
Impaired Autophagy in Krabbe Disease: The Role of BCL2 and Beclin-1 Phosphorylation

 ,
,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Autophagosomes Formation Is Reduced in KD Fibroblasts under Starvation
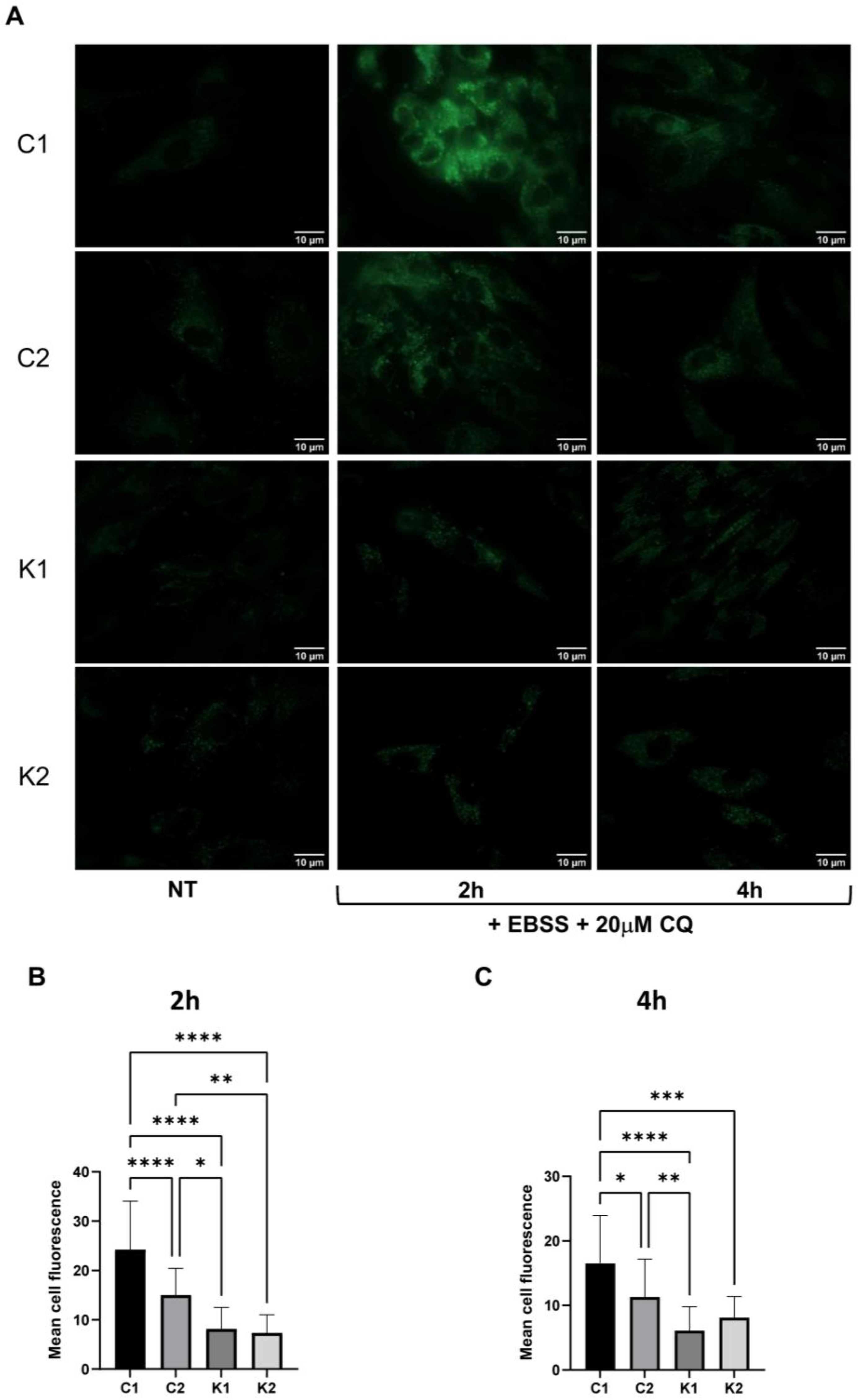
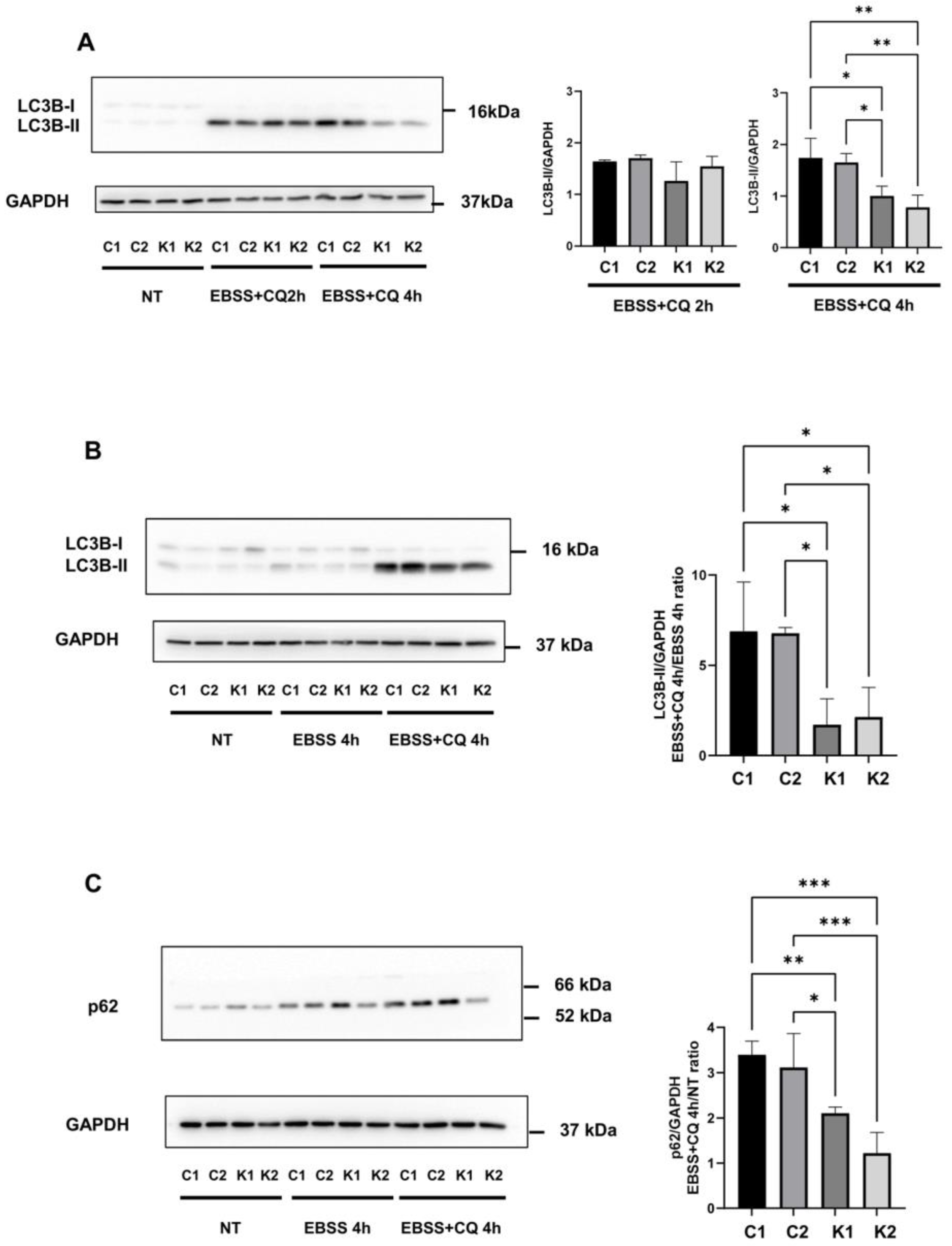
2.2. The Autophagic Response Is Impaired in KD Fibroblasts
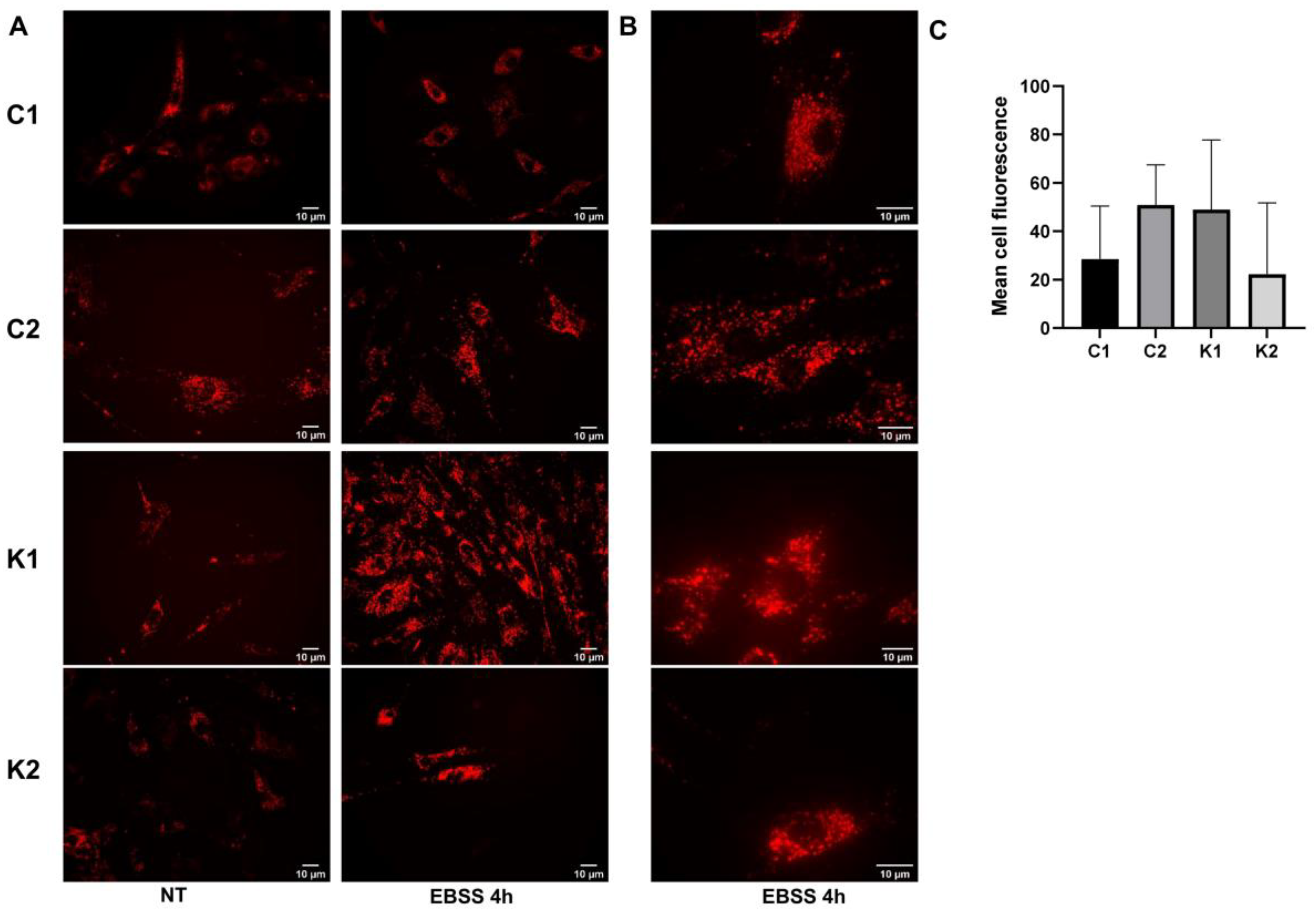
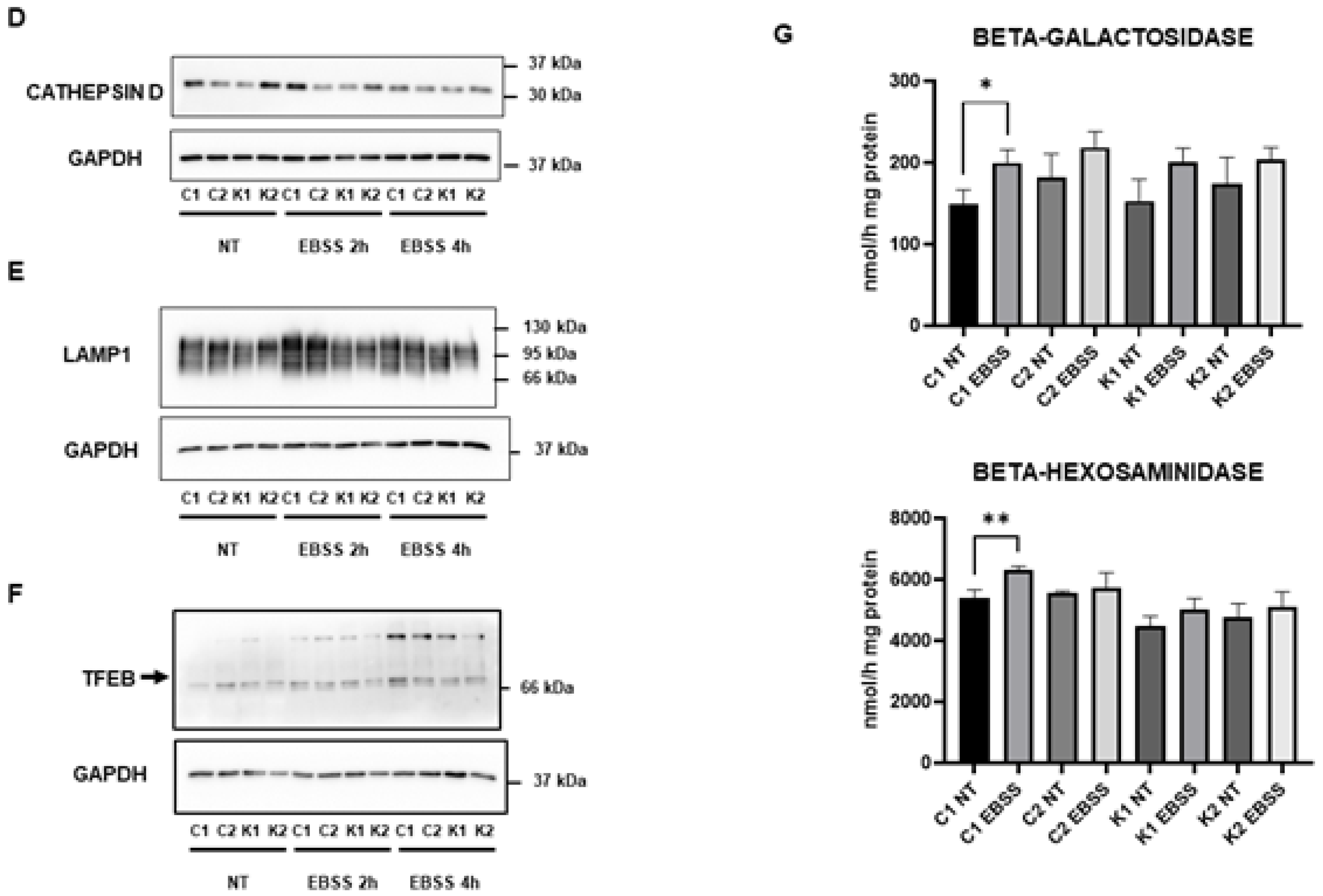
2.3. Lysosomal Alterations during Starvation-Induced Autophagy in KD Fibroblasts
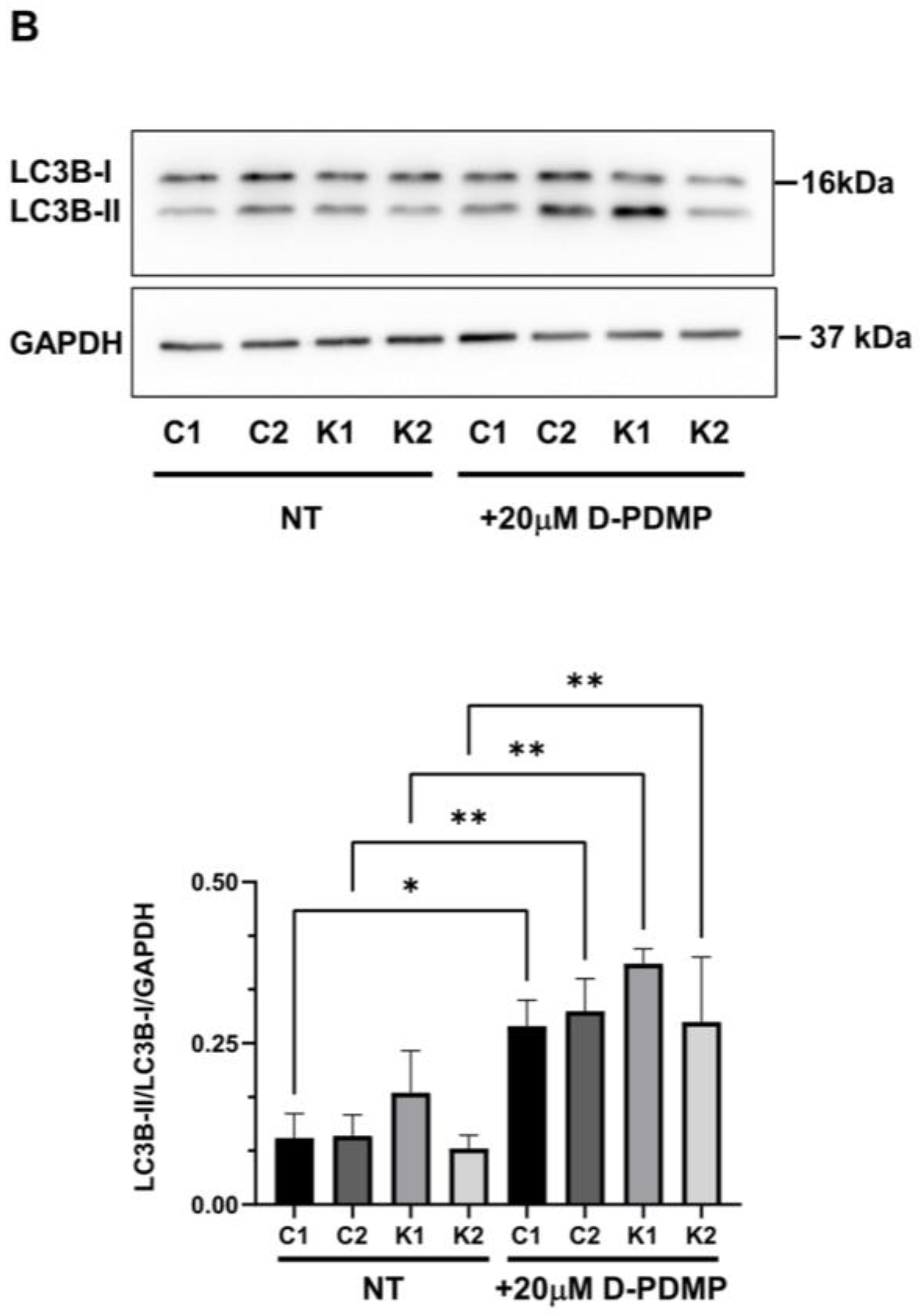
2.4. Cell Sphingolipids and Autophagy
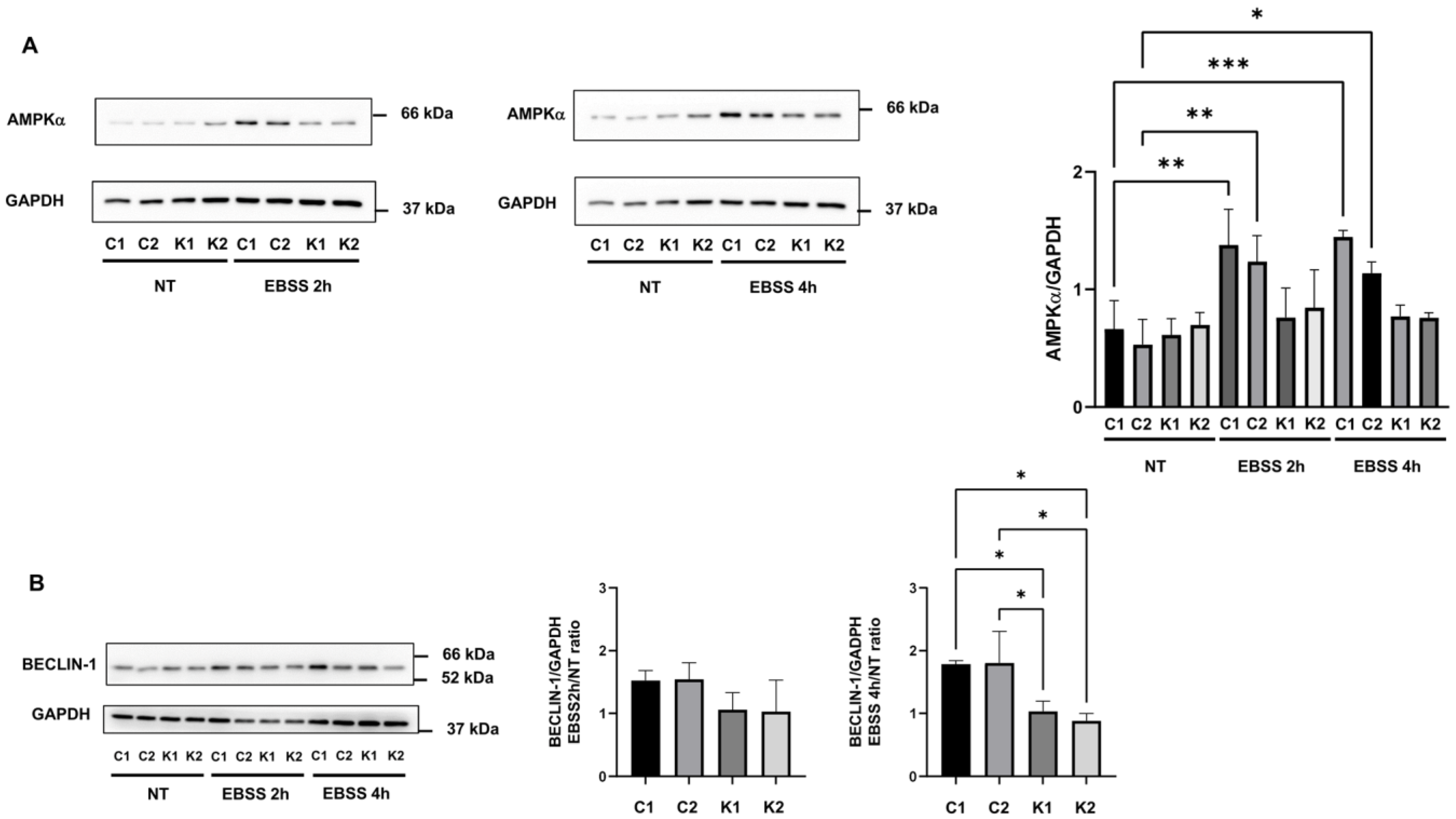
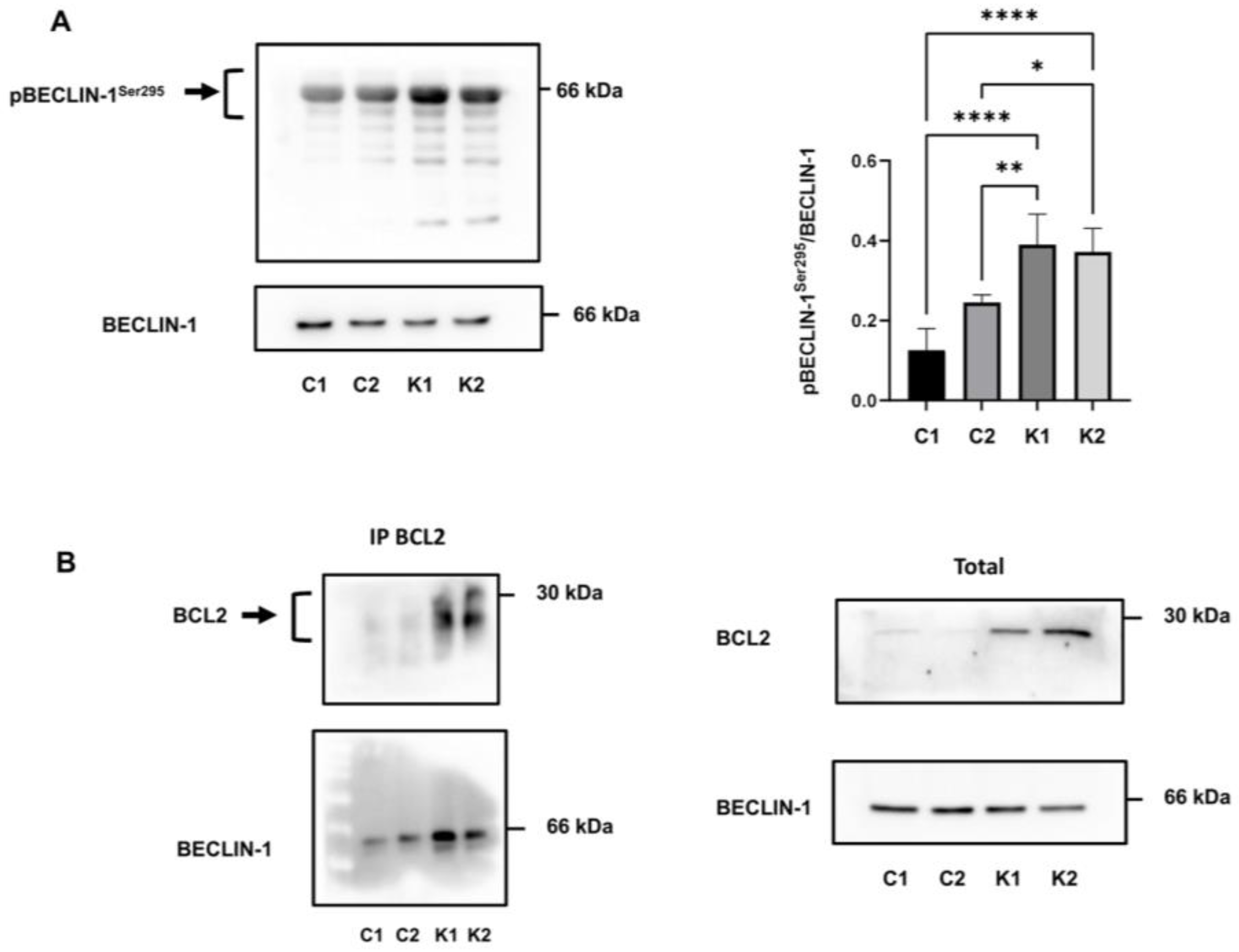
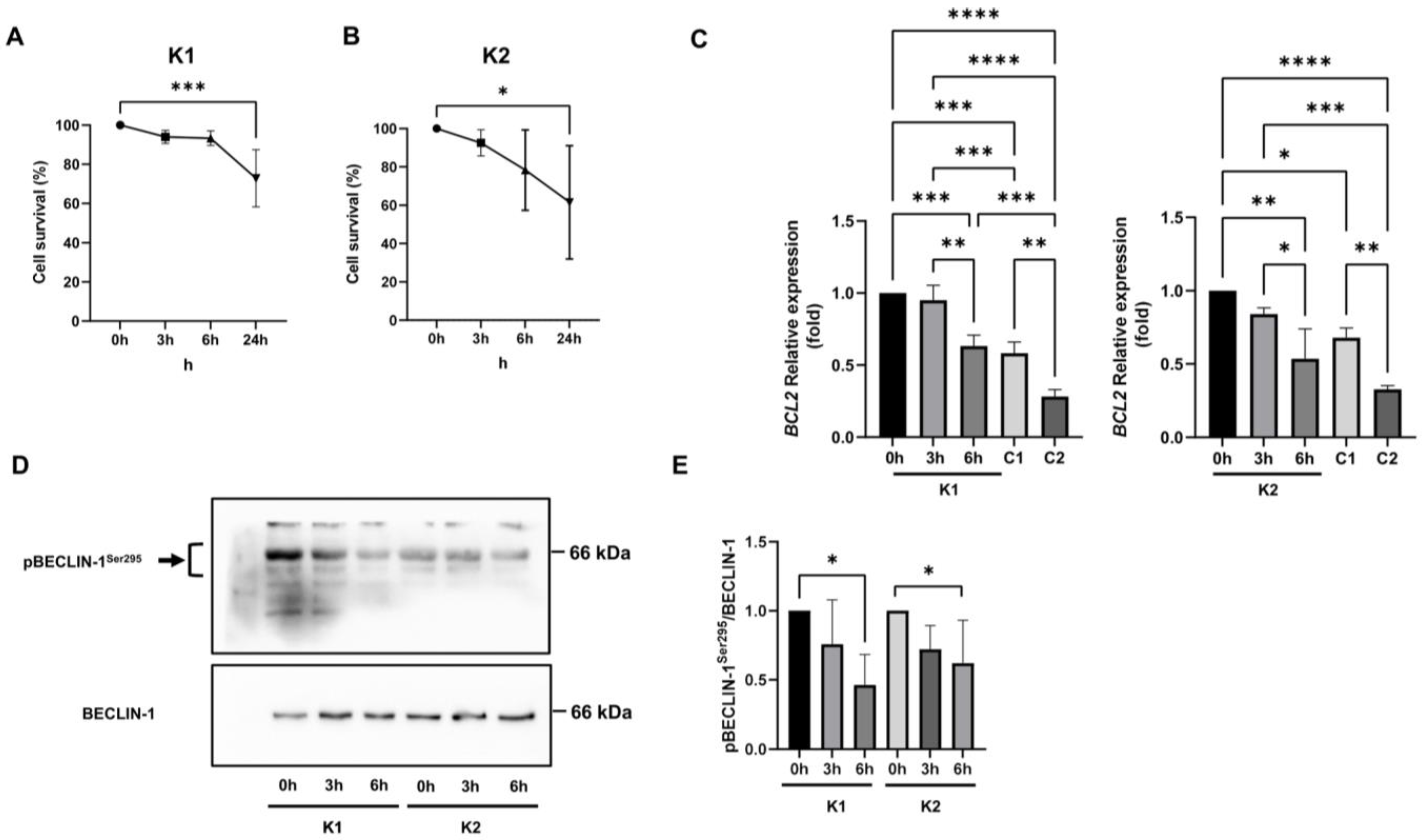
2.5. Involvement of Beclin-1 and BCL2 in Autophagy Impairment
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Treatments
4.2. Whole-Cell Lysate Preparation
4.3. Western Blotting Analysis
4.4. Beta-Galactosidase and Beta-Hexosaminidase Enzymatic Activities
4.5. RNA Extraction and Real-Time PCR
4.6. Fluorescence Microscopy
4.6.1. Monodansylcadaverine (MDC) Staining
4.6.2. CYTO-ID® Autophagy Detection Kit
4.6.3. LysoTracker Red DND-99 Staining
4.7. Immunoprecipitation
4.8. KD Fibroblasts Treatment with LY294002
4.9. Sphingolipid Quantification by LC-MS/MS
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Parenti, G.; Medina, D.L.; Ballabio, A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 2021, 13, e12836. [Google Scholar] [CrossRef]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci. 2016, 39, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribbens, J.J.; Moser, A.B.; Hubbard, W.C.; Bongarzone, E.R.; Maegawa, G.H. Characterization and application of a disease-cell model for a neurodegenerative lysosomal disease. Mol. Genet. Metab. 2014, 111, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Grosso, A.; Antonini, S.; Angella, L.; Tonazzini, I.; Signore, G.; Cecchini, M. Lithium improves cell viability in psychosine-treated MO3.13 human oligodendrocyte cell line via autophagy activation. J. Neurosci. Res. 2016, 94, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Del Grosso, A.; Angella, L.; Tonazzini, I.; Moscardini, A.; Giordano, N.; Caleo, M.; Rocchiccioli, S.; Cecchini, M. Dysregulated autophagy as a new aspect of the molecular pathogenesis of Krabbe disease. Neurobiol. Dis. 2019, 129, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.S.; Ho, C.S.; Huang, Y.W.; Wu, T.Y.; Lee, T.H.; Huang, Z.D.; Wang, T.J.; Yang, S.J.; Chiang, M.F. Impairment of Proteasome and Autophagy Underlying the Pathogenesis of Leukodystrophy. Cells 2020, 9, 1124. [Google Scholar] [CrossRef] [PubMed]
- Hatton, C.; Ghanem, S.S.; Koss, D.J.; Abdi, I.Y.; Gibbons, E.; Guerreiro, R.; Bras, J.; Walker, L.; Gelpi, E.; Heywood, W.; et al. Prion-like α-synuclein pathology in the brain of infants with Krabbe disease. Brain 2022, 145, 1257–1263. [Google Scholar] [CrossRef]
- Marshall, M.S.; Issa, Y.; Heller, G.; Nguyen, D.; Bongarzone, E.R. AAV-Mediated GALC Gene Therapy Rescues Alpha-Synucleinopathy in the Spinal Cord of a Leukodystrophic Lysosomal Storage Disease Mouse Model. Front. Cell. Neurosci. 2020, 14, 619712. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.R.; Santos, M.B.; Marshall, M.S.; Cantuti-Castelvetri, L.; Lopez-Rosas, A.; Li, G.; van Breemen, R.; Claycomb, K.I.; Gallea, J.I.; Celej, M.S.; et al. Neuronal inclusions of α-synuclein contribute to the pathogenesis of Krabbe disease. J. Pathol. 2014, 232, 509–521. [Google Scholar] [CrossRef] [Green Version]
- Del Grosso, A.; Parlanti, G.; Angella, L.; Giordano, N.; Tonazzini, I.; Ottalagana, E.; Carpi, S.; Pellegrino, R.M.; Alabed, H.B.R.; Emiliani, C.; et al. Chronic lithium administration in a mouse model for Krabbe disease. JIMD Rep. 2022, 63, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Papini, N.; Giallanza, C.; Brioschi, L.; Ranieri, F.R.; Giussani, P.; Mauri, L.; Ciampa, M.G.; Viani, P.; Tringali, C. Galactocerebrosidase deficiency induces an increase in lactosylceramide content: A new hallmark of Krabbe disease? Int. J. Biochem. Cell Biol. 2022, 145, 106184. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Hirata, N.; Suizu, F. The links between AKT and two intracellular proteolytic cascades: Ubiquitination and autophagy. Biochim.Biophys. Acta-Rev. Cancer 2014, 1846, 342–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Levine, B. Bcl-2 inhibition of autophagy: A new route to cancer? Cancer Res. 2006, 66, 2885–2888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, A.O.; Proud, C.G. Chloroquine and bafilomycin A mimic lysosomal storage disorders and impair mTORC1 signalling. Biosci. Rep. 2020, 40, BSR20200905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.E.; Johansen, T. Following autophagy step by step. BMC Biol. 2011, 9, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awojoodu, A.O.; Keegan, P.M.; Lane, A.R.; Zhang, Y.; Lynch, K.R.; Platt, M.O.; Botchwey, E.A. Acid sphingomyelinase is activated in sickle cell erythrocytes and contributes to inflammatory microparticle generation in SCD. Blood 2014, 124, 1941–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, T.; Komatsu, M. Monitoring Autophagy Flux and Activity: Principles and Applications. Bioessays 2020, 42, e2000122. [Google Scholar] [CrossRef]
- Weidberg, H.; Shvets, E.; Shpilka, T.; Shimron, F.; Shinder, V.; Elazar, Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29, 1792–1802. [Google Scholar] [CrossRef] [Green Version]
- Abdel Karim, N.; Gaber, O.; Aljohani, H.M.; Eldessouki, I.; Bahassi, E.M.; Morris, J. Exosomes as a Surrogate Marker for Autophagy in Peripheral Blood, Correlative Data from Phase I Study of Chloroquine in Combination with Carboplatin/Gemcitabine in Advanced Solid Tumors. Asian Pac. J. Cancer Prev. 2019, 20, 3789–3796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, Y.; Koksal, A.R.; Reddy, V.; Lin, D.; Osman, H.; Heidari, Z.; Rhadhi, S.M.; Wimley, W.C.; Parsi, M.A.; Dash, S. Extracellular Vesicle Release Promotes Viral Replication during Persistent HCV Infection. Cells 2021, 10, 984. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.V.; Mills, J.; Lapierre, L.R. Selective Autophagy Receptor p62/SQSTM1, a Pivotal Player in Stress and Aging. Front. Cell Dev. Biol. 2022, 10, 793328. [Google Scholar] [CrossRef]
- Yamane, M.; Miyazawa, K.; Moriya, S.; Abe, A.; Yamane, S. D,L-Threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (DL-PDMP) increases endoplasmic reticulum stress, autophagy and apoptosis accompanying ceramide accumulation via ceramide synthase 5 protein expression in A549 cells. Biochimie 2011, 93, 1446–1459. [Google Scholar] [CrossRef]
- Ode, T.; Podyma-Inoue, K.A.; Terasawa, K.; Inokuchi, J.I.; Kobayashi, T.; Watabe, T.; Izumi, Y.; Hara-Yokoyama, M. PDMP, a ceramide analogue, acts as an inhibitor of mTORC1 by inducing its translocation from lysosome to endoplasmic reticulum. Exp. Cell Res. 2017, 350, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Changotra, H. The beclin 1 interactome: Modification and roles in the pathology of autophagy-related disorders. Biochimie 2020, 175, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation–at the Center of Autophagy Regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciechomska, I.A.; Goemans, G.C.; Skepper, J.N.; Tolkovsky, A.M. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene 2009, 28, 2128–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquez, R.T.; Xu, L. Bcl-2:Beclin 1 complex: Multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am. J. Cancer Res. 2012, 2, 214–221. [Google Scholar]
- Scerra, G.; De Pasquale, V.; Scarcella, M.; Caporaso, M.G.; Pavone, L.M.; D’Agostino, M. Lysosomal positioning diseases: Beyond substrate storage. Open Biol. 2022, 12, 220155. [Google Scholar] [CrossRef] [PubMed]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Mack, H.I.; Zheng, B.; Asara, J.M.; Thomas, S.M. AMPK-dependent phosphorylation of ULK1 regulates ATG9 localization. Autophagy 2012, 8, 1197–1214. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 12637. [Google Scholar] [CrossRef] [Green Version]
- Giri, S.; Khan, M.; Nath, N.; Singh, I.; Singh, A.K. The role of AMPK in psychosine mediated effects on oligodendrocytes and astrocytes: Implication for Krabbe disease. J. Neurochem. 2008, 105, 1820–1833. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.S.; Zhuang, L.; Gan, B. Spatial control of AMPK signaling at subcellular compartments. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Sahani, M.H.; Itakura, E.; Mizushima, N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy 2014, 10, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [Green Version]
- De Araujo, M.E.G.; Liebscher, G.; Hess, M.W.; Huber, L.A. Lysosomal size matters. Traffic 2020, 21, 60–75. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, C.D.; Kunkel, R.; Lieberman, A.P. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 2007, 16, 1495–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filocamo, M.; Mazzotti, R.; Corsolini, F.; Stroppiano, M.; Stroppiana, G.; Grossi, S.; Lualdi, S.; Tappino, B.; Lanza, F.; Galotto, S.; et al. Cell Line and DNA Biobank from Patients Affected by Genetic Diseases. Open J. Bioresour. 2014, 1, 2. [Google Scholar]
- Morano, C.; Zulueta, A.; Caretti, A.; Roda, G.; Paroni, R.; Dei Cas, M. An Update on Sphingolipidomics: Is Something Still Missing? Some Considerations on the Analysis of Complex Sphingolipids and Free-Sphingoid Bases in Plasma and Red Blood Cells. Metabolites 2022, 12, 450. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papini, N.; Todisco, R.; Giussani, P.; Dei Cas, M.; Paroni, R.; Giallanza, C.; Tringali, C. Impaired Autophagy in Krabbe Disease: The Role of BCL2 and Beclin-1 Phosphorylation. Int. J. Mol. Sci. 2023, 24, 5984. https://doi.org/10.3390/ijms24065984
Papini N, Todisco R, Giussani P, Dei Cas M, Paroni R, Giallanza C, Tringali C. Impaired Autophagy in Krabbe Disease: The Role of BCL2 and Beclin-1 Phosphorylation. International Journal of Molecular Sciences. 2023; 24(6):5984. https://doi.org/10.3390/ijms24065984
Chicago/Turabian StylePapini, Nadia, Roberta Todisco, Paola Giussani, Michele Dei Cas, Rita Paroni, Chiara Giallanza, and Cristina Tringali. 2023. "Impaired Autophagy in Krabbe Disease: The Role of BCL2 and Beclin-1 Phosphorylation" International Journal of Molecular Sciences 24, no. 6: 5984. https://doi.org/10.3390/ijms24065984
APA StylePapini, N., Todisco, R., Giussani, P., Dei Cas, M., Paroni, R., Giallanza, C., & Tringali, C. (2023). Impaired Autophagy in Krabbe Disease: The Role of BCL2 and Beclin-1 Phosphorylation. International Journal of Molecular Sciences, 24(6), 5984. https://doi.org/10.3390/ijms24065984