
Clinical Consequences and Functional Impact of the Rare S737F CFTR Variant and Its Responsiveness to CFTR Modulators

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
2.1. Genotype, Clinical Data and Clinical Course
- −
- one had a diagnosis of CF in presence of positive NBS and pathological SC level (66 mEq/L);
- −
- six were diagnosed with CRMS/CFSPID on the grounds of SC in intermediate or normal range and another CFTR variant on the second allele;
- −
- three were labelled as having an inconclusive diagnosis in presence of negative result to CF NBS, isolated episode of hypochloremic alkalosis and SC testing in the intermediate or pathological range (subject 1 and 3 of Table 1) and no CF related respiratory symptoms and pathological SC testing (subject 10 of Table 1).
- −
- 4 out of 6 asymptomatic CRMS/CFSPID progressed to a CF diagnosis justified by pathological SC values at a mean age of 2.2 years (range 1.2–3.4);
- −
- 2 asymptomatic subjects maintained the CRMS/CFSPID diagnosis, after a mean follow up of 18 months, in presence of SC value in normal or intermediate range;
- −
- 3 subjects still had an inconclusive diagnosis, in presence of pathological SC values but neither additional symptoms nor bronchiectasis at chest CT scan.
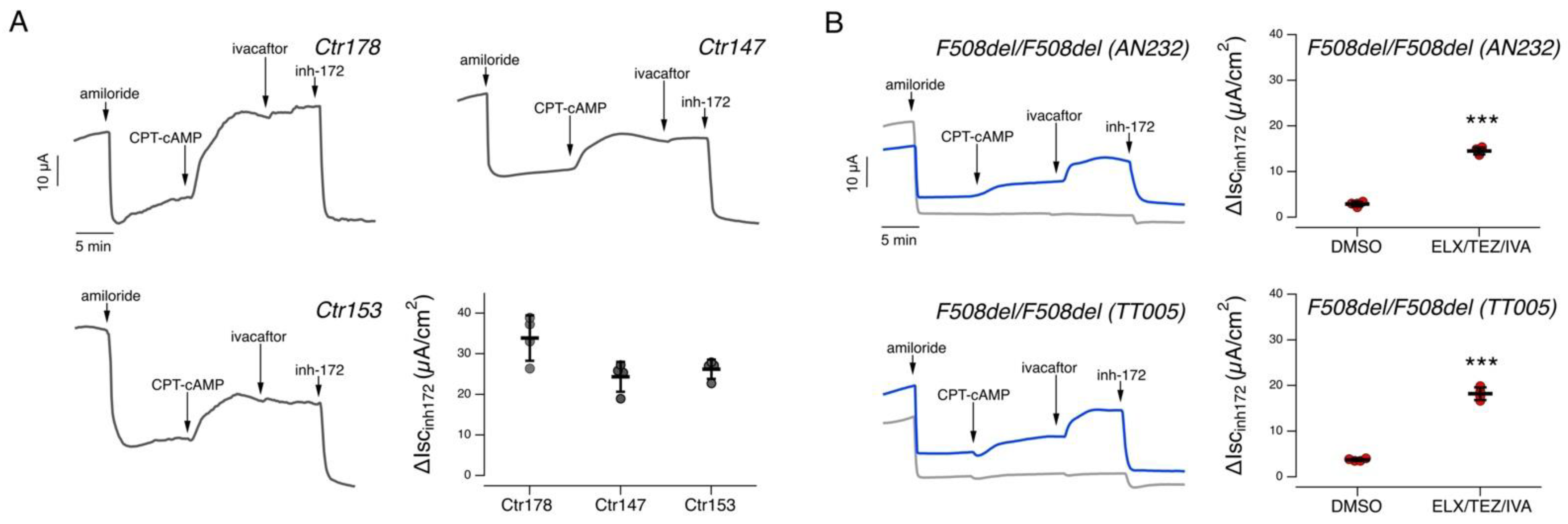
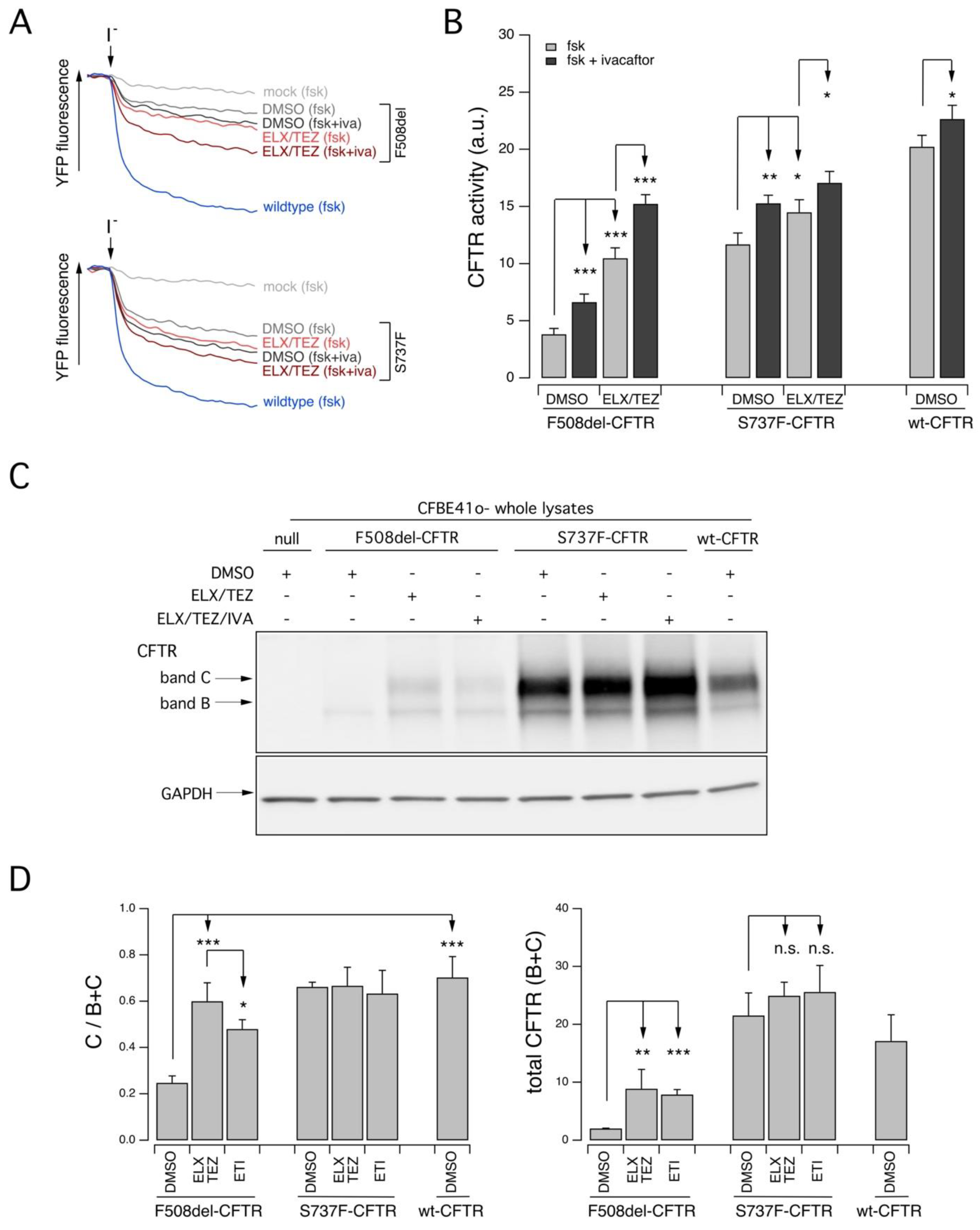
2.2. Functional Evaluation of CFTR Activity in Patient-Derived Nasal Epithelial Cells
3. Discussion
4. Methods
4.1. Patients Population
4.2. Primary Nasal Epithelial Cell Culture
4.3. Short-Circuit Current Recordings
4.4. Cell Culture
4.5. Chemicals, Antibodies and Vectors
4.6. Transient Transfection of CFBE41o-Cells
4.7. YFP-Based Assay for CFTR Activity
4.8. Western Blot
4.9. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.-R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [PubMed] [Green Version]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellani, C.; Cuppens, H.; Macek, M., Jr.; Cassiman, J.J.; Kerem, E.; Durie, P.; Tullis, E.; Assael, B.M.; Bombieri, C.; Brown, A.; et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 2008, 7, 179–196. [Google Scholar] [CrossRef] [Green Version]
- Dequeker, E.; Stuhrmann, M.; Morris, M.A.; Casals, T.; Castellani, C.; Claustres, M.; Cuppens, H.; Georges, M.D.; Ferec, C.; Macek, M.; et al. Best practice guidelines for molecular genetic diagnosis of cystic fibrosis and CFTR-related disorders—Updated European recommendations. Eur. J. Hum. Genet. 2009, 17, 51–65. [Google Scholar] [CrossRef]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Terlizzi, V.; Claut, L.; Tosco, A.; Colombo, C.; Raia, V.; Fabrizzi, B.; Lucarelli, M.; Angeloni, A.; Cimino, G.; Castaldo, A.; et al. A survey of the prevalence, management and outcome of infants with an inconclusive diagnosis following newborn bloodspot screening for cystic fibrosis (CRMS/CFSPID) in six Italian centres. J. Cyst. Fibros. 2021, 20, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; Swiatecka-Urban, A.; Brautigan, D.L.; Jordan, P. Regulatory Crosstalk by Protein Kinases on CFTR Trafficking and Activity. Front. Chem. 2016, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Terlizzi, V.; Di Lullo, A.M.; Comegna, M.; Centrone, C.; Pelo, E.; Castaldo, G.; Raia, V.; Braggion, C. S737F is a new CFTR mutation typical of patients originally from the Tuscany region in Italy. Ital. J. Pediatr. 2018, 44, 2. [Google Scholar] [CrossRef] [Green Version]
- Bianchimani, C.; Dolce, D.; Centrone, C.; Campana, S.; Ravenni, N.; Orioli, T.; Camera, E.; Mergni, G.; Fevola, C.; Bonomi, P.; et al. Impact of Pancreatitis-Associated Protein on Newborn Screening Outcomes and Detection of CFTR-Related Metabolic Syndrome (CRMS)/Cystic Fibrosis Screen Positive, Inconclusive Diagnosis (CFSPID): A Monocentric Prospective Pilot Experience. Int. J. Neonatal. Screen. 2022, 8, 46. [Google Scholar] [CrossRef]
- Tomati, V.; Costa, S.; Capurro, V.; Pesce, E.; Pastorino, C.; Lena, M.; Sondo, E.; Di Duca, M.; Cresta, F.; Cristadoro, S.; et al. Rescue by elexacaftor-tezacaftor-ivacaftor of the G1244E cystic fibrosis mutation’s stability and gating defects are dependent on cell background. J. Cyst. Fibros. 2022. [Google Scholar] [CrossRef]
- Sondo, E.; Cresta, F.; Pastorino, C.; Tomati, V.; Capurro, V.; Pesce, E.; Lena, M.; Iacomino, M.; Baffico, A.M.; Coviello, D.; et al. The L467F-F508del Complex Allele Hampers Pharmacological Rescue of Mutant CFTR by Elexacaftor/Tezacaftor/Ivacaftor in Cystic Fibrosis Patients: The Value of the Ex Vivo Nasal Epithelial Model to Address Non-Responders to CFTR-Modulating Drugs. Int. J. Mol. Sci. 2022, 23, 3175. [Google Scholar] [CrossRef] [PubMed]
- Capurro, V.; Tomati, V.; Sondo, E.; Renda, M.; Borrelli, A.; Pastorino, C.; Guidone, D.; Venturini, A.; Giraudo, A.; Bertozzi, S.M.; et al. Partial Rescue of F508del-CFTR Stability and Trafficking Defects by Double Corrector Treatment. Int. J. Mol. Sci. 2021, 22, 5262. [Google Scholar] [CrossRef]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef]
- Ooi, C.Y.; Castellani, C.; Keenan, K.; Avolio, J.; Volpi, S.; Boland, M.; Kovesi, T.; Bjornson, C.; Chilvers, M.; Morgan, L.; et al. Inconclusive diagnosis of cystic fibrosis after newborn screening. Pediatrics 2015, 135, e1377–e1385. [Google Scholar] [CrossRef] [Green Version]
- Terlizzi, V.; Mergni, G.; Centrone, C.; Festini, F.; Taccetti, G. Trend of sweat chloride values in a cohort of patients carrying CFTR mutations of varying clinical consequence: Is there a risk of increasing sweat chloride over time? Pediatr. Pulmonol. 2020, 55, 1089–1093. [Google Scholar] [CrossRef]
- Tosco, A.; Carnovale, V.; Claut, L.; Fabrizzi, B.; Majo, F.; Castellani, C.; Sepe, A.; Castaldo, G.; Terlizzi, V. Clinical outcome of individuals carrying 5T;TG12 in trans with CFTR variants with varying clinical consequences. Pediatr. Pulmonol. 2023, 58, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Bombieri, C.; Claustres, M.; De Boeck, K.; Derichs, N.; Dodge, J.; Girodon, E.; Sermet, I.; Schwarz, M.; Tzetis, M.; Bareil, C.; et al. Recommendations for the classification of diseases as CFTR-related disorders. J. Cyst. Fibros. 2011, 10 (Suppl. S2), S86–S102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellani, C.; De Boeck, K.; De Wachter, E.; Sermet-Gaudelus, I.; Simmonds, N.J.; Southern, K.W.; ECFS Diagnostic Network Working Group. ECFS standards of care on CFTR-related disorders: Updated diagnostic criteria. J. Cyst. Fibros. 2022, 21, 908–921. [Google Scholar] [CrossRef]
- Sermet-Gaudelus, I.; Girodon, E.; Vermeulen, F.; Solomon, G.M.; Melotti, P.; Graeber, S.Y.; Bronsveld, I.; Rowe, S.; Wilschanski, M.; Tümmler, B.; et al. ECFS standards of care on CFTR-related disorders: Diagnostic criteria of CFTR dysfunction. J. Cyst. Fibros. 2022, 21, 922–936. [Google Scholar] [CrossRef]
- Terlizzi, V.; Castaldo, G.; Salvatore, D.; Lucarelli, M.; Raia, V.; Angioni, A.; Carnovale, V.; Cirilli, N.; Casciaro, R.; Colombo, C.; et al. Genotype-phenotype correlation and functional studies in patients with cystic fibrosis bearing CFTR complex alleles. J. Med. Genet. 2017, 54, 224–235. [Google Scholar] [CrossRef] [Green Version]
- Hatton, A.; Bergougnoux, A.; Zybert, K.; Chevalier, B.; Mesbahi, M.; Altéri, J.P.; Walicka-Serzysko, K.; Postek, M.; Taulan-Cadars, M.; Edelman, A.; et al. Reclassifying inconclusive diagnosis after newborn screening for cystic fibrosis. Moving forward. J. Cyst. Fibros. 2022, 21, 448–455. [Google Scholar] [CrossRef]
- Davis, P.B. Cystic fibrosis. Pediatr. Rev. 2001, 22, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.L.; Borowitz, D.S.; Gonska, T.; Howenstine, M.S.; Levy, H.; Massie, J.; Milla, C.; Munck, A.; Southern, K.W. Cystic Fibrosis Transmembrane Conductance Regulator-Related Metabolic Syndrome and Cystic Fibrosis Screen Positive, Inconclusive Diagnosis. J. Pediatr. 2017, 181, S45–S51.e1. [Google Scholar] [CrossRef] [Green Version]
- LeGrys, V.A.; Yankaskas, J.R.; Quittell, L.M.; Marshall, B.C.; Mogayzel, P.J. Cystic Fibrosis Foundation. Diagnostic sweat testing: The Cystic Fibrosis Foundation guidelines. J. Pediatr. 2007, 151, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Borowitz, D. Update on the evolution of pancreatic exocrine status in cystic fibrosis. Curr. Opin. Pulm. Med. 2005, 11, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Quanjer, P.H.; Stanojevic, S.; Cole, T.J.; Baur, X.; Hall, G.L.; Culver, B.H.; Enright, P.L.; Hankinson, J.L.; Ip, M.S.M.; Zheng, J. Multiethnic reference values for spirometry for the 3–95 year age range: The global lung function 2012 equations. Eur. Respir. J. 2012, 40, 1324–1343. [Google Scholar] [CrossRef]
- Lee, T.W.; Brownlee, K.G.; Conway, S.P.; Denton, M.; Littlewood, J.M. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J. Cyst. Fibros. 2003, 2, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flume, P.A.; Mogayzel, P.J., Jr.; Robinson, K.A.; Goss, C.H.; Rosenblatt, R.L.; Kuhn, R.J.; Marshall, B.C.; Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: Treatment of pulmonary exacerbations. Am. J. Respir. Crit. Care Med. 2009, 180, 802–808. [Google Scholar] [CrossRef] [Green Version]
- Mou, H.; Vinarsky, V.; Tata, P.R.; Brazauskas, K.; Choi, S.; Crooke, A.K.; Zhang, B.; Solomon, G.M.; Turner, B.; Bihler, H.; et al. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell Stem Cell 2016, 19, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Sondo, E.; Tomati, V.; Caci, E.; Esposito, A.I.; Pfeffer, U.; Pedemonte, N.; Galietta, L.J.V. Rescue of the mutant CFTR chloride channel by pharmacological correctors and low temperature analyzed by gene expression profiling. Am. J. Physiol. Cell Physiol. 2011, 301, C872–C885. [Google Scholar] [CrossRef] [Green Version]
- Sondo, E.; Falchi, F.; Caci, E.; Ferrera, L.; Giacomini, E.; Pesce, E.; Tomati, V.; Bertozzi, S.M.; Goldoni, L.; Armirotti, A.; et al. Pharmacological Inhibition of the Ubiquitin Ligase RNF5 Rescues F508del-CFTR in Cystic Fibrosis Airway Epithelia. Cell Chem. Biol. 2018, 25, 891–905.e8. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Subject | IRT ^ (ng/mL) | Age at First Evaluation (m/y) | Reason for Sweat Testing | Second CFTR Variant | First SC Value | Diagnosis/Label at First Evaluation | Age at 31 September 2022 (Years) | Microbiological | Last BMI-Weight for Length Pc × | Last FEV1 | Last SC Value | Diagnosis/Label at Study End |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 ˜ | 31 | 20 m | Hypochloremic alkalosis | S737F | 45 | Inconclusive diagnosis | 14.5 | MSSA | 21.0 | 111 | 98 | Inconclusive diagnosis |
| 2 | 76 | 1 m | Positive NBS | F508del | 35 | CRMS/CFSPID | 15 | No pathogenic bacteria | 21.5 | 108 | 78 | CF |
| 3 ˜ | 108 | 1 m | Positive NBS | 541delC | 48 | CRMS/CFSPID | 13 | MSSA | 19.0 | 107 | 94 | CF |
| 4 ˜,# | 37 | 10 m | Hypochloremic alkalosis | 22, 23, 24 del | 71 | Inconclusive diagnosis | 20.5 | MSSA | 21.9 | 102 | 89 | Inconclusive diagnosis |
| 5 ˜,# | 64 | 1 m | Positive NBS | W1282X | 51 | CRMS/CFSPID | 20.8 | MSSA | 20.5 | 102 | 121 | CF |
| 6 | 65 | 1 m | Positive NBS | F508del | 51 | CRMS/CFSPID | 1.0 | No pathogenic bacteria | 50° | n.a | 48 | CRMS/CFSPID |
| 7 | 61 | 1 m | Positive NBS | G1069R | 25 | CRMS/CFSPID | 2.3 | No pathogenic bacteria | 16.0 | n.a | 20 | CRMS/CFSPID |
| 8 | 48 | 1 m | Positive NBS | F508del | 66 | CF | 2.5 | MSSA | 14.0 | n.a | 66 | CF |
| 9 | 76 | 1 m | Positive NBS | F508del | 51 | CRMS/CFSPID | 7.0 | MSSA | 15.4 | 116 | 69 | CF |
| 10 ˜,# | n.a | 40 y | Respiratory Symptoms | Exon scanning negative | 62 | Inconclusive diagnosis | 44.0 | No pathogenic bacteria | 29.0 | 95 | 82 | Inconclusive diagnosis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terlizzi, V.; Pesce, E.; Capurro, V.; Tomati, V.; Lena, M.; Pastorino, C.; Bocciardi, R.; Zara, F.; Centrone, C.; Taccetti, G.; et al. Clinical Consequences and Functional Impact of the Rare S737F CFTR Variant and Its Responsiveness to CFTR Modulators. Int. J. Mol. Sci. 2023, 24, 6576. https://doi.org/10.3390/ijms24076576
Terlizzi V, Pesce E, Capurro V, Tomati V, Lena M, Pastorino C, Bocciardi R, Zara F, Centrone C, Taccetti G, et al. Clinical Consequences and Functional Impact of the Rare S737F CFTR Variant and Its Responsiveness to CFTR Modulators. International Journal of Molecular Sciences. 2023; 24(7):6576. https://doi.org/10.3390/ijms24076576
Chicago/Turabian StyleTerlizzi, Vito, Emanuela Pesce, Valeria Capurro, Valeria Tomati, Mariateresa Lena, Cristina Pastorino, Renata Bocciardi, Federico Zara, Claudia Centrone, Giovanni Taccetti, and et al. 2023. "Clinical Consequences and Functional Impact of the Rare S737F CFTR Variant and Its Responsiveness to CFTR Modulators" International Journal of Molecular Sciences 24, no. 7: 6576. https://doi.org/10.3390/ijms24076576