagic astroglial death (Clasmatodendrosis) following status epilepticus. Cell. Mol. Neurobiol. 2011, 31, 1071–1078. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12, 1528–1534. [Google Scholar] [CrossRef] [Green Version]
- Bursch, W.; Hochegger, K.; Torok, L.; Marian, B.; Ellinger, A.; Hermann, R.S. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J. Cell. Sci. 2000, 113, 1189–1198. [Google Scholar] [CrossRef]
- Tomimoto, H.; Akiguchi, I.; Wakita, H.; Suenaga, T.; Nakamura, S.; Kimura, J. Regressive changes of astroglia in white matter lesions in cerebrovascular disease and Alzheimer’s disease patients. Acta Neuropathol. 1997, 94, 146–152. [Google Scholar] [CrossRef]
- Bouchat, J.; Gilloteaux, J.; Suain, V.; Van Vlaender, D.; Brion, J.P.; Nicaise, C. Ultrastructural Analysis of Thalamus Damages in a Mouse Model of Osmotic-Induced Demyelination. Neurotox. Res. 2019, 36, 144–162. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Kim, J.E. P2X7 Receptor Inhibits Astroglial Autophagy via Regulating FAK- and PHLPP1/2-Mediated AKT-S473 Phosphorylation Following Kainic Acid-Induced Seizures. Int. J. Mol. Sci. 2020, 21, 6476. [Google Scholar] [CrossRef]
- Ko, A.R.; Hyun, H.W.; Min, S.J.; Kim, J.E. The Differential DRP1 Phosphorylation and Mitochondrial Dynamics in the Regional Specific Astroglial Death Induced by Status Epilepticus. Front. Cell. Neurosci. 2016, 10, 124. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.S.; Kim, T.H.; Park, H.; Kim, J.E. CDDO-Me attenuates clasmatodendrosis in CA1 astrocyte by inhibiting HSP25-AKT mediated DRP1-S637 phosphorylation in chronic epilepsy rats. Biomedicine 2022, 23, 4569. [Google Scholar] [CrossRef]
- Camargo, L.L.; Babelova, A.; Mieth, A.; Weigert, A.; Mooz, J.; Rajalingam, K.; Heide, H.; Wittig, I.; Lopes, L.R.; Brandes, R.P. Endo-PDI is required for TNFα-induced angiogenesis. Free Radic. Biol. Med. 2013, 65, 1398–1407. [Google Scholar] [CrossRef]
- Lee, D.S.; Kim, J.E. PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis. 2018, 9, 869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, A.R.; Kim, J.Y.; Hyun, H.W.; Kim, J.E. Endoplasmic reticulum (ER) stress protein responses in relation to spatio-temporal dynamics of astroglial responses to status epilepticus in rats. Neuroscience 2015, 307, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Yang, Y.X.; Zhe, H.; He, Z.X.; Zhou, S.F. Bardoxolone methyl (CDDO-Me) as a therapeutic agent: An update on its pharmacokinetic and pharmacodynamic properties. Drug Des. Devel. Ther. 2014, 8, 2075–2088. [Google Scholar] [PubMed] [Green Version]
- Kang, T.C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617. [Google Scholar] [CrossRef]
- Ahmad, R.; Raina, D.; Meyer, C.; Kharbanda, S.; Kufe, D. Triterpenoid CDDO-Me blocks the NF-kappaB pathway by direct inhibition of IKKbeta on Cys-179. J. Biol. Chem. 2006, 281, 35764–35769. [Google Scholar] [CrossRef] [Green Version]
- Yore, M.M.; Liby, K.T.; Honda, T.; Gribble, G.W.; Sporn, M.B. The synthetic triterpenoid 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole blocks nuclear factor-kappaB activation through direct inhibition of IkappaB kinase beta. Mol. Cancer Ther. 2006, 5, 3232–3239. [Google Scholar] [CrossRef] [Green Version]
- Cribbs, J.T.; Strack, S. Functional characterization of phosphorylation sites in dynamin-related protein. Methods Enzymol. 2009, 457, 231–253. [Google Scholar]
- Merrill, R.A.; Dagda, R.K.; Dickey, A.S.; Cribbs, J.T.; Green, S.H.; Usachev, Y.M.; Strack, S. Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP. PLoS Biol. 2011, 9, e1000612. [Google Scholar] [CrossRef] [Green Version]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Ryu, H.J.; Kim, M.J.; Kang, T.C. LIM kinase-2 induces programmed necrotic neuronal death via dysfunction of DRP1-mediated mitochondrial fission. Cell Death Differ. 2014, 21, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 2014, 74, 129–144. [Google Scholar] [CrossRef]
- Kallakunta, V.M.; Slama-Schwok, A.; Mutus, B. Protein disulfide isomerase may facilitate the efflux of nitrite derived S-nitrosothiols from red blood cells. Redox Biol. 2013, 1, 373–380. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Bursch, W.; Ellinger, A.; Kienzl, H.; Török, L.; Pandey, S.; Sikorska, M.; Walker, R.; Hermann, R.S. Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells (MCF-7) in culture: The role of autophagy. Carcinogenesis 1996, 17, 1595–1607. [Google Scholar] [CrossRef] [Green Version]
- Nivon, M.; Richet, E.; Codogno, P.; Arrigo, A.P.; Kretz-Remy, C. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy 2009, 5, 766–783. [Google Scholar] [CrossRef] [Green Version]
- Liby, K.T.; Sporn, M.B. Synthetic oleanane triterpenoids: Multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol. Rev. 2012, 64, 972–1003. [Google Scholar] [CrossRef] [Green Version]
- Bird, T.A.; Schooley, K.; Dower, S.K.; Hagen, H.; Virca, G.D. Activation of nuclear transcription factor NF-kappaB by interleukin-1 is accompanied by casein kinase II-mediated phosphorylation of the p65 subunit. J. Biol. Chem. 1997, 272, 32606–32612. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Westerheide, S.D.; Hanson, J.L.; Baldwin, A.S., Jr. Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J. Biol. Chem. 2000, 275, 32592–32597. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Sabharwal, L.; Ota, M.; Nakagawa, I.; Jiang, J.J.; Arima, Y.; Ogura, H.; Okochi, M.; Ishii, M.; Kamimura, D.; et al. Presenilin 1 Regulates NF-κB Activation via Association with Breakpoint Cluster Region and Casein Kinase II. J. Immunol. 2018, 201, 2256–2263. [Google Scholar] [CrossRef] [Green Version]
- Lyles, M.M.; Gilbert, H.F. Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: Dependence of the rate on the composition of the redox buffer. Biochemistry 1991, 30, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Ali Khan, H.; Mutus, B. Protein disulfide isomerase a multifunctional protein with multiple physiological roles. Front. Chem. 2014, 2, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perri, E.R.; Thomas, C.J.; Parakh, S.; Spencer, D.M.; Atkin, J.D. The Unfolded Protein Response and the Role of Protein Disulfide Isomerase in Neurodegeneration. Front. Cell Dev. Biol. 2016, 3, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Russelakis-Carneiro, M.; Wälchli, S.; Carboni, S.; Vial-Knecht, E.; Maundrell, K.; Castilla, J.; Soto, C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J. Neurosci. 2005, 25, 2793–2802. [Google Scholar] [CrossRef] [Green Version]
- Ko, H.S.; Uehara, T.; Nomura, Y. Role of ubiquilin associated with protein-disulfide isomerase in the endoplasmic reticulum in stress-induced apoptotic cell death. J. Biol. Chem. 2002, 277, 35386–35392. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Lu, H.; Li, C. Proapoptotic activities of protein disulfide isomerase (PDI) and PDIA3 protein, a role of the Bcl-2 protein Bak. J. Biol. Chem. 2015, 290, 8949–8963. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Kanekura, K.; Suzuki, H.; Aiso, S.; Matsuoka, M. ER stress and unfolded protein response in amyotrophic lateral sclerosis. Mol. Neurobiol. 2009, 39, 81–89. [Google Scholar] [CrossRef]
- Wang, W.T.; Sun, L.; Sun, C.H. PDIA3-regulted inflammation and oxidative stress contribute to the traumatic brain injury (TBI) in mice. Biochem. Biophys. Res. Commun. 2019, 518, 657–663. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, C.; Gu, M.; Wang, H.; Chen, W.; Luo, G.; Yang, G.; Zhang, Z.; Zhang, Y.; Xian, G.; et al. Protein Disulfide Isomerase Silence Inhibits Inflammatory Functions of Macrophages by Suppressing Reactive Oxygen Species and NF-κB Pathway. Inflammation 2018, 41, 614–625. [Google Scholar] [CrossRef]
- Higuchi, T.; Watanabe, Y.; Waga, I. Protein disulfide isomerase suppresses the transcriptional activity of NF-kappaB. Biochem. Biophys. Res. Commun. 2004, 318, 46–52. [Google Scholar] [CrossRef]
- Tsai, K.L.; Wang, S.M.; Chen, C.C.; Fong, T.H.; Wu, M.L. Mechanism of oxidative stress-induced intracellular acidosis in rat cerebellar astrocytes and C6 glioma cells. J. Physiol. 1997, 502, 161–174. [Google Scholar] [CrossRef]
- Muranyi, M.; Ding, C.; He, Q.; Lin, Y.; Li, P.A. Streptozotocin-induced diabetes causes astrocyte death after ischemia and reperfusion injury. Diabetes 2006, 55, 349–355. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Dargusch, R.; Maher, P. Lactacidosis modulates glutathione metabolism and oxidative glutamate toxicity. J. Neurochem. 2010, 113, 502–514. [Google Scholar] [CrossRef]
- Pandya, J.D.; Nukala, V.N.; Sullivan, P.G. Concentration dependent effect of calcium on brain mitochondrial bioenergetics and oxidative stress parameters. Front. Neuroenergetics 2013, 5, 10. [Google Scholar] [CrossRef]
- Lezi, E.; Swerdlow, R.H. Mitochondria in neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar]
- Chan, D.C. Mitochondria: Dynamic organelles in disease, aging, and development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Campello, S.; Scorrano, L. Mitochondrial shape changes: Orchestrating cell pathophysiology. EMBO Rep. 2010, 11, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Terman, A.; Gustafsson, B.; Brunk, U.T. Mitochondrial damage and intralysosomal degradation in cellular aging. Mol. Aspects Med. 2006, 27, 471–482. [Google Scholar] [CrossRef]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Uehara, T.; Nakamura, T.; Yao, D.; Shi, Z.Q.; Gu, Z.; Ma, Y.; Masliah, E.; Nomura, Y.; Lipton, S.A. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006, 441, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guan, T.; Li, C.; Shang, H.; Cui, L.; Li, X.M.; Kong, J. SOD1 aggregation in astrocytes following ischemia/reperfusion injury: A role of NO-mediated S-nitrosylation of protein disulfide isomerase (PDI). J. Neuroinflammation 2012, 9, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Park, H.; Kang, T.C. Peroxiredoxin 6 Regulates Glutathione Peroxidase 1-Medited Glutamine Synthase Preservation in the Hippocampus of Chronic Epilepsy Rats. Antioxidants 2023, 12, 156. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Lee, D.S.; Kang, T.C. Sp1-Mediated Prdx6 Upregulation Leads to Clasmatodendrosis by Increasing Its aiPLA2 Activity in the CA1 Astrocytes in Chronic Epilepsy Rats. Antioxidants 2022, 11, 1883. [Google Scholar] [CrossRef]
- Kim, J.E.; Lee, D.S.; Kim, T.H.; Kang, T.C. Glutathione Regulates GPx1 Expression during CA1 Neuronal Death and Clasmatodendrosis in the Rat Hippocampus following Status Epilepticus. Antioxidants 2022, 11, 756. [Google Scholar] [CrossRef]
- Eskelinen, E.L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Aspects Med. 2006, 27, 495–502. [Google Scholar] [CrossRef]
- Ma, X.; Godar, R.J.; Liu, H.; Diwan, A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 2012, 8, 297–309. [Google Scholar] [CrossRef] [Green Version]
- Sakai, K.; Fukuda, T.; Iwadate, K. Beading of the astrocytic processes (clasmatodendrosis) following head trauma is associated with protein degradation pathways. Brain Inj. 2013, 27, 1692–1697. [Google Scholar] [CrossRef]
- Qin, A.P.; Liu, C.F.; Qin, Y.Y.; Hong, L.Z.; Xu, M.; Yang, L.; Liu, J.; Qin, Z.H.; Zhang, H.L. Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy 2010, 6, 738–753. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Ko, A.R.; Hyun, H.W.; Min, S.J.; Kim, J.E. PDI regulates seizure activity via NMDA receptor redox in rats. Sci. Rep. 2017, 7, 42491. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Park, H.; Lee, J.E.; Kang, T.C. CDDO-Me Inhibits Microglial Activation and Monocyte Infiltration by Abrogating NFκB- and p38 MAPK-Mediated Signaling Pathways Following Status Epilepticus. Cells 2020, 9, 1123. [Google Scholar] [CrossRef]
- Lee, D.S.; Kim, J.E. Protein disulfide isomerase-mediated S-nitrosylation facilitates surface expression of P2X7 receptor following status epilepticus. J. Neuroinflammation 2021, 18, 14. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, Y.J.; Kim, J.Y.; Kang, T.C. PARP1 activation/expression modulates regional-specific neuronal and glial responses to seizure in a hemodynamic-independent manner. Cell Death Dis. 2014, 5, e1362. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Park, H.; Kim, T.H.; Kang, T.C. LONP1 Regulates Mitochondrial Accumulations of HMGB1 and Caspase-3 in CA1 and PV Neurons Following Status Epilepticus. Int. J. Mol. Sci. 2021, 22, 2275. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Host | Manufacturer (Catalog Number) | Dilution Used |
|---|---|---|---|
| DRP1 | Rabbit | Thermo (#PA1-16987) | 1:1000 (WB) |
| DRP1 S616 | Rabbit | Cell Signaling (#4494) | 1:200 (IH) |
| GFAP | Rabbit Mouse | Abcam (#ab7260) Millipore (#MAB3402) | 1:500 (IH) 1:2000 (IH) |
| LAMP1 | Rabbit | Lifespan (#LS-B580) | 1:200 (IH) 1:1000 (WB) |
| LC3 | Rabbit | Abgent (#AP1802a) | 1:100 (IH) 1:1000 (WB) |
| Mitochondrial marker (Mitochondrial complex IV subunit 1, MTCO1) | Mouse | Abcam (#ab14705) | 1:500 (IH) |
| NF-κB S529 | Rabbit | Abcam (#ab47395) | 1:100 (IH) |
| PDI | Mouse | Abcam (#ab2792) | 1:100 (IH) 1:1000 (WB) |
| β-actin | Mouse | Sigma (#A5316) | 1:6000 (WB) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, D.-S.; Kim, T.-H.; Park, H.; Kim, J.-E. CDDO-Me Abrogates Aberrant Mitochondrial Elongation in Clasmatodendritic Degeneration by Regulating NF-κB-PDI-Mediated S-Nitrosylation of DRP1. Int. J. Mol. Sci. 2023, 24, 5875. https://doi.org/10.3390/ijms24065875
Lee D-S, Kim T-H, Park H, Kim J-E. CDDO-Me Abrogates Aberrant Mitochondrial Elongation in Clasmatodendritic Degeneration by Regulating NF-κB-PDI-Mediated S-Nitrosylation of DRP1. International Journal of Molecular Sciences. 2023; 24(6):5875. https://doi.org/10.3390/ijms24065875
Chicago/Turabian StyleLee, Duk-Shin, Tae-Hyun Kim, Hana Park, and Ji-Eun Kim. 2023. "CDDO-Me Abrogates Aberrant Mitochondrial Elongation in Clasmatodendritic Degeneration by Regulating NF-κB-PDI-Mediated S-Nitrosylation of DRP1" International Journal of Molecular Sciences 24, no. 6: 5875. https://doi.org/10.3390/ijms24065875