
The Labyrinthine Landscape of APP Processing: State of the Art and Possible Novel Soluble APP-Related Molecular Players in Traumatic Brain Injury and Neurodegeneration
 , , , and
, , , and
Abstract
:1. Introduction
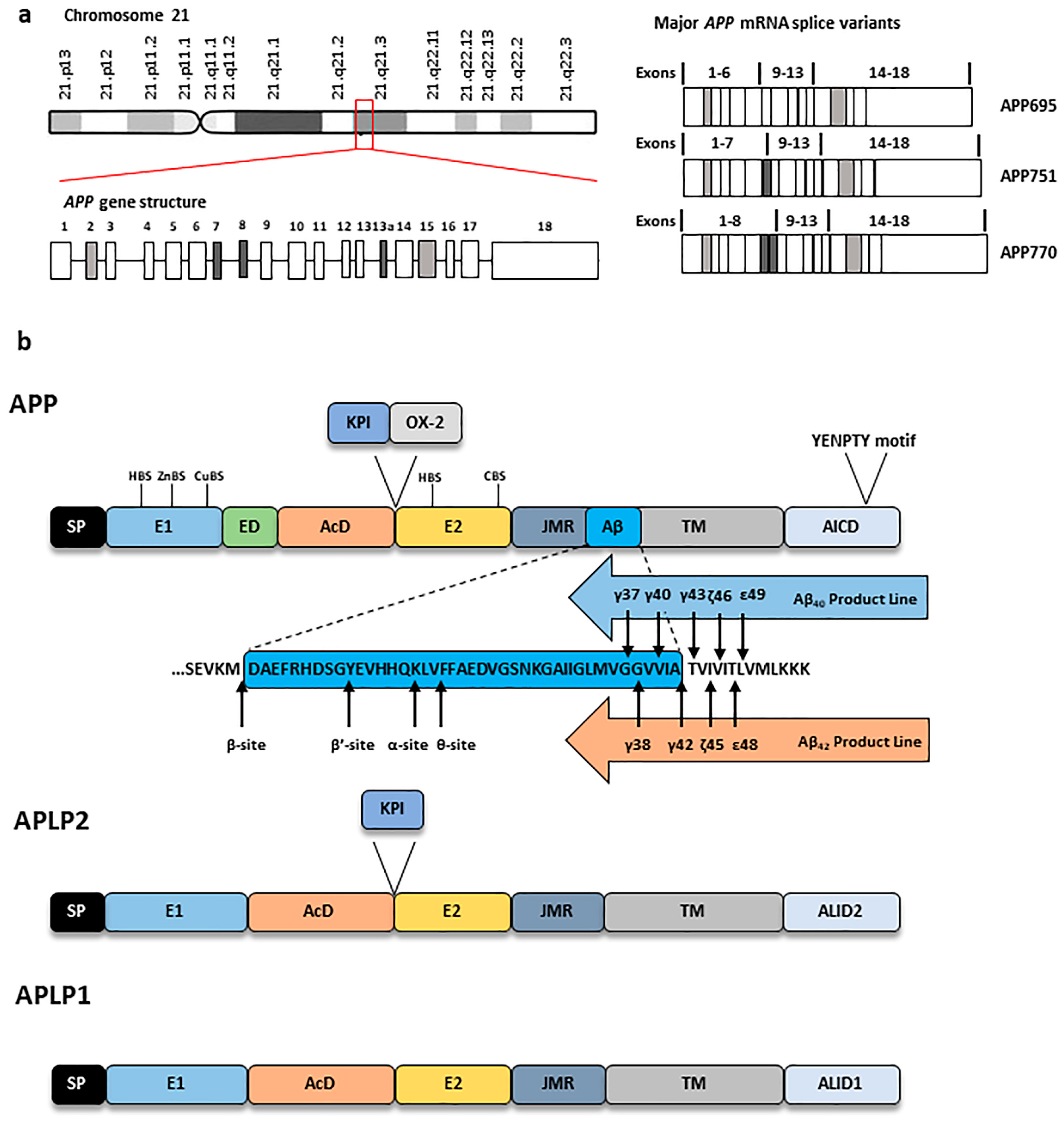
2. Amyloid Precursor Protein: Structure, Expression and Processing
2.1. APP Structure, Expression, Trafficking and Modification
2.1.1. APP Structure and Expression
2.1.2. APP Trafficking and Post-Translational Modifications (PTMs)
- Glycosylation and phosphorylation
- Palmitoylation, ubiquitination, SUMOylation and sulphation
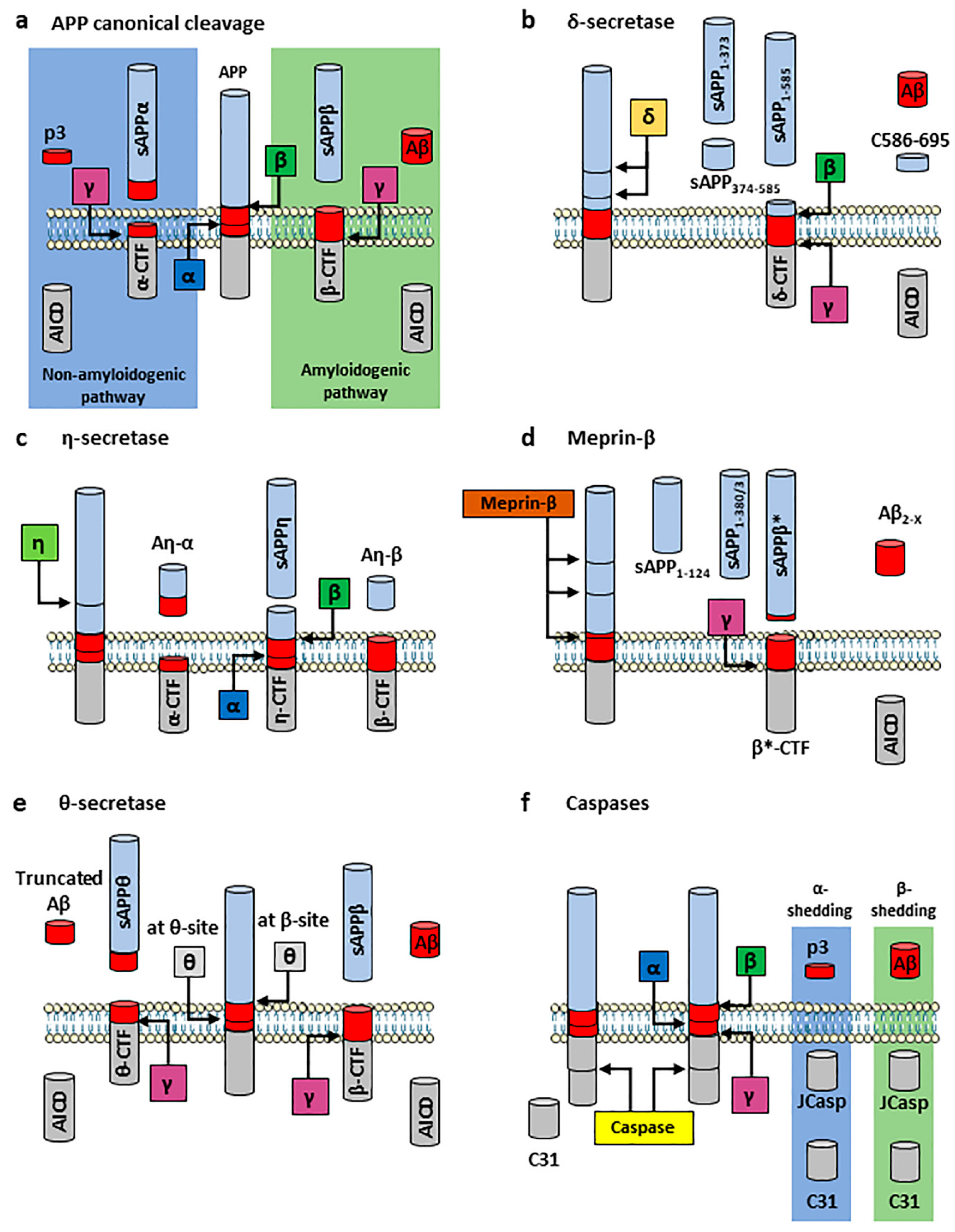
2.2. APP Processing
2.2.1. APP Canonical Cleavage: Amyloidogenic and Non-Amyloidogenic Pathways
2.2.2. APP Non-Canonical Cleavage
- δ-secretase
- η-secretase
- Meprin-β
- θ-secretase
- Other secretases
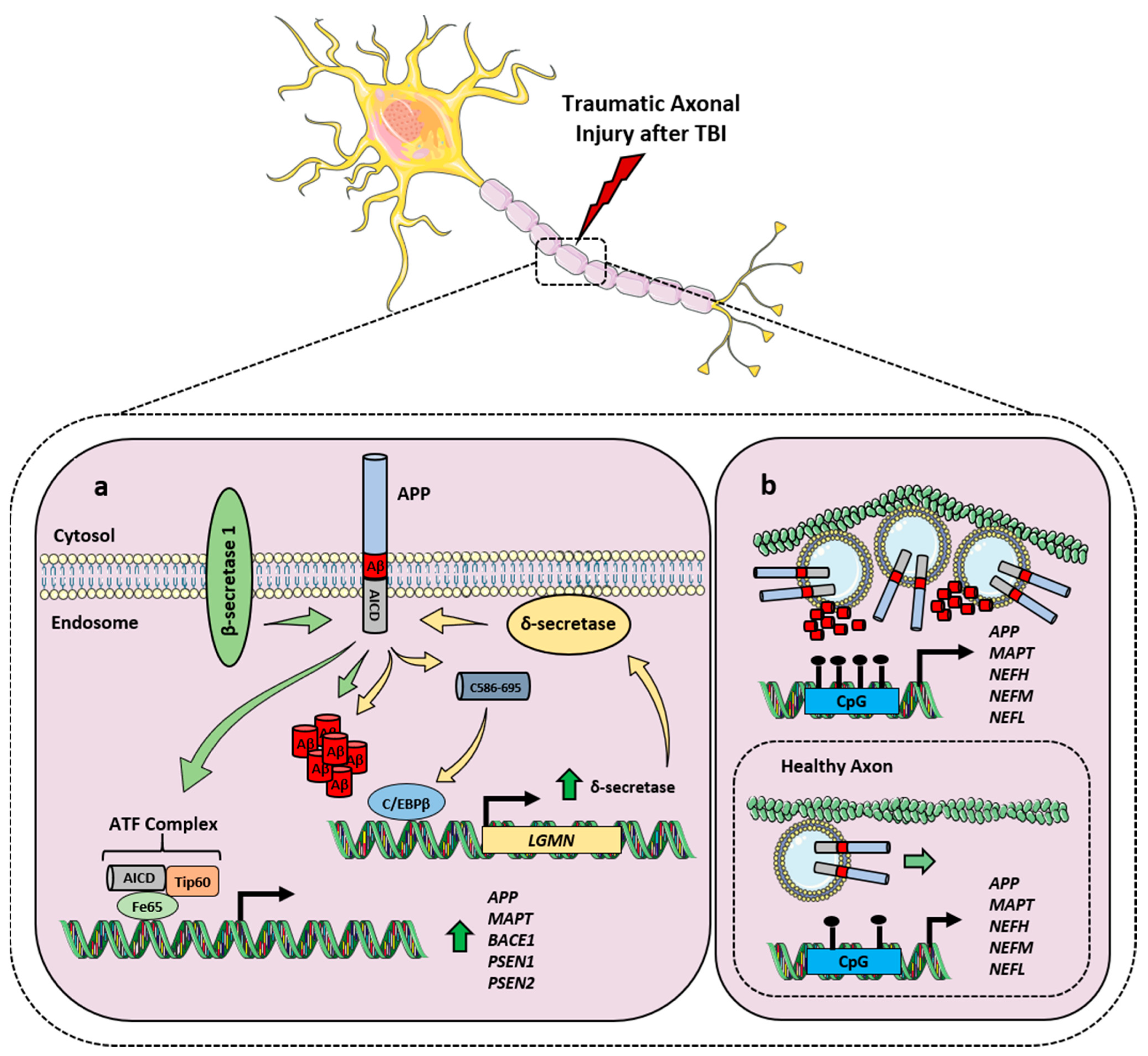
2.3. APP and Its Processing in TBI
3. Physiological and Pathological Roles of APP and Its Cleavage Fragments
4. Results
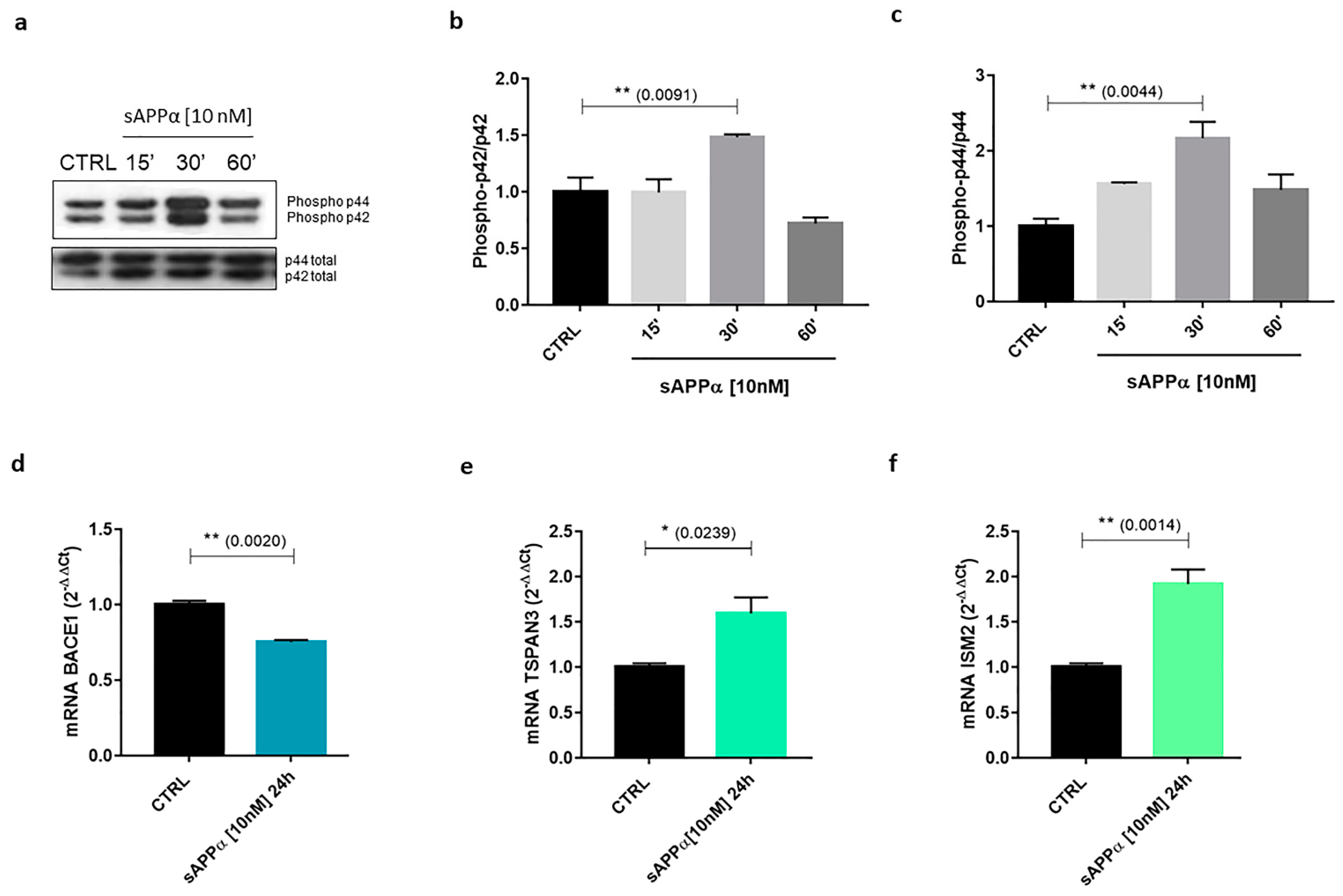
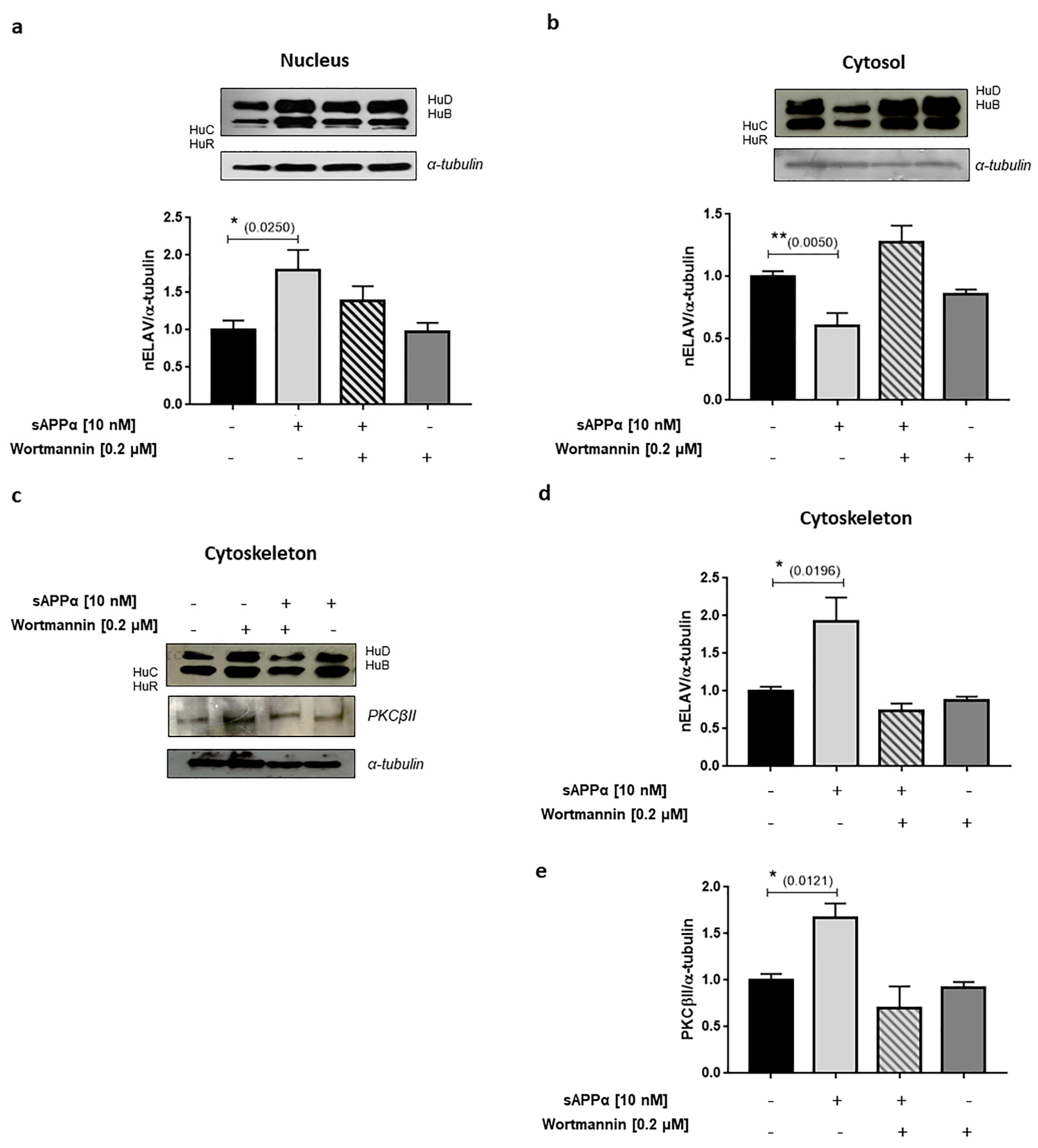
4.1. Neuron-Related Differential sAPPα-Mediated Gene Regulation In Vitro
4.2. BACE1
4.3. TSPAN3
4.4. ISM2
4.5. VEGFA
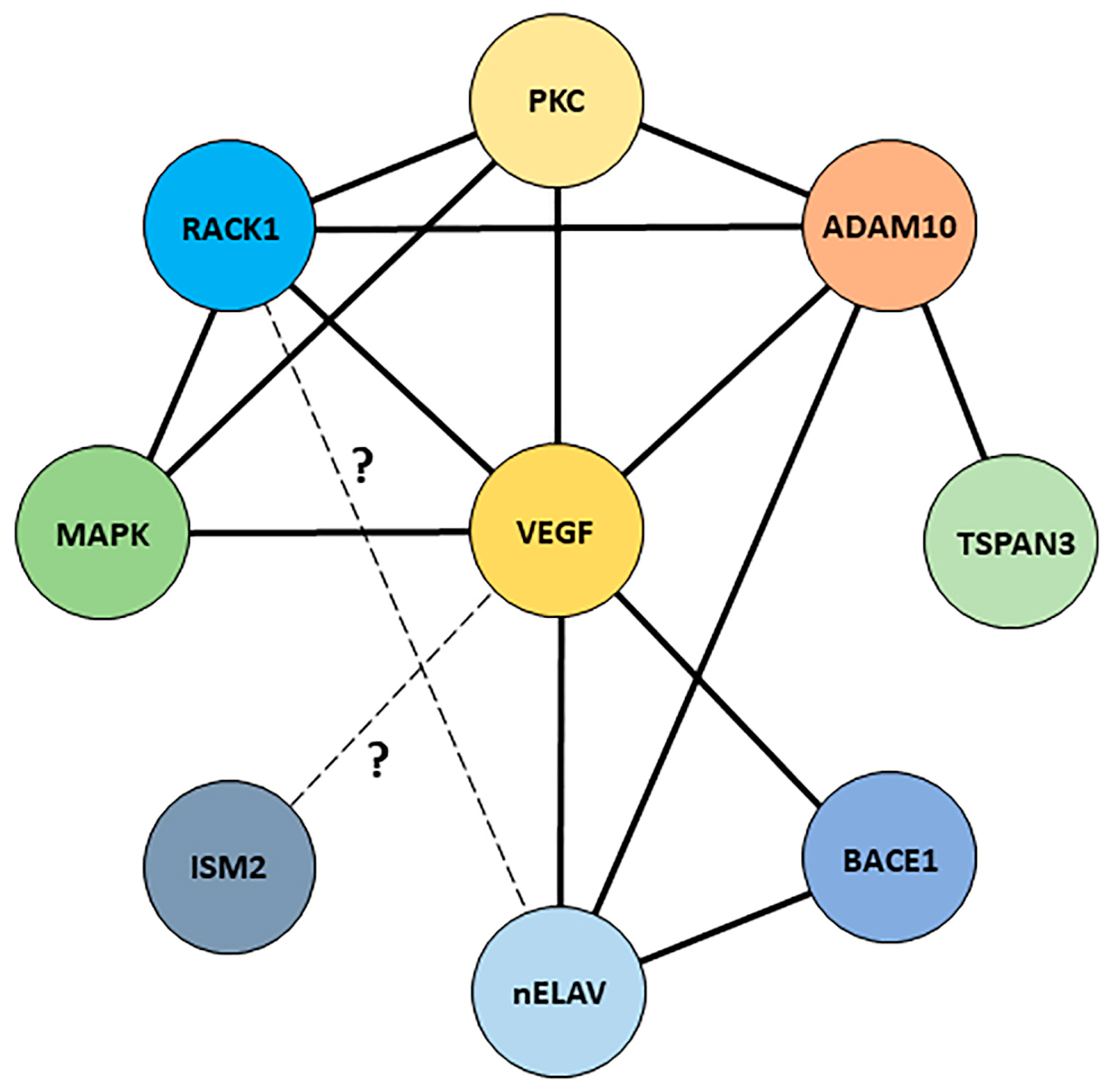
5. Discussion
5.1. A Possible Therapeutic Intervention via sAPPα?
5.2. A Putative Interaction Network for the Receptor for Activated C Kinase 1?
6. Materials and Methods
6.1. Chemicals
6.2. Cell Culture and Treatments
6.3. Subcellular Fractionation
6.4. Plasmid DNA Preparation, Transient Transfections and Luciferase Assays
6.5. qPCR
6.6. mRNA Stability Analysis
6.7. Immunoblot
6.8. Densitometry and Statistics
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef]
- Masi, M.; Attanzio, A.; Racchi, M.; Wolozin, B.; Borella, S.; Biundo, F.; Buoso, E. Proteostasis Deregulation in Neurodegeneration and Its Link with Stress Granules: Focus on the Scaffold and Ribosomal Protein RACK1. Cells 2022, 11, 2590. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Koo, E.H. Biology and pathophysiology of the amyloid precursor protein. Mol. Neurodegener. 2011, 6, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racchi, M.; Mazzucchelli, M.; Porrello, E.; Lanni, C.; Govoni, S. Acetylcholinesterase inhibitors: Novel activities of old molecules. Pharmacol. Res. 2004, 50, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Esiri, M.M. Ageing and the brain. J. Pathol. 2007, 211, 181–187. [Google Scholar] [CrossRef]
- Mattson, M.P.; Maudsley, S.; Martin, B. BDNF and 5-HT: A dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004, 27, 589–594. [Google Scholar] [CrossRef]
- Lanni, C.; Masi, M.; Racchi, M.; Govoni, S. Cancer and Alzheimer’s disease inverse relationship: An age-associated diverging derailment of shared pathways. Mol. Psychiatry 2021, 26, 280–295. [Google Scholar] [CrossRef]
- Green, R.C.; Cupples, L.A.; Kurz, A.; Auerbach, S.; Go, R.; Sadovnick, D.; Duara, R.; Kukull, W.A.; Chui, H.; Edeki, T.; et al. Depression as a risk factor for Alzheimer disease: The MIRAGE Study. Arch. Neurol. 2003, 60, 753–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diniz, B.S.; Butters, M.A.; Albert, S.M.; Dew, M.A.; Reynolds, C.F., 3rd. Late-life depression and risk of vascular dementia and Alzheimer’s disease: Systematic review and meta-analysis of community-based cohort studies. Br. J. Psychiatry 2013, 202, 329–335. [Google Scholar] [CrossRef]
- Gorelick, P.B. Risk factors for vascular dementia and Alzheimer disease. Stroke 2004, 35 (Suppl. S1), 2620–2622. [Google Scholar] [CrossRef] [Green Version]
- Danna-Dos-Santos, A.; Mohapatra, S.; Santos, M.; Degani, A.M. Long-term effects of mild traumatic brain injuries to oculomotor tracking performances and reaction times to simple environmental stimuli. Sci. Rep. 2018, 8, 4583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Freitas Cardoso, M.G.; Faleiro, R.M.; de Paula, J.J.; Kummer, A.; Caramelli, P.; Teixeira, A.L.; de Souza, L.C.; Miranda, A.S. Cognitive Impairment Following Acute Mild Traumatic Brain Injury. Front. Neurol. 2019, 10, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LoBue, C.; Wadsworth, H.; Wilmoth, K.; Clem, M.; Hart, J., Jr.; Womack, K.B.; Didehbani, N.; Lacritz, L.H.; Rossetti, H.C.; Cullum, C.M. Traumatic brain injury history is associated with earlier age of onset of Alzheimer disease. Clin. Neuropsychol. 2017, 31, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacono, D.; Raiciulescu, S.; Olsen, C.; Perl, D.P. Traumatic Brain Injury Exposure Lowers Age of Cognitive Decline in AD and Non-AD Conditions. Front. Neurol. 2021, 12, 573401. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel, C.; Blumbergs, P.C.; Finnie, J.W.; Manavis, J.; Jones, N.R.; Reilly, P.L.; Pereira, R.A. Upregulation of amyloid precursor protein messenger RNA in response to traumatic brain injury: An ovine head impact model. Exp. Neurol. 1999, 159, 441–450. [Google Scholar] [CrossRef]
- Nadler, Y.; Alexandrovich, A.; Grigoriadis, N.; Hartmann, T.; Rao, K.S.; Shohami, E.; Stein, R. Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 2008, 56, 552–567. [Google Scholar] [CrossRef]
- Blasko, I.; Beer, R.; Bigl, M.; Apelt, J.; Franz, G.; Rudzki, D.; Ransmayr, G.; Kampfl, A.; Schliebs, R. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer’s disease beta-secretase (BACE-1). J. Neural. Transm. 2004, 111, 523–536. [Google Scholar] [CrossRef]
- Galvão, F., Jr.; Grokoski, K.C.; da Silva, B.B.; Lamers, M.L.; Siqueira, I.R. The amyloid precursor protein (APP) processing as a biological link between Alzheimer’s disease and cancer. Ageing Res. Rev. 2019, 49, 83–91. [Google Scholar] [CrossRef]
- Corrigan, F.; Pham, C.L.; Vink, R.; Blumbergs, P.C.; Masters, C.L.; van den Heuvel, C.; Cappai, R. The neuroprotective domains of the amyloid precursor protein, in traumatic brain injury, are located in the two growth factor domains. Brain Res. 2011, 1378, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, Q.; Chen, S.; Xu, C. Functions of amyloid precursor protein in metabolic diseases. Metabolism 2021, 115, 154454. [Google Scholar] [CrossRef]
- Baumkötter, F.; Schmidt, N.; Vargas, C.; Schilling, S.; Weber, R.; Wagner, K.; Fiedler, S.; Klug, W.; Radzimanowski, J.; Nickolaus, S.; et al. Amyloid precursor protein dimerization and synaptogenic function depend on copper binding to the growth factor-like domain. J. Neurosci. 2014, 34, 11159–11172. [Google Scholar] [CrossRef] [Green Version]
- Sisodia, S.S.; Koo, E.H.; Hoffman, P.N.; Perry, G.; Price, D.L. Identification and transport of full-length amyloid precursor proteins in rat peripheral nervous system. J. Neurosci. 1993, 13, 3136–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludewig, S.; Korte, M. Novel Insights into the Physiological Function of the APP (Gene) Family and Its Proteolytic Fragments in Synaptic Plasticity. Front. Mol. Neurosci. 2017, 9, 161. [Google Scholar] [CrossRef] [Green Version]
- Erdinger, S.; Amrein, I.; Back, M.; Ludewig, S.; Korte, M.; von Engelhardt, J.; Wolfer, D.P.; Müller, U.C. Lack of APLP1 leads to subtle alter-ations in neuronal morphology but does not affect learning and memory. Front. Mol. Neurosci. 2022, 15, 1028836. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kang, J.; Ho, A.; Watanabe, H.; Bolshakov, V.Y.; Shen, J. APP Family Regulates Neuronal Excitability and Synaptic Plasticity but Not Neuronal Survival. Neuron 2020, 108, 676–690.e8. [Google Scholar] [CrossRef] [PubMed]
- Onodera, W.; Asahi, T.; Sawamura, N. Rapid evolution of mammalian APLP1 as a synaptic adhesion molecule. Sci. Rep. 2021, 11, 11305. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.; Mehr, A.; Ludewig, S.; Stephan, J.; Zimmermann, M.; August, A.; Strecker, P.; Korte, M.; Koo, E.H.; Müller, U.C.; et al. APLP1 Is a Synaptic Cell Adhesion Molecule, Supporting Maintenance of Dendritic Spines and Basal Synaptic Transmission. J. Neurosci. 2017, 37, 5345–5365. [Google Scholar] [CrossRef] [Green Version]
- Lim, B.; Tsolaki, M.; Soosaipillai, A.; Brown, M.; Zilakaki, M.; Tagaraki, F.; Fotiou, D.; Koutsouraki, E.; Grosi, E.; Prassas, I.; et al. Liquid biopsy of cerebrospinal fluid identifies neuronal pentraxin receptor (NPTXR) as a biomarker of progression of Alz-heimer’s disease. Clin. Chem. Lab. Med. 2019, 57, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, X.; Li, G.; Zhang, Y.; Wu, Y.; Song, W. Modifications and Trafficking of APP in the Pathogenesis of Alzheimer’s Disease. Front. Mol. Neurosci. 2017, 10, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, Y.S.; Park, Y.; Oh, H.G.; Kim, T.W.; Yang, H.O.; Park, M.K.; Chung, S. O-GlcNAcylation promotes non-amyloidogenic processing of amyloid-? protein precursor via inhibition of endocytosis from the plasma membrane. J. Alzheimers Dis. 2015, 44, 261–275. [Google Scholar] [CrossRef]
- Bhattacharyya, R.; Barren, C.; Kovacs, D.M. Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J. Neurosci. 2013, 33, 11169–11183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, E.; Chamoun, Z.; Lasiecka, Z.M.; Chan, R.B.; Williamson, R.L.; Vetanovetz, C.; Dall’Armi, C.; Simoes, S.; Point Du Jour, K.S.; McCabe, B.D.; et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 2013, 4, 2250. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Hikichi, Y.; Willuweit, A.; Shintani, Y.; Horiguchi, T. FBL2 regulates amyloid precursor protein (APP) metabolism by promoting ubiquitination-dependent APP degradation and inhibition of APP endocytosis. J. Neurosci. 2012, 32, 3352–3365. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Q.; Sarge, K.D. Sumoylation of amyloid precursor protein negatively regulates Abeta aggregate levels. Biochem. Biophys. Res. Commun. 2008, 374, 673–678. [Google Scholar] [CrossRef] [Green Version]
- Andrew, R.J.; Kellett, K.A.; Thinakaran, G.; Hooper, N.M. A Greek Tragedy: The Growing Complexity of Alzheimer Amyloid Precursor Protein Proteolysis. J. Biol. Chem. 2016, 291, 19235–19244. [Google Scholar] [CrossRef] [Green Version]
- Edbauer, D.; Winkler, E.; Regula, J.T.; Pesold, B.; Steiner, H.; Haass, C. Reconstitution of gamma-secretase activity. Nat. Cell Biol. 2003, 5, 486–488. [Google Scholar] [CrossRef]
- Kimura, A.; Hata, S.; Suzuki, T. Alternative Selection of β-Site APP-Cleaving Enzyme 1 (BACE1) Cleavage Sites in Amyloid β-Protein Precursor (APP) Harboring Protective and Pathogenic Mutations within the Aβ Sequence. J. Biol. Chem. 2016, 291, 24041–24053. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Tsai, L.H. Bridging physiology and pathology in AD. Cell 2009, 137, 997–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Olsen, O.; Tzvetkova-Robev, D.; Tessier-Lavigne, M.; Nikolov, D.B. The crystal structure of DR6 in complex with the amyloid precursor protein provides insight into death receptor activation. Genes Dev. 2015, 29, 785–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buoso, E.; Lanni, C.; Schettini, G.; Govoni, S.; Racchi, M. Beta-Amyloid precursor protein metabolism: Focus on the functions and degradation of its intracellular domain. Pharmacol. Res. 2010, 62, 308–317. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Hamdane, M.; Bégard, S.; Loyens, A.; Delacourte, A.; Beauvillain, J.C.; Buée, L.; Marambaud, P.; Sergeant, N. Intracellular pH regulates amyloid precursor protein intracellular domain accumulation. Neurobiol. Dis. 2007, 25, 686–696. [Google Scholar] [CrossRef]
- Edbauer, D.; Willem, M.; Lammich, S.; Steiner, H.; Haass, C. Insulin-degrading enzyme rapidly removes the beta-amyloid precursor protein intracellular domain (AICD). J. Biol. Chem. 2002, 277, 13389–13393. [Google Scholar] [CrossRef] [Green Version]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Südhof, T.C. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 2001, 293, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Grösgen, S.; Haupenthal, V.J.; Blümel, T.; Hundsdörfer, B.; Zimmer, V.C.; Mylonas, N.T.; Tanila, H.; et al. APP intracellular domain derived from amyloidogenic β- and γ-secretase cleavage regulates neprilysin expression. Front. Aging Neurosci. 2015, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Konietzko, U. AICD nuclear signaling and its possible contribution to Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 200–216. [Google Scholar] [CrossRef]
- Von Rotz, R.C.; Kohli, B.M.; Bosset, J.; Meier, M.; Suzuki, T.; Nitsch, R.M.; Konietzko, U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J. Cell Sci. 2004, 117, 4435–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheinfeld, M.H.; Ghersi, E.; Laky, K.; Fowlkes, B.J.; D’Adamio, L. Processing of beta-amyloid precursor-like protein-1 and -2 by gamma-secretase regulates transcription. J. Biol. Chem. 2002, 277, 44195–44201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, H.J.; Shen, J.; Kang, J. Regulation of gene expression by the APP family in the adult cerebral cortex. Sci. Rep. 2022, 12, 66. [Google Scholar] [CrossRef]
- Xu, X. Gamma-secretase catalyzes sequential cleavages of the AbetaPP transmembrane domain. J. Alzheimers Dis. 2009, 16, 211–224. [Google Scholar] [CrossRef] [Green Version]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. gamma-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hur, J.Y. γ-Secretase in Alzheimer’s disease. Exp. Mol. Med. 2022, 54, 433–446. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Nhan, H.S.; Chiang, K.; Koo, E.H. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol. 2015, 129, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Song, M.; Liu, X.; Kang, S.S.; Kwon, I.S.; Duong, D.M.; Seyfried, N.T.; Hu, W.T.; Liu, Z.; Wang, J.Z.; et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat. Med. 2014, 20, 1254–1262. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Song, M.; Liu, X.; Su Kang, S.; Duong, D.M.; Seyfried, N.T.; Cao, X.; Cheng, L.; Sun, Y.E.; Ping, Y.S.; et al. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat. Commun. 2015, 6, 8762. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Kang, S.S.; Xia, Y.; Wang, Z.H.; Liu, X.; Muller, T.; Sun, Y.E.; Ye, K. A delta-secretase-truncated APP fragment activates CEBPB, mediating Alzheimer’s disease pathologies. Brain 2021, 144, 1833–1852. [Google Scholar] [CrossRef] [PubMed]
- Baranger, K.; Khrestchatisky, M.; Rivera, S. MT5-MMP, just a new APP processing proteinase in Alzheimer’s disease? J. Neuroinflammation 2016, 13, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranger, K.; Marchalant, Y.; Bonnet, A.E.; Crouzin, N.; Carrete, A.; Paumier, J.M.; Py, N.A.; Bernard, A.; Bauer, C.; Charrat, E.; et al. MT5-MMP is a new pro-amyloidogenic proteinase that promotes amyloid pathology and cognitive decline in a transgenic mouse model of Alzheimer’s disease. Cell Mol. Life Sci. 2016, 73, 217–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willem, M.; Tahirovic, S.; Busche, M.A.; Ovsepian, S.V.; Chafai, M.; Kootar, S.; Hornburg, D.; Evans, L.D.; Moore, S.; Daria, A.; et al. η-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 2015, 526, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Guillamon, M.; Mawhirt, S.; Blais, S.; Montaner, J.; Neubert, T.A.; Rostagno, A.; Ghiso, J. Sequential Amyloid-β Degradation by the Matrix Metalloproteases MMP-2 and MMP-9. J. Biol. Chem. 2015, 290, 15078–15091. [Google Scholar] [CrossRef] [Green Version]
- Fragkouli, A.; Tsilibary, E.C.; Tzinia, A.K. Neuroprotective role of MMP-9 overexpression in the brain of Alzheimer’s 5xFAD mice. Neurobiol. Dis. 2014, 70, 179–189. [Google Scholar] [CrossRef]
- Schönherr, C.; Bien, J.; Isbert, S.; Wichert, R.; Prox, J.; Altmeppen, H.; Kumar, S.; Walter, J.; Lichtenthaler, S.F.; Weggen, S.; et al. Generation of aggregation prone N-terminally truncated amyloid β peptides by meprin β depends on the sequence specificity at the cleavage site. Mol. Neurodegener. 2016, 11, 19. [Google Scholar] [CrossRef] [Green Version]
- Bien, J.; Jefferson, T.; Causević, M.; Jumpertz, T.; Munter, L.; Multhaup, G.; Weggen, S.; Becker-Pauly, C.; Pietrzik, C.U. The metalloprotease meprin β generates amino terminal-truncated amyloid β peptide species. J. Biol. Chem. 2012, 287, 33304–33313. [Google Scholar] [CrossRef] [Green Version]
- Jefferson, T.; Čaušević, M.; auf dem Keller, U.; Schilling, O.; Isbert, S.; Geyer, R.; Maier, W.; Tschickardt, S.; Jumpertz, T.; Weggen, S.; et al. Metalloprotease meprin beta generates nontoxic N-terminal amyloid precursor protein fragments in vivo. J. Biol. Chem. 2011, 286, 27741–27750. [Google Scholar] [CrossRef] [Green Version]
- Jäckle, F.; Schmidt, F.; Wichert, R.; Arnold, P.; Prox, J.; Mangold, M.; Ohler, A.; Pietrzik, C.U.; Koudelka, T.; Tholey, A.; et al. Metalloprotease meprin β is activated by transmembrane serine protease matriptase-2 at the cell surface thereby enhancing APP shedding. Biochem. J. 2015, 470, 91–103. [Google Scholar] [CrossRef]
- Sun, X.; He, G.; Song, W. BACE2, as a novel APP theta-secretase, is not responsible for the pathogenesis of Alzheimer’s disease in Down syndrome. FASEB J. 2006, 20, 1369–1376. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, Q.; Cai, F.; Liu, X.; Wu, Y.; Song, W. BACE2, a conditional β-secretase, contributes to Alzheimer’s disease pathogenesis. JCI Insight 2019, 4, e123431. [Google Scholar] [CrossRef] [Green Version]
- Qiu, K.; Liang, W.; Wang, S.; Kong, T.; Wang, X.; Li, C.; Wang, Z.; Wu, Y. BACE2 degradation is mediated by both the proteasome and lysosome pathways. BMC Mol. Cell Biol. 2020, 21, 13. [Google Scholar] [CrossRef] [PubMed]
- Portelius, E.; Mattsson, N.; Andreasson, U.; Blennow, K.; Zetterberg, H. Novel Aβisoforms in Alzheimer’s disease-their role in diagnosis and treatment. Curr. Pharm. Des. 2011, 17, 2594–2602. [Google Scholar] [CrossRef] [PubMed]
- Welzel, A.T.; Maggio, J.E.; Shankar, G.M.; Walker, D.E.; Ostaszewski, B.L.; Li, S.; Klyubin, I.; Rowan, M.J.; Seubert, P.; Walsh, D.M.; et al. Secreted amyloid β-proteins in a cell culture model include N-terminally extended peptides that impair synaptic plasticity. Biochemistry 2014, 53, 3908–3921. [Google Scholar] [CrossRef] [PubMed]
- Portelius, E.; Brinkmalm, G.; Tran, A.; Andreasson, U.; Zetterberg, H.; Westman-Brinkmalm, A.; Blennow, K.; Ohrfelt, A. Identification of novel N-terminal fragments of amyloid precursor protein in cerebrospinal fluid. Exp. Neurol. 2010, 223, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Dawkins, E.; Gasperini, R.; Hu, Y.; Cui, H.; Vincent, A.J.; Bolós, M.; Young, K.M.; Foa, L.; Small, D.H. The N-terminal fragment of the β-amyloid precursor protein of Alzheimer’s disease (N-APP) binds to phosphoinositide-rich domains on the surface of hippocampal neurons. J. Neurosci. Res. 2014, 92, 1478–1489. [Google Scholar] [CrossRef]
- Fanutza, T.; Del Prete, D.; Ford, M.J.; Castillo, P.E.; D’Adamio, L. APP and APLP2 interact with the synaptic release machinery and facilitate transmitter release at hippocampal synapses. eLife 2015, 4, e09743. [Google Scholar] [CrossRef]
- García-González, L.; Pilat, D.; Baranger, K.; Rivera, S. Emerging Alternative Proteinases in APP Metabolism and Alzheimer’s Disease Pathogenesis: A Focus on MT1-MMP and MT5-MMP. Front. Aging Neurosci. 2019, 11, 244. [Google Scholar] [CrossRef]
- Sun, B.; Zhou, Y.; Halabisky, B.; Lo, I.; Cho, S.H.; Mueller-Steiner, S.; Devidze, N.; Wang, X.; Grubb, A.; Gan, L. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron 2008, 60, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Hook, V.Y.; Kindy, M.; Hook, G. Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J. Biol. Chem. 2008, 283, 7745–7753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; De Camilli, P.; Ferguson, S.M. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hook, G.; Yu, J.; Toneff, T.; Kindy, M.; Hook, V. Brain pyroglutamate amyloid-β is produced by cathepsin B and is reduced by the cysteine protease inhibitor E64d, representing a potential Alzheimer’s disease therapeutic. J Alzheimer’s Dis. 2014, 41, 129–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, A.Z.; Nestor, P.J.; Cumming, P.; Nasrallah, F.A.; Alzheimer’s Disease Neuroimaging Initiative. Traumatic brain injury fast-forwards Alzheimer’s pathology: Evidence from amyloid positron emission tomorgraphy imaging. J. Neurol. 2022, 269, 873–884. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Peltz, C.B.; Mustapic, M.; Kapogiannis, D.; Yaffe, K. Neuron-Derived Plasma Exosome Proteins after Remote Traumatic Brain Injury. J. Neurotrauma. 2020, 37, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Bogoslovsky, T.; Wilson, D.; Chen, Y.; Hanlon, D.; Gill, J.; Jeromin, A.; Song, L.; Moore, C.; Gong, Y.; Kenney, K.; et al. Increases of Plasma Levels of Glial Fibrillary Acidic Protein, Tau, and Amyloid β up to 90 Days after Traumatic Brain Injury. J. Neurotrauma. 2017, 34, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Tung, Y.C.; Zhang, Y.; Liu, F.; Iqbal, K. Involvement of Activation of Asparaginyl Endopeptidase in Tau Hyperphosphorylation in Repetitive Mild Traumatic Brain Injury. J. Alzheimer’s Dis. 2018, 64, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Wang, Z.H.; Liu, X.; Zhang, Z.; Gu, X.; Yu, S.P.; Keene, C.D.; Cheng, L.; Ye, K. Traumatic brain injury triggers APP and Tau cleavage by delta-secretase, mediating Alzheimer’s disease pathology. Prog. Neurobiol. 2020, 185, 101730. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, C.; Kang, S.S.; Liu, X.; Gu, X.; Yu, S.P.; Keene, C.D.; Cheng, L.; Ye, K. Neurotrophic signaling deficiency exacerbates environmental risks for Alzheimer’s disease pathogenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2100986118. [Google Scholar] [CrossRef]
- Hook, G.; Reinheckel, T.; Ni, J.; Wu, Z.; Kindy, M.; Peters, C.; Hook, V. Cathepsin B Gene Knockout Improves Behavioral Deficits and Reduces Pathology in Models of Neurologic Disorders. Pharmacol. Rev. 2022, 74, 600–629. [Google Scholar] [CrossRef]
- Stone, J.R.; Okonkwo, D.O.; Singleton, R.H.; Mutlu, L.K.; Helm, G.A.; Povlishock, J.T. Caspase-3-mediated cleavage of amyloid precursor protein and formation of amyloid Beta peptide in traumatic axonal injury. J. Neurotrauma 2002, 19, 601–614. [Google Scholar] [CrossRef]
- Abu Hamdeh, S.; Ciuculete, D.M.; Sarkisyan, D.; Bakalkin, G.; Ingelsson, M.; Schiöth, H.B.; Marklund, N. Differential DNA Methylation of the Genes for Amyloid Precursor Protein, Tau, and Neurofilaments in Human Traumatic Brain Injury. J. Neurotrauma 2021, 38, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarraj, S.; Troakes, C.; Rutty, G.N. Axonal injury is detected by βAPP immunohistochemistry in rapid death from head injury following road traffic collision. Int. J. Leg. Med. 2022, 136, 1321–1339. [Google Scholar] [CrossRef] [PubMed]
- Chaves, R.S.; Tran, M.; Holder, A.R.; Balcer, A.M.; Dickey, A.M.; Roberts, E.A.; Bober, B.G.; Gutierrez, E.; Head, B.P.; Groisman, A.; et al. Amyloidogenic Processing of Amyloid Precursor Protein Drives Stretch-Induced Disruption of Axonal Transport in hiPSC-Derived Neurons. J. Neurosci. 2021, 41, 10034–10053. [Google Scholar] [CrossRef] [PubMed]
- Freude, K.K.; Penjwini, M.; Davis, J.L.; LaFerla, F.M.; Blurton-Jones, M. Soluble amyloid precursor protein induces rapid neural differentiation of human embryonic stem cells. J. Biol. Chem. 2011, 286, 24264–24274. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, K.; Sopher, B.L.; Rydel, R.E.; Begley, J.G.; Pham, D.G.; Martin, G.M.; Fox, M.; Mattson, M.P. Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J. Neurochem. 1996, 67, 1882–1896. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.J.; Ireland, D.R.; Ballagh, I.; Bourne, K.; Marechal, N.M.; Turner, P.R.; Bilkey, D.K.; Tate, W.P.; Abraham, W.C. Endogenous secreted amyloid precursor protein-alpha regulates hippocampal NMDA receptor function, long-term potentiation and spatial memory. Neurobiol. Dis. 2008, 31, 250–260. [Google Scholar] [CrossRef]
- Austin, S.A.; Combs, C.K. Mechanisms of microglial activation by amyloid precursor protein and its proteolytic fragments. In Central Nervous System Diseases and Inflammation, 2008th ed.; Lane, T.E., Carson, M., Bergmann, C., Wyss-Coray, T., Eds.; Springer: New York, NY, USA, 2008; pp. 13–32. [Google Scholar]
- Barger, S.W.; Harmon, A.D. Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 1997, 388, 878–881. [Google Scholar] [CrossRef]
- Tamayev, R.; Matsuda, S.; Arancio, O.; D’Adamio, L. β- but not γ-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol. Med. 2012, 4, 171–179. [Google Scholar] [CrossRef]
- Bittner, T.; Fuhrmann, M.; Burgold, S.; Jung, C.K.; Volbracht, C.; Steiner, H.; Mitteregger, G.; Kretzschmar, H.A.; Haass, C.; Herms, J. Gamma-secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J. Neurosci. 2009, 29, 10405–10409. [Google Scholar] [CrossRef] [Green Version]
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012, 32, 16243–16255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitani, Y.; Yarimizu, J.; Saita, K.; Uchino, H.; Akashiba, H.; Shitaka, Y.; Ni, K.; Matsuoka, N. Differential effects between β-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J. Neurosci. 2012, 32, 2037–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yankner, B.A.; Dawes, L.R.; Fisher, S.; Villa-Komaroff, L.; Oster-Granite, M.L.; Neve, R.L. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science 1989, 245, 417–420. [Google Scholar] [CrossRef]
- Neve, R.L.; Kammesheidt, A.; Hohmann, C.F. Brain transplants of cells expressing the carboxyl-terminal fragment of the Alzheimer amyloid protein precursor cause specific neuropathology in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 3448–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.K.; Won, M.H.; Jung, J.S.; Lee, J.C.; Kang, T.C.; Suh, H.W.; Huh, S.O.; Paek, S.H.; Kim, Y.H.; Kim, S.H.; et al. Behavioral and neuropathologic changes induced by central injection of carboxyl-terminal fragment of beta-amyloid precursor protein in mice. J. Neurochem. 1998, 71, 875–878. [Google Scholar] [CrossRef]
- Fukuchi, K.I.; Kunkel, D.D.; Schwartzkroin, P.A.; Kamino, K.; Ogburn, C.E.; Furlong, C.E.; Martin, G.M. Overexpression of a C-terminal portion of the beta-amyloid precursor protein in mouse brains by transplantation of transformed neuronal cells. Exp. Neurol. 1994, 127, 253–264. [Google Scholar] [CrossRef]
- Multhaup, G.; Huber, O.; Buée, L.; Galas, M.C. Amyloid Precursor Protein (APP) Metabolites APP Intracellular Fragment (AICD), Aβ42, and Tau in Nuclear Roles. J. Biol. Chem. 2015, 290, 23515–23522. [Google Scholar] [CrossRef] [Green Version]
- Beckett, C.; Nalivaeva, N.N.; Belyaev, N.D.; Turner, A.J. Nuclear signalling by membrane protein intracellular domains: The AICD enigma. Cell Signal. 2012, 24, 402–409. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, E.M.; Lee, J.P.; Park, C.H.; Kim, S.; Seo, J.H.; Chang, K.A.; Yu, E.; Jeong, S.J.; Chong, Y.H.; et al. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3beta expression. FASEB J. 2003, 17, 1951–1953. [Google Scholar] [CrossRef]
- Ozaki, T.; Li, Y.; Kikuchi, H.; Tomita, T.; Iwatsubo, T.; Nakagawara, A. The intracellular domain of the amyloid precursor protein (AICD) enhances the p53-mediated apoptosis. Biochem. Biophys. Res. Commun. 2006, 351, 57–63. [Google Scholar] [CrossRef]
- Passer, B.; Pellegrini, L.; Russo, C.; Siegel, R.M.; Lenardo, M.J.; Schettini, G.; Bachmann, M.; Tabaton, M.; D’Adamio, L. Generation of an apoptotic intracellular peptide by gamma-secretase cleavage of Alzheimer’s amyloid beta protein precursor. J. Alzheimers Dis. 2000, 2, 289–301. [Google Scholar] [CrossRef]
- Milosch, N.; Tanriöver, G.; Kundu, A.; Rami, A.; François, J.C.; Baumkötter, F.; Weyer, S.W.; Samanta, A.; Jäschke, A.; Brod, F.; et al. Holo-APP and G-protein-mediated signaling are required for sAPPα-induced activation of the Akt survival pathway. Cell Death Dis. 2014, 5, e1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chasseigneaux, S.; Allinquant, B. Functions of Aβ, sAPPα and sAPPβ: Similarities and differences. J. Neurochem. 2012, 120 (Suppl. S1), 99–108. [Google Scholar] [CrossRef] [PubMed]
- Small, D.H.; Nurcombe, V.; Reed, G.; Clarris, H.; Moir, R.; Beyreuther, K.; Masters, C.L. A heparin-binding domain in the amyloid protein precursor of Alzheimer’s disease is involved in the regulation of neurite outgrowth. J. Neurosci. 1994, 14, 2117–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrigan, F.; Vink, R.; Blumbergs, P.C.; Masters, C.L.; Cappai, R.; van den Heuvel, C. sAPPα rescues deficits in amyloid precursor protein knockout mice following focal traumatic brain injury. J. Neurochem. 2012, 122, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Fol, R.; Braudeau, J.; Ludewig, S.; Abel, T.; Weyer, S.W.; Roederer, J.P.; Brod, F.; Audrain, M.; Bemelmans, A.P.; Buchholz, C.J.; et al. Viral gene transfer of APPsα rescues synaptic failure in an Alzheimer’s disease mouse model. Acta Neuropathol. 2016, 131, 247–266. [Google Scholar] [CrossRef]
- Hick, M.; Herrmann, U.; Weyer, S.W.; Mallm, J.P.; Tschäpe, J.A.; Borgers, M.; Mercken, M.; Roth, F.C.; Draguhn, A.; Slomianka, L.; et al. Acute function of secreted amyloid precursor protein fragment APPsα in synaptic plasticity. Acta Neuropathol. 2015, 129, 21–37. [Google Scholar] [CrossRef]
- Jäger, S.; Leuchtenberger, S.; Martin, A.; Czirr, E.; Wesselowski, J.; Dieckmann, M.; Waldron, E.; Korth, C.; Koo, E.H.; Heneka, M.; et al. alpha-secretase mediated conversion of the amyloid precursor protein derived membrane stub C99 to C83 limits Abeta generation. J. Neurochem. 2009, 111, 1369–1382. [Google Scholar] [CrossRef]
- Tian, Y.; Crump, C.J.; Li, Y.M. Dual role of alpha-secretase cleavage in the regulation of gamma-secretase activity for amyloid production. J. Biol. Chem. 2010, 285, 32549–32556. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Azimova, R.; Kagan, B.L.; Nussinov, R.; Lal, R. Truncated beta-amyloid peptide channels provide an alternative mechanism for Alzheimer’s Disease and Down syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 6538–6543. [Google Scholar] [CrossRef] [Green Version]
- Gowing, E.; Roher, A.E.; Woods, A.S.; Cotter, R.J.; Chaney, M.; Little, S.P.; Ball, M.J. Chemical characterization of A beta 17-42 peptide, a component of diffuse amyloid deposits of Alzheimer disease. J. Biol. Chem. 1994, 269, 10987–10990. [Google Scholar] [CrossRef] [PubMed]
- Szczepanik, A.M.; Rampe, D.; Ringheim, G.E. Amyloid-beta peptide fragments p3 and p4 induce pro-inflammatory cytokine and chemokine production in vitro and in vivo. J. Neurochem. 2001, 77, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Rice, H.C.; de Malmazet, D.; Schreurs, A.; Frere, S.; Van Molle, I.; Volkov, A.N.; Creemers, E.; Vertkin, I.; Nys, J.; Ranaivoson, F.M.; et al. Secreted amyloid-β precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science 2019, 363, eaao4827. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sang, N.; Zhang, C.; Raghupathi, R.; Tanzi, R.E.; Saunders, A.; Cathepsin, L. Mediates the Degradation of Novel APP C-Terminal Fragments. Biochemistry 2015, 54, 2806–2816. [Google Scholar] [CrossRef] [Green Version]
- Armbrust, F.; Bickenbach, K.; Marengo, L.; Pietrzik, C.; Becker-Pauly, C. The Swedish dilemma-the almost exclusive use of APPswe-based mouse models impedes adequate evaluation of alternative β-secretases. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119164. [Google Scholar] [CrossRef]
- Vella, L.J.; Cappai, R. Identification of a novel amyloid precursor protein processing pathway that generates secreted N-terminal fragments. FASEB J. 2012, 26, 2930–2940. [Google Scholar] [CrossRef]
- Lu, D.C.; Rabizadeh, S.; Chandra, S.; Shayya, R.F.; Ellerby, L.M.; Ye, X.; Salvesen, G.S.; Koo, E.H.; Bredesen, D.E. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat. Med. 2000, 6, 397–404. [Google Scholar] [CrossRef]
- Bertrand, E.; Brouillet, E.; Caillé, I.; Bouillot, C.; Cole, G.M.; Prochiantz, A.; Allinquant, B. A short cytoplasmic domain of the amyloid precursor protein induces apoptosis in vitro and in vivo. Mol. Cell Neurosci. 2001, 18, 503–511. [Google Scholar] [CrossRef]
- De Chiara, G.; Marcocci, M.E.; Civitelli, L.; Argnani, R.; Piacentini, R.; Ripoli, C.; Manservigi, R.; Grassi, C.; Garaci, E.; Palamara, A.T. APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PLoS ONE 2010, 5, e13989. [Google Scholar] [CrossRef] [Green Version]
- Racchi, M.; Govoni, S. Rationalizing a pharmacological intervention on the amyloid precursor protein metabolism. Trends Pharmacol. Sci. 1999, 20, 418–423. [Google Scholar] [CrossRef]
- Racchi, M.; Govoni, S. The pharmacology of amyloid precursor protein processing. Exp. Gerontol. 2003, 38, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Kögel, D.; Deller, T.; Behl, C. Roles of amyloid precursor protein family members in neuroprotection, stress signaling and aging. Exp. Brain Res. 2012, 217, 471–479. [Google Scholar] [CrossRef]
- Westmark, C.J. What’s hAPPening at synapses? The role of amyloid β-protein precursor and β-amyloid in neurological disorders. Mol. Psychiatry 2013, 18, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, B.; Long, J.M.; Sokol, D.K.; Lahiri, D.K. Increased secreted amyloid precursor protein-α (sAPPα) in severe autism: Proposal of a specific, anabolic pathway and putative biomarker. PLoS ONE 2011, 6, e20405. [Google Scholar] [CrossRef] [Green Version]
- Sokol, D.K.; Chen, D.; Farlow, M.R.; Dunn, D.W.; Maloney, B.; Zimmer, J.A.; Lahiri, D.K. High levels of Alzheimer beta-amyloid precursor protein (APP) in children with severely autistic behavior and aggression. J. Child Neurol. 2006, 21, 444–449. [Google Scholar] [CrossRef]
- Palmert, M.R.; Usiak, M.; Mayeux, R.; Raskind, M.; Tourtellotte, W.W.; Younkin, S.G. Soluble derivatives of the beta amyloid protein precursor in cerebrospinal fluid: Alterations in normal aging and in Alzheimer’s disease. Neurology 1990, 40, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, W.E.; Wagner, S.L.; Shankle, W.R.; Farrow, J.S.; Dick, M.; Rozemuller, J.M.; Kuiper, M.A.; Wolters, E.C.; Zimmerman, J.; Cotman, C.W.; et al. Decreased levels of soluble amyloid beta-protein precursor in cerebrospinal fluid of live Alzheimer disease patients. Proc. Natl. Acad. Sci. USA 1992, 89, 2551–2555. [Google Scholar] [CrossRef] [Green Version]
- Bergamaschi, S.; Binetti, G.; Govoni, S.; Wetsel, W.C.; Battaini, F.; Trabucchi, M.; Bianchetti, A.; Racchi, M. Defective phorbol ester-stimulated secretion of beta-amyloid precursor protein from Alzheimer’s disease fibroblasts. Neurosci. Lett. 1995, 201, 1–5. [Google Scholar] [CrossRef]
- Mattson, M.P. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol. Rev. 1997, 77, 1081–1132. [Google Scholar] [CrossRef] [Green Version]
- Buoso, E.; Biundo, F.; Lanni, C.; Aiello, S.; Grossi, S.; Schettini, G.; Govoni, S.; Racchi, M. Modulation of Rack-1/PKCβII signalling by soluble AβPPα in SH-SY5Y cells. Curr. Alzheimer Res. 2013, 10, 697–705. [Google Scholar] [CrossRef]
- Thornton, E.; Vink, R.; Blumbergs, P.C.; Van Den Heuvel, C. Soluble amyloid precursor protein alpha reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res. 2006, 1094, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Kovalevich, J.; Santerre, M.; Langford, D. Considerations for the Use of SH-SY5Y Neuroblastoma Cells in Neurobiology. Methods Mol. Biol. 2021, 2311, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Skotak, M.; Wang, F.; Chandra, N. An in vitro injury model for SH-SY5Y neuroblastoma cells: Effect of strain and strain rate. J. Neurosci. Methods 2012, 205, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Gakhar-Koppole, N.; Hundeshagen, P.; Mandl, C.; Weyer, S.W.; Allinquant, B.; Müller, U.; Ciccolini, F. Activity requires soluble amyloid precursor protein alpha to promote neurite outgrowth in neural stem cell-derived neurons via activation of the MAPK pathway. Eur. J. Neurosci. 2008, 28, 871–882. [Google Scholar] [CrossRef]
- Dong, Y.; Li, T.; Ma, Z.; Zhou, C.; Wang, X.; Li, J. HSPA1A, HSPA2, and HSPA8 Are Potential Molecular Biomarkers for Prognosis among HSP70 Family in Alzheimer’s Disease. Dis. Markers 2022, 2022, 9480398. [Google Scholar] [CrossRef]
- Hoffman, J.L.; Faccidomo, S.; Kim, M.; Taylor, S.M.; Agoglia, A.E.; May, A.M.; Smith, E.N.; Wong, L.C.; Hodge, C.W. Alcohol drinking exacerbates neural and behavioral pathology in the 3xTg-AD mouse model of Alzheimer’s disease. Int. Rev. Neurobiol. 2019, 148, 169–230. [Google Scholar] [CrossRef]
- Ramos, E.; Romero, A.; Marco-Contelles, J.; López-Muñoz, F.; Del Pino, J. Modulation of Heat Shock Response Proteins by ASS234, Targeted for Neurodegenerative Diseases Therapy. Chem. Res. Toxicol. 2018, 31, 839–842. [Google Scholar] [CrossRef]
- Von Wittgenstein, J.; Zheng, F.; Wittmann, M.T.; Balta, E.A.; Ferrazzi, F.; Schäffner, I.; Häberle, B.M.; Valero-Aracama, M.J.; Koehl, M.; Miranda, C.J.; et al. Sox11 is an Activity-Regulated Gene with Dentate-Gyrus-Specific Expression Upon General Neural Activation. Cereb. Cortex 2020, 30, 3731–3743. [Google Scholar] [CrossRef]
- Li, Y.; Struebing, F.L.; Wang, J.; King, R.; Geisert, E.E. Different Effect of Sox11 in Retinal Ganglion Cells Survival and Axon Regeneration. Front. Genet. 2018, 9, 633. [Google Scholar] [CrossRef] [Green Version]
- Jankowski, M.P.; McIlwrath, S.L.; Jing, X.; Cornuet, P.K.; Salerno, K.M.; Koerber, H.R.; Albers, K.M. Sox11 transcription factor modulates peripheral nerve regeneration in adult mice. Brain Res. 2009, 1256, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Salerno, K.M.; Jing, X.; Diges, C.M.; Cornuet, P.K.; Glorioso, J.C.; Albers, K.M. Sox11 modulates brain-derived neurotrophic factor expression in an exon promoter-specific manner. J. Neurosci. Res. 2012, 90, 1011–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Liu, S.; Zhang, X.; Wang, L.; Zhang, X.; Hao, A.; Han, A.; Yang, J. Sox11 promotes endogenous neurogenesis and locomotor recovery in mice spinal cord injury. Biochem. Biophys. Res. Commun. 2014, 446, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Sato, T.; Aoki, T.; Yamamoto, A.; Okada, T.; Hirai, R.; Harada, R.; Mori, K.; Tagaya, M.; Harada, A. p31 deficiency influences endoplasmic reticulum tubular morphology and cell survival. Mol. Cell Biol. 2009, 29, 1869–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnell, E.; Shapiro, L.P.; Voorn, R.A.; Forrest, M.P.; Jalloul, H.A.; Loizzo, D.D.; Penzes, P. KALRN: A central regulator of synaptic function and synaptopathies. Gene 2021, 768, 145306. [Google Scholar] [CrossRef]
- Russo-Savage, L.; Rao, V.K.S.; Eipper, B.A.; Mains, R.E. Role of Kalirin and mouse strain in retention of spatial memory training in an Alzheimer’s disease model mouse line. Neurobiol. Aging 2020, 95, 69–80. [Google Scholar] [CrossRef]
- Xie, Z.; Shapiro, L.P.; Cahill, M.E.; Russell, T.A.; Lacor, P.N.; Klein, W.L.; Penzes, P. Kalirin-7 prevents dendritic spine dysgenesis induced by amyloid beta-derived oligomers. Eur. J. Neurosci. 2019, 49, 1091–1101. [Google Scholar] [CrossRef]
- Liu, M.; Zhong, W.; Li, C.; Su, W. Fluoxetine attenuates apoptosis in early brain injury after subarachnoid hemorrhage through Notch1/ASK1/p38 MAPK signaling pathway. Bioengineered 2022, 13, 8396–8411. [Google Scholar] [CrossRef]
- Yeo, E.J.; Eum, W.S.; Yeo, H.J.; Choi, Y.J.; Sohn, E.J.; Kwon, H.J.; Kim, D.W.; Kim, D.S.; Cho, S.W.; Park, J.; et al. Protective Role of Transduced Tat-Thioredoxin1 (Trx1) against Oxidative Stress-Induced Neuronal Cell Death via ASK1-MAPK Signal Pathway. Biomol. Ther. 2021, 29, 321–330. [Google Scholar] [CrossRef]
- Gómora-García, J.C.; Gerónimo-Olvera, C.; Pérez-Martínez, X.; Massieu, L. IRE1α RIDD activity induced under ER stress drives neuronal death by the degradation of 14-3-3 θ mRNA in cortical neurons during glucose deprivation. Cell Death Discov. 2021, 7, 131. [Google Scholar] [CrossRef]
- Zou, D.; Li, R.; Huang, X.; Chen, G.; Liu, Y.; Meng, Y.; Wang, Y.; Wu, Y.; Mao, Y. Identification of molecular correlations of RBM8A with autophagy in Alzheimer’s disease. Aging 2019, 11, 11673–11685. [Google Scholar] [CrossRef]
- Maekawa, M.; Iwayama, Y.; Ohnishi, T.; Toyoshima, M.; Shimamoto, C.; Hisano, Y.; Toyota, T.; Balan, S.; Matsuzaki, H.; Iwata, Y.; et al. Investigation of the fatty acid transporter-encoding genes SLC27A3 and SLC27A4 in autism. Sci. Rep. 2015, 5, 16239. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, N.; Buchan, J.R. RPS28B mRNA acts as a scaffold promoting cis-translational interaction of proteins driving P-body assembly. Nucleic Acids Res. 2020, 48, 6265–6279. [Google Scholar] [CrossRef] [PubMed]
- Riggs, C.L.; Kedersha, N.; Ivanov, P.; Anderson, P. Mammalian stress granules and P bodies at a glance. J. Cell Sci. 2020, 133, jcs242487. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Vatsa, N.; Kumar, V.; Shekhar, S.; Sharma, A.; Jana, N.R. Ube3a deficiency inhibits amyloid plaque formation in APPswe/PS1δE9 mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 4042–4054. [Google Scholar] [CrossRef] [Green Version]
- Olabarria, M.; Pasini, S.; Corona, C.; Robador, P.; Song, C.; Patel, H.; Lefort, R. Dysfunction of the ubiquitin ligase E3A Ube3A/E6-AP contributes to synaptic pathology in Alzheimer’s disease. Commun. Biol. 2019, 2, 111. [Google Scholar] [CrossRef] [Green Version]
- Sacco, A.; Martelli, F.; Pal, A.; Saraceno, C.; Benussi, L.; Ghidoni, R.; Rongioletti, M.; Squitti, R. Regulatory miRNAs in Cardiovascular and Alzheimer’s Disease: A Focus on Copper. Int. J. Mol. Sci. 2022, 23, 3327. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.A.; Przemylska, L.; Clavane, E.M.; Meakin, P.J. BACE1, More than just a β-secretase. Obes. Rev. 2022, 23, e13430. [Google Scholar] [CrossRef]
- Peters-Libeu, C.; Campagna, J.; Mitsumori, M.; Poksay, K.S.; Spilman, P.; Sabogal, A.; Bredesen, D.E.; John, V. sAβPPα is a Potent Endogenous Inhibitor of BACE1. J. Alzheimers Dis. 2015, 47, 545–555. [Google Scholar] [CrossRef]
- Obregon, D.; Hou, H.; Deng, J.; Giunta, B.; Tian, J.; Darlington, D.; Shahaduzzaman, M.; Zhu, Y.; Mori, T.; Mattson, M.P.; et al. Soluble amyloid precursor protein-β modulates β-secretase activity and amyloid-β generation. Nat. Commun. 2012, 3, 777. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Habib, A.; Obregon, D.F.; Barger, S.W.; Giunta, B.; Wang, Y.J.; Hou, H.; Sawmiller, D.; Tan, J. Soluble amyloid precursor protein alpha inhibits tau phosphorylation through modulation of GSK3β signaling pathway. J. Neurochem. 2015, 135, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Tiwari-Woodruff, S.K.; Kaplan, R.; Kornblum, H.I.; Bronstein, J.M. Developmental expression of OAP-1/Tspan-3, a member of the tetraspanin superfamily. J. Neurosci. Res. 2004, 77, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005, 6, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Boucheix, C.; Rubinstein, E. Tetraspanins. Cell Mol. Life Sci. 2001, 58, 1189–1205. [Google Scholar] [CrossRef]
- Charrin, S.; le Naour, F.; Silvie, O.; Milhiet, P.E.; Boucheix, C.; Rubinstein, E. Lateral organization of membrane proteins: Tetraspanins spin their web. Biochem. J. 2009, 420, 133–154. [Google Scholar] [CrossRef] [Green Version]
- Hemler, M.E. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu. Rev. Cell Dev. Biol. 2003, 19, 397–422. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Barreiro, O.; Gordon-Alonso, M.; Sala-Valdés, M.; Sánchez-Madrid, F. Tetraspanin-enriched microdomains: A functional unit in cell plasma membranes. Trends Cell Biol. 2009, 19, 434–446. [Google Scholar] [CrossRef]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a glance. J. Cell Sci. 2014, 127, 3641–3648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassani, S.; Cingolani, L.A.; Valnegri, P.; Folci, A.; Zapata, J.; Gianfelice, A.; Sala, C.; Goda, Y.; Passafaro, M. The X-linked intellectual disability protein TSPAN7 regulates excitatory synapse development and AMPAR trafficking. Neuron 2012, 73, 1143–1158. [Google Scholar] [CrossRef] [Green Version]
- Murru, L.; Vezzoli, E.; Longatti, A.; Ponzoni, L.; Falqui, A.; Folci, A.; Moretto, E.; Bianchi, V.; Braida, D.; Sala, M.; et al. Pharmacological Modulation of AMPAR Rescues Intellectual Disability-Like Phenotype in Tm4sf2-/y Mice. Cereb. Cortex 2017, 27, 5369–5384. [Google Scholar] [CrossRef] [Green Version]
- Murru, L.; Moretto, E.; Martano, G.; Passafaro, M. Tetraspanins shape the synapse. Mol. Cell Neurosci. 2018, 91, 76–81. [Google Scholar] [CrossRef]
- Tiwari-Woodruff, S.K.; Buznikov, A.G.; Vu, T.Q.; Micevych, P.E.; Chen, K.; Kornblum, H.I.; Bronstein, J.M. OSP/claudin-11 forms a complex with a novel member of the tetraspanin super family and beta1 integrin and regulates proliferation and migration of oligodendrocytes. J. Cell Biol. 2001, 153, 295–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiede-Stan, N.K.; Tews, B.; Albrecht, D.; Ristic, Z.; Ewers, H.; Schwab, M.E. Tetraspanin-3 is an organizer of the multi-subunit Nogo-A signaling complex. J. Cell Sci. 2015, 128, 3583–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seipold, L.; Damme, M.; Prox, J.; Rabe, B.; Kasparek, P.; Sedlacek, R.; Altmeppen, H.; Willem, M.; Boland, B.; Glatzel, M.; et al. Tetraspanin 3, A central endocytic membrane component regulating the expression of ADAM10, presenilin and the amyloid precursor protein. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.; González-Ramírez, J.; Marín, M.E.; Martínez-Coronilla, G.; Meza-Reyna, V.I.; Mora, R.; Díaz-Molina, R. Isthmin 2 is decreased in preeclampsia and highly expressed in choriocarcinoma. Heliyon 2020, 6, e05096. [Google Scholar] [CrossRef]
- Hu, M.; Zhang, X.; Hu, C.; Teng, T.; Tang, Q.Z. A brief overview about the adipokine: Isthmin-1. Front. Cardiovasc. Med. 2022, 9, 939757. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Katayama, K.; Suzuki, T.; Dohmae, N.; Simizu, S. Regulation of N-glycosylation and secretion of Isthmin-1 by its C-mannosylation. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129840. [Google Scholar] [CrossRef]
- Xiang, W.; Ke, Z.; Zhang, Y.; Cheng, G.H.; Irwan, I.D.; Sulochana, K.N.; Potturi, P.; Wang, Z.; Yang, H.; Wang, J.; et al. Isthmin is a novel secreted angiogenesis inhibitor that inhibits tumour growth in mice. J. Cell Mol. Med. 2011, 15, 359–374. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, M.; Venugopal, S.; Zhou, Y.; Xiang, W.; Li, Y.H.; Lin, Q.; Kini, R.M.; Chong, Y.S.; Ge, R. Isthmin exerts pro-survival and death-promoting effect on endothelial cells through alphavbeta5 integrin depending on its physical state. Cell Death Dis. 2011, 2, e153. [Google Scholar] [CrossRef] [Green Version]
- Yuan, B.; Xian, R.; Ma, J.; Chen, Y.; Lin, C.; Song, Y. Isthmin inhibits glioma growth through antiangiogenesis in vivo. J. Neurooncol. 2012, 109, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Zhao, M.; Voilquin, L.; Jung, Y.; Aikio, M.A.; Sahai, T.; Dou, F.Y.; Roche, A.M.; Carcamo-Orive, I.; Knowles, J.W.; et al. Isthmin-1 is an adipokine that promotes glucose uptake and improves glucose tolerance and hepatic steatosis. Cell Metab. 2021, 33, 1836–1852.e11. [Google Scholar] [CrossRef]
- GeneCards. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=ISM2 (accessed on 10 October 2022).
- Kundu, B.; Brock, A.A.; Englot, D.J.; Butson, C.R.; Rolston, J.D. Deep brain stimulation for the treatment of disorders of consciousness and cognition in traumatic brain injury patients: A review. Neurosurg. Focus 2018, 45, E14. [Google Scholar] [CrossRef] [Green Version]
- Aronson, J.P.; Katnani, H.A.; Huguenard, A.; Mulvaney, G.; Bader, E.R.; Yang, J.C.; Eskandar, E.N. Phasic stimulation in the nucleus accumbens enhances learning after traumatic brain injury. Cereb. Cortex Commun. 2022, 3, tgac016. [Google Scholar] [CrossRef] [PubMed]
- Osera, C.; Fassina, L.; Amadio, M.; Venturini, L.; Buoso, E.; Magenes, G.; Govoni, S.; Ricevuti, G.; Pascale, A. Cytoprotective response induced by electromagnetic stimulation on SH-SY5Y human neuroblastoma cell line. Tissue Eng. Part A 2011, 17, 2573–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolatai, A.; He, Y.; Wu, N. Vascular endothelial growth factor and its receptors regulation in gestational diabetes mellitus and eclampsia. J. Transl. Med. 2022, 20, 400. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Hu, Y.; Cui, B.; Zhuang, S.; Liu, N. Vascular endothelial growth factor-mediated peritoneal neoangiogenesis in peritoneal dialysis. Perit. Dial. Int. 2022, 42, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Theis, V.; Theiss, C. VEGF—A Stimulus for Neuronal Development and Regeneration in the CNS and PNS. Curr. Protein. Pept. Sci. 2018, 19, 589–597. [Google Scholar] [CrossRef]
- Okabe, K.; Fukada, H.; Tai-Nagara, I.; Ando, T.; Honda, T.; Nakajima, K.; Takeda, N.; Fong, G.H.; Ema, M.; Kubota, Y. Neuron-derived VEGF contributes to cortical and hippocampal development independently of VEGFR1/2-mediated neurotrophism. Dev. Biol. 2020, 459, 65–71. [Google Scholar] [CrossRef]
- Ureña-Guerrero, M.E.; Castañeda-Cabral, J.L.; Rivera-Cervantes, M.C.; Macias-Velez, R.J.; Jarero-Basulto, J.J.; Gudiño-Cabrera, G.; Beas-Zárate, C. Neuroprotective and Neurorestorative Effects of Epo and VEGF: Perspectives for New Therapeutic Approaches to Neurological Diseases. Curr. Pharm. Des. 2020, 26, 1263–1276. [Google Scholar] [CrossRef]
- Carmeliet, P.; Ruiz de Almodovar, C. VEGF ligands and receptors: Implications in neurodevelopment and neurodegeneration. Cell Mol. Life Sci. 2013, 70, 1763–1778. [Google Scholar] [CrossRef]
- Chiappelli, M.; Borroni, B.; Archetti, S.; Calabrese, E.; Corsi, M.M.; Franceschi, M.; Padovani, A.; Licastro, F. VEGF gene and phenotype relation with Alzheimer’s disease and mild cognitive impairment. Rejuvenation Res. 2006, 9, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, C.L.; Wang, Z.X.; Wang, H.J.; Yin, F.J.; Li, W.D.; Liu, C.C.; Fan, H.N. VEGF Family Gene Expression as Prognostic Biomarkers for Alzheimer’s Disease and Primary Liver Cancer. Comput. Math Methods Med. 2021, 2021, 3422393. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Bates, D.O. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.W.; Bentel, J.M.; Tilley, W.D. Vascular endothelial growth factor (VEGF) expression in prostate cancer and benign prostatic hyperplasia. J. Urol. 1997, 157, 2323–2328. [Google Scholar] [CrossRef]
- Bucolo, C.; Barbieri, A.; Viganò, I.; Marchesi, N.; Bandello, F.; Drago, F.; Govoni, S.; Zerbini, G.; Pascale, A. Short-and Long-Term Expression of Vegf: A Temporal Regulation of a Key Factor in Diabetic Retinopathy. Front. Pharmacol. 2021, 12, 707909. [Google Scholar] [CrossRef] [PubMed]
- Fahmideh, F.; Marchesi, N.; Campagnoli, L.I.M.; Landini, L.; Caramella, C.; Barbieri, A.; Govoni, S.; Pascale, A. Effect of troxerutin in counteracting hyperglycemia-induced VEGF upregulation in endothelial cells: A new option to target early stages of diabetic retinopathy? Front. Pharmacol. 2022, 13, 951833. [Google Scholar] [CrossRef] [PubMed]
- Amadio, M.; Scapagnini, G.; Lupo, G.; Drago, F.; Govoni, S.; Pascale, A. PKCbetaII/HuR/VEGF: A new molecular cascade in retinal pericytes for the regulation of VEGF gene expression. Pharmacol Res. 2008, 57, 60–66. [Google Scholar] [CrossRef]
- Pascale, A.; Govoni, S. The complex world of post-transcriptional mechanisms: Is their deregulation a common link for diseases? Focus on ELAV-like RNA-binding proteins. Cell Mol. Life Sci. 2012, 69, 501–517. [Google Scholar] [CrossRef]
- Deschênes-Furry, J.; Perrone-Bizzozero, N.; Jasmin, B.J. The RNA-binding protein HuD: A regulator of neuronal differentiation, maintenance and plasticity. Bioessays 2006, 28, 822–833. [Google Scholar] [CrossRef]
- Quattrone, A.; Pascale, A.; Nogues, X.; Zhao, W.; Gusev, P.; Pacini, A.; Alkon, D.L. Posttranscriptional regulation of gene expression in learning by the neuronal ELAV-like mRNA-stabilizing proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 11668–11673. [Google Scholar] [CrossRef] [Green Version]
- Pascale, A.; Gusev, P.A.; Amadio, M.; Dottorini, T.; Govoni, S.; Alkon, D.L.; Quattrone, A. Increase of the RNA-binding protein HuD and posttranscriptional up-regulation of the GAP-43 gene during spatial memory. Proc. Natl. Acad. Sci. USA 2004, 101, 1217–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascale, A.; Amadio, M.; Scapagnini, G.; Lanni, C.; Racchi, M.; Provenzani, A.; Govoni, S.; Alkon, D.L.; Quattrone, A. Neuronal ELAV proteins enhance mRNA stability by a PKCalpha-dependent pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 12065–12070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadio, M.; Pascale, A.; Wang, J.; Ho, L.; Quattrone, A.; Gandy, S.; Haroutunian, V.; Racchi, M.; Pasinetti, G.M. nELAV proteins alteration in Alzheimer’s disease brain: A novel putative target for amyloid-beta reverberating on AbetaPP processing. J. Alzheimers Dis. 2009, 16, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, N.; Amadio, M.; Colombrita, C.; Govoni, S.; Ratti, A.; Pascale, A. PKC Activation Counteracts ADAM10 Deficit in HuD-Silenced Neuroblastoma Cells. J. Alzheimers Dis. 2016, 54, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Xia, D.; Liao, S.; Niu, B.; Tang, J.; Hu, H.; Qian, H.; Cao, B. Vascular endothelial growth factor improves the cognitive decline of Alzheimer’s disease via concurrently inducing the expression of ADAM10 and reducing the expression of β-site APP cleaving enzyme 1 in Tg2576 mice. Neurosci. Res. 2019, 142, 49–57. [Google Scholar] [CrossRef]
- Baker, T.L.; Agoston, D.V.; Brady, R.D.; Major, B.; McDonald, S.J.; Mychasiuk, R.; Wright, D.K.; Yamakawa, G.R.; Sun, M.; Shultz, S.R. Targeting the Cerebrovascular System: Next-Generation Biomarkers and Treatment for Mild Traumatic Brain Injury. Neuroscientist 2021, 28, 594–612. [Google Scholar] [CrossRef]
- Wang, K.; Jing, Y.; Xu, C.; Zhao, J.; Gong, Q.; Chen, S. HIF-1α and VEGF Are Involved in Deferoxamine-Ameliorated Traumatic Brain Injury. J. Surg. Res. 2020, 246, 419–426. [Google Scholar] [CrossRef]
- Xu, Y.Q.; Sun, Z.Q.; Wang, Y.T.; Xiao, F.; Chen, M.W. Function of Nogo-A/Nogo-A receptor in Alzheimer’s disease. CNS Neurosci. Ther. 2015, 21, 479–485. [Google Scholar] [CrossRef]
- Corrigan, F.; Thornton, E.; Roisman, L.C.; Leonard, A.V.; Vink, R.; Blumbergs, P.C.; van den Heuvel, C.; Cappai, R. The neuroprotective activity of the amyloid precursor protein against traumatic brain injury is mediated via the heparin binding site in residues 96–110. J. Neurochem. 2014, 128, 196–204. [Google Scholar] [CrossRef]
- Plummer, S.L.; Corrigan, F.; Thornton, E.; Woenig, J.A.; Vink, R.; Cappai, R.; Van Den Heuvel, C. The amyloid precursor protein derivative, APP96-110, is efficacious following intravenous administration after traumatic brain injury. PLoS ONE 2018, 13, e0190449. [Google Scholar] [CrossRef] [Green Version]
- Hodgetts, S.I.; Lovett, S.J.; Baron-Heeris, D.; Fogliani, A.; Sturm, M.; Van den Heuvel, C.; Harvey, A.R. Effects of amyloid precursor protein peptide APP96-110, alone or with human mesenchymal stromal cells, on recovery after spinal cord injury. Neural. Regen. Res. 2022, 17, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Mockett, B.G.; Richter, M.; Abraham, W.C.; Müller, U.C. Therapeutic Potential of Secreted Amyloid Precursor Protein APPsα. Front. Mol. Neurosci. 2017, 10, 30. [Google Scholar] [CrossRef] [Green Version]
- Mockett, B.G.; Ryan, M.M. The therapeutic potential of the neuroactive peptides of soluble amyloid precursor protein-alpha in Alzheimer’s disease and related neurological disorders. Semin. Cell Dev. Biol. 2022, 139, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Buoso, E.; Lanni, C.; Molteni, E.; Rousset, F.; Corsini, E.; Racchi, M. Opposing effects of cortisol and dehydroepiandrosterone on the expression of the receptor for Activated C Kinase 1: Implications in immunosenescence. Exp. Gerontol. 2011, 46, 877–883. [Google Scholar] [CrossRef]
- Buoso, E.; Galasso, M.; Ronfani, M.; Serafini, M.M.; Lanni, C.; Corsini, E.; Racchi, M. Role of spliceosome proteins in the regulation of glucocorticoid receptor isoforms by cortisol and dehydroepiandrosterone. Pharmacol. Res. 2017, 120, 180–187. [Google Scholar] [CrossRef]
- Buoso, E.; Galasso, M.; Ronfani, M.; Papale, A.; Galbiati, V.; Eberini, I.; Marinovich, M.; Racchi, M.; Corsini, E. The scaffold protein RACK1 is a target of endocrine disrupting chemicals (EDCs) with important implication in immunity. Toxicol. Appl. Pharmacol. 2017, 325, 37–47. [Google Scholar] [CrossRef]
- Racchi, M.; Buoso, E.; Ronfani, M.; Serafini, M.M.; Galasso, M.; Lanni, C.; Corsini, E. Role of Hormones in the Regulation of RACK1 Expression as a Signaling Checkpoint in Immunosenescence. Int. J. Mol. Sci. 2017, 18, 1453. [Google Scholar] [CrossRef] [Green Version]
- Buoso, E.; Masi, M.; Galbiati, V.; Maddalon, A.; Iulini, M.; Kenda, M.; Sollner Dolenc, M.; Marinovich, M.; Racchi, M.; Corsini, E. Effect of estrogen-active compounds on the expression of RACK1 and immunological implications. Arch. Toxicol. 2020, 94, 2081–2095. [Google Scholar] [CrossRef]
- Corsini, E.; Buoso, E.; Galbiati, V.; Racchi, M. Role of Protein Kinase C in Immune Cell Activation and Its Implication Chemical-Induced Immunotoxicity. Adv. Exp. Med. Biol. 2021, 1275, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Buoso, E.; Kenda, M.; Masi, M.; Linciano, P.; Galbiati, V.; Racchi, M.; Dolenc, M.S.; Corsini, E. Effects of Bisphenols on RACK1 Expression and Their Immunological Implications in THP-1 Cells. Front. Pharmacol. 2021, 12, 743991. [Google Scholar] [CrossRef]
- Galbiati, V.; Buoso, E.; d’Emmanuele di Villa Bianca, R.; Paola, R.D.; Morroni, F.; Nocentini, G.; Racchi, M.; Viviani, B.; Corsini, E. Immune and Nervous Systems Interaction in Endocrine Disruptors Toxicity: The Case of Atrazine. Front. Toxicol. 2021, 3, 649024. [Google Scholar] [CrossRef] [PubMed]
- Maddalon, A.; Masi, M.; Iulini, M.; Linciano, P.; Galbiati, V.; Marinovich, M.; Racchi, M.; Buoso, E.; Corsini, E. Effects of endocrine active contaminating pesticides on RACK1 expression and immunological consequences in THP-1 cells. Environ. Toxicol. Pharmacol. 2022, 95, 103971. [Google Scholar] [CrossRef] [PubMed]
- Masi, M.; Maddalon, A.; Iulini, M.; Linciano, P.; Galbiati, V.; Marinovich, M.; Racchi, M.; Corsini, E.; Buoso, E. Effects of endocrine disrupting chemicals on the expression of RACK1 and LPS-induced THP-1 cell activation. Toxicology 2022, 480, 153321. [Google Scholar] [CrossRef] [PubMed]
- Buoso, E.; Ronfani, M.; Galasso, M.; Ventura, D.; Corsini, E.; Racchi, M. Cortisol-induced SRSF3 expression promotes GR splicing, RACK1 expression and breast cancer cells migration. Pharmacol. Res. 2019, 143, 17–26. [Google Scholar] [CrossRef]
- Buoso, E.; Masi, M.; Long, A.; Chiappini, C.; Travelli, C.; Govoni, S.; Racchi, M. Ribosomes as a nexus between translation and cancer progression: Focus on ribosomal Receptor for Activated C Kinase 1 (RACK1) in breast cancer. Br. J. Pharmacol. 2022, 179, 2813–2828. [Google Scholar] [CrossRef]
- Buoso, E.; Masi, M.; Racchi, M.; Corsini, E. Endocrine-Disrupting Chemicals’ (EDCs) Effects on Tumour Microenvironment and Cancer Progression: Emerging Contribution of RACK1. Int. J. Mol. Sci. 2020, 21, 9229. [Google Scholar] [CrossRef]
- Masi, M.; Garattini, E.; Bolis, M.; Di Marino, D.; Maraccani, L.; Morelli, E.; Grolla, A.A.; Fagiani, F.; Corsini, E.; Travelli, C.; et al. OXER1 and RACK1-associated pathway: A promising drug target for breast cancer progression. Oncogenesis 2020, 9, 105. [Google Scholar] [CrossRef]
- Masi, M.; Racchi, M.; Travelli, C.; Corsini, E.; Buoso, E. Molecular Characterization of Membrane Steroid Receptors in Hormone-Sensitive Cancers. Cells 2021, 10, 2999. [Google Scholar] [CrossRef]
- Cheng, Z.F.; Cartwright, C.A. Rack1 maintains intestinal homeostasis by protecting the integrity of the epithelial barrier. Am. J. Physiol. Gastrointest Liver Physiol. 2018, 314, G263–G274. [Google Scholar] [CrossRef]
- Cheng, Z.F.; Pai, R.K.; Cartwright, C.A. Rack1 function in intestinal epithelia: Regulating crypt cell proliferation and regeneration and promoting differentiation and apoptosis. Am. J. Physiol. Gastrointest Liver Physiol. 2018, 314, G1–G13. [Google Scholar] [CrossRef]
- Kershner, L.; Welshhans, K. RACK1 regulates neural development. Neural. Regen. Res. 2017, 12, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Buoso, E.; Biundo, F.; Lanni, C.; Schettini, G.; Govoni, S.; Racchi, M. AβPP intracellular C-terminal domain function is related to its degradation processes. J. Alzheimers Dis. 2012, 30, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Battaini, F.; Pascale, A.; Paoletti, R.; Govoni, S. The role of anchoring protein RACK1 in PKC activation in the ageing rat brain. Trends Neurosci. 1997, 20, 410–415. [Google Scholar] [CrossRef]
- Dwane, S.; Durack, E.; O’Connor, R.; Kiely, P.A. RACK1 promotes neurite outgrowth by scaffolding AGAP2 to FAK. Cell Signal. 2014, 26, 9–18. [Google Scholar] [CrossRef]
- Kershner, L.; Welshhans, K. RACK1 is necessary for the formation of point contacts and regulates axon growth. Dev. Neurobiol. 2017, 77, 1038–1056. [Google Scholar] [CrossRef] [PubMed]
- Romano, N.; Di Giacomo, B.; Nobile, V.; Borreca, A.; Willems, D.; Tilesi, F.; Catalani, E.; Agrawal, M.; Welshhans, K.; Ricciardi, S.; et al. Ribosomal RACK1 Regulates the Dendritic Arborization by Repressing FMRP Activity. Int. J. Mol. Sci. 2022, 23, 11857. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Chen, L.; Li, Y.; Huang, M.; Shao, J.; Li, S.; Cheng, J.; Yang, H.; Wu, Y.; Zhang, J.; et al. Rack1 is essential for corticogenesis by preventing p21-dependent senescence in neural stem cells. Cell. Rep. 2021, 36, 109639. [Google Scholar] [CrossRef]
- Yaka, R.; Thornton, C.; Vagts, A.J.; Phamluong, K.; Bonci, A.; Ron, D. NMDA receptor function is regulated by the inhibitory scaffolding protein, RACK1. Proc. Natl. Acad. Sci. USA 2002, 99, 5710–5715. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Wu, R.; Zhang, Q.; Wu, J.B.; Lou, J.; Zheng, Z.; Ding, J.Q.; Yuan, Z. DJ-1 interacts with RACK1 and protects neurons from oxidative-stress-induced apoptosis. Biochem. J. 2014, 462, 489–497. [Google Scholar] [CrossRef]
- He, D.Y.; Neasta, J.; Ron, D. Epigenetic regulation of BDNF expression via the scaffolding protein RACK1. J. Biol. Chem. 2010, 285, 19043–19050. [Google Scholar] [CrossRef] [Green Version]
- Neasta, J.; Kiely, P.A.; He, D.Y.; Adams, D.R.; O’Connor, R.; Ron, D. Direct interaction between scaffolding proteins RACK1 and 14-3-3? regulates brain-derived neurotrophic factor (BDNF) transcription. J. Biol. Chem. 2012, 287, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Brivio, P.; Buoso, E.; Masi, M.; Gallo, M.T.; Gruca, P.; Lason, M.; Litwa, E.; Papp, M.; Fumagalli, F.; Racchi, M.; et al. The coupling of RACK1 with the beta isoform of the glucocorticoid receptor promotes resilience to chronic stress exposure. Neurobiol. Stress 2021, 15, 100372. [Google Scholar] [CrossRef] [PubMed]
- Pascale, A.; Fortino, I.; Govoni, S.; Trabucchi, M.; Wetsel, W.C.; Battaini, F. Functional impairment in protein kinase C by RACK1 (receptor for activated C kinase 1) deficiency in aged rat brain cortex. J. Neurochem. 1996, 67, 2471–2477. [Google Scholar] [CrossRef] [PubMed]
- Battaini, F.; Pascale, A.; Lucchi, L.; Pasinetti, G.M.; Govoni, S. Protein kinase C anchoring deficit in postmortem brains of Alzheimer’s disease patients. Exp. Neurol. 1999, 159, 559–564. [Google Scholar] [CrossRef]
- Battaini, F.; Pascale, A. Protein kinase C signal transduction regulation in physiological and pathological aging. Ann. N. Y. Acad Sci. 2005, 1057, 177–192. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Dou, F.; Feng, J.; Yan, Z. RACK1 is involved in β-amyloid impairment of muscarinic regulation of GABAergic transmission. Neurobiol. Aging 2011, 32, 1818–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Chen, X.; Song, Y.; Zhang, Y.; Zhou, L.; Wan, L. Deficit of RACK1 contributes to the spatial memory impairment via upregulating BECLIN1 to induce autophagy. Life Sci. 2016, 151, 115–121. [Google Scholar] [CrossRef]
- Ni, H.; Rui, Q.; Xu, Y.; Zhu, J.; Gao, F.; Dang, B.; Li, D.; Gao, R.; Chen, G. RACK1 upregulation induces neuroprotection by activating the IRE1-XBP1 signaling pathway following traumatic brain injury in rats. Exp. Neurol. 2018, 304, 102–113. [Google Scholar] [CrossRef]
- Amiri, S.; Azadmanesh, K.; Dehghan Shasaltaneh, M.; Mayahi, V.; Naghdi, N. The Implication of Androgens in the Presence of Protein Kinase C to Repair Alzheimer’s Disease-Induced Cognitive Dysfunction. Iran. Biomed. J. 2020, 24, 64–80. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Tu, M.; Du, Y.; Li, J.; Pang, Y.; Dong, Z. Nicotine Promotes AβPP Nonamyloidogenic Processing via RACK1-Dependent Activation of PKC in SH-SY5Y-AβPP695 Cells. J. Alzheimers Dis. 2020, 75, 451–460. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, M.; Yang, X.; Wang, Y.; Qin, B.; Cui, C.; Chen, H.; Sang, A. Inhibition of RACK1 ameliorates choroidal neovascularization formation in vitro and in vivo. Exp. Mol. Pathol. 2016, 100, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yamauchi, M.; Muramatsu, M.; Osawa, T.; Tsuchida, R.; Shibuya, M. RACK1 regulates VEGF/Flt1-mediated cell migration via activation of a PI3K/Akt pathway. J. Biol. Chem. 2011, 286, 9097–9106. [Google Scholar] [CrossRef] [Green Version]
- Almeida, J.; Costa, J.; Coelho, P.; Cea, V.; Galesio, M.; Noronha, J.P.; Diniz, M.S.; Prudêncio, C.; Soares, R.; Sala, C.; et al. Adipocyte proteome and secretome influence inflammatory and hormone pathways in glioma. Metab. Brain Dis. 2019, 34, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, N.; Ma, D.; Liu, L.; Jiang, L.; Zhou, Y.; Zeng, X.; Li, J.; Chen, Q. Receptor for activated C kinase 1 (RACK1) promotes the progression of OSCC via the AKT/mTOR pathway. Int. J. Oncol. 2016, 49, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcelo, A.; Koppenol, R.; de Almeida, L.P.; Matos, C.A.; Nóbrega, C. Stress granules, RNA-binding proteins and polyglutamine diseases: Too much aggregation? Cell Death Dis. 2021, 12, 592. [Google Scholar] [CrossRef]
- Jung, Y.D.; Kim, M.S.; Shin, B.A.; Chay, K.O.; Ahn, B.W.; Liu, W.; Bucana, C.D.; Gallick, G.E.; Ellis, L.M. EGCG, a major component of green tea, inhibits tumour growth by inhibiting VEGF induction in human colon carcinoma cells. Br. J. Cancer 2001, 84, 844–850. [Google Scholar] [CrossRef] [Green Version]
- Sartippour, M.R.; Shao, Z.M.; Heber, D.; Beatty, P.; Zhang, L.; Liu, C.; Ellis, L.; Liu, W.; Go, V.L.; Brooks, M.N. Green tea inhibits vascular endothelial growth factor (VEGF) induction in human breast cancer cells. J. Nutr. 2002, 132, 2307–2311. [Google Scholar] [CrossRef] [Green Version]
- Lanni, C.; Necchi, D.; Pinto, A.; Buoso, E.; Buizza, L.; Memo, M.; Uberti, D.; Govoni, S.; Racchi, M. Zyxin is a novel target for β-amyloid peptide: Characterization of its role in Alzheimer’s pathogenesis. J. Neurochem. 2013, 125, 790–799. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| APP Processing | APP Fragment | Cell Line/Model | Characteristics and Functions | Ref. |
|---|---|---|---|---|
| - | APP full-length | PC12 pheochromocytoma cells; APP- B103 cells; embryonic carcinoma P19 and NT2 cell lines; NB-1 neuroblastoma cell line; SK-N-MC cells (human neuroblastoma cell line); rat hippocampal neurons; in vivo (hAPP751 or hAPP695 over-expressing mice, APP−/− mice, Down’s Syndrome (DS) Ts65Dn and 1YeY mice) | Acting as cell surface receptor; involved in neuronal adhesion and iron transport; promotes cell division, neurite outgrowth, axonogenesis, synapse formation and maintenance and synaptic plasticity; neuroprotective role against Aβ and glutamate toxicity | [58] |
| Canonical—Amyloidogenic | sAPPβ | Human Embryonic Stem Cells (hESCs) | Induction of stem cells neural differentiation | [95] |
| Rat hippocampal cells | 100-fold less active neurotrophic effects compared to sAPPα in protecting hippocampal neurons against excitotoxicity, Aβ-induced toxicity, and glucose deprivation | [96] | ||
| Sprague–Dawley rats brain slices | No marked changes in Long-Term Potentiation (LTP) compared to sAPPα | [97] | ||
| N9 cells (myc-immortalized murine microglial cell line), | Stimulation of microglia activation through MAP kinase signaling pathways (i.e., ERKs, p38 kinase, JNKs) and NF-κB activity; production of proinflammatory and neurotoxic products (e.g., iNOS, IL-1β, ROS) | [98,99] | ||
| N-APP | E13 rat dorsal spinal cord explant; mouse sensory and motor neuron explants; dissociated sensory neuron cultures; In vivo (DR6 KO, Bax KO and p75NTR KO mice) | Interaction with DR6 to recruit caspase-3 and caspase-6 (in cell bodies and axons respectively) and triggering of axonal pruning, but also neuronal death and AD development | [42,43] | |
| β-CTF (C99) | Neuro-2a cells (neuroblast cell line); in vivo (mouse) | LTP disruption | [100] | |
| In vivo (APP−/− and APP+/− mice) | Synaptotoxicity induction and spine density reduction | [101] | ||
| In vivo (3xTgAD with PS1M146V, βAPPswe and TauP301L transgenes); APP695-H4 cells (human glioma cell line) | Early pathological accumulation and learning and memory deficits | [102,103] | ||
| PC12 cells (pheochromocytoma cell line); SK-N-MC cells (human neuroblastoma cell line); rat neuronal cultures; transgenic mouse models | Selective neurotoxicity, cortical atrophy, loss of hippocampal granule cells, astrogliosis, Aβ and APP immunoreactivity, impaired working memory, neocortical and hippocampal neurodegeneration and gliosis | [104,105,106,107] | ||
| β′-CTF (C89) | - | Unknown physiologic and pathologic properties | - | |
| Aβ | PC12 pheochromocytoma cells; APP- B103 cells; embryonic carcinoma P19 and NT2 cell lines; NB-1 neuroblastoma cell line; SK-N-MC cells (human neuroblastoma cell line); rat hippocampal neurons; in vivo (hAPP751 or hAPP695 over-expressing mice, APP−/− mice, Down’s Syndrome (DS) Ts65Dn and 1YeY mice) | Major APP metabolic fragment involved in AD development and progression
| [58] | |
| AICD | SH-SY5Y cells wild type (wt) and APP/APPswe); Mouse Embryonic Fibroblasts (MEF) and mouse brains (wt, PS1/2−/−, APP/APLP2−/− and APPΔCT15) | Transcriptional regulation, together with Tip60 and Fe65 and forming the ATF complex, of APP and AD-related genes | [108,109] | |
| Differentiated PC12 cells; rat primary cortical neurons | Induction of neurotoxicity by up-regulating the expression of GSK-3β and activating p53 | [110,111] | ||
| AICD-transfected Jurkat cells | Apoptosis induction through Fas-Associated protein with Death Domain (FADD)-induced programmed cell death | [112] | ||
| Canonical—Non-amyloidogenic | sAPPα | SH-SY5Y cells; B103 cells; rat primary cortical neurons; murine hippocampal neurons; MEFs; in vivo (wt and APP−/− mice) | Induction of Akt neuronal cell survival-correlated pathway, facilitation of normal neurophysiological functions (e.g., memory functions) and promotion of neurite outgrowth | [113,114,115] |
| In vivo (wt, APPswe/PS1ΔE9 and APP−/− mice) | Neuroprotective effect against synaptic dysfunction and TBI | [116,117] | ||
| In vivo (APP−/− and APLP2−/− mice); Sprague–Dawley rats brain slices | Contribution to the regulation of synaptic plasticity and LTP induction | [97,118] | ||
| α-CTF (C83) | In vivo (APP−/− and APP+/− mice) | Induction of synaptotoxicity and reduction of spine density | [101] | |
| CHO cells | Indirect promotion of survival by lowering C99 levels | [119] | ||
| In vitro assay; HEK293-APP cells | Hypothesised to be a γ-secretase inhibitor | [120] | ||
| p3 | In vitro assay; APP−/− MEFs; human cortical neurons | Induction of neuronal excitotoxicity by contributing to the formation of Ca2+-permeable ion channels | [121] | |
| AD brain human samples | Accumulation in amyloid plaques | [122] | ||
| Differentiated THP-1 cells (human monocyte line); MG7 cells (microglia cell line); D30 (murine astrocyte line); U373 cells (human astrocyte line) | Induction of apoptosis, inflammatory responses and neurotoxic effects by producing proinflammatory cytokines (e.g., Interleukin (IL)-1α, IL-1β, IL-6, Tumour Necrosis Factor-α (TNF-α), chemokine MCP-1) | [123] | ||
| Non-canonical—δ-secretase | sAPP1–373 sAPP1–585 sAPP374–585 | GFP-APP- and GST-APP-HEK293 cells; primary cultured neurons; in vivo (wt and AEP−/− 5xFAD and APP/PS1 mice) | sAPP1–373, but not sAPP1–585 nor sAPP374–585, exhibited neurotoxic properties | [60] |
| δ-CTF | AD brain human samples | Accumulation in brain lysates from AD patients | [60] | |
| Non-canonical—η-secretase | sAPPη (sAPP95) η-CTF (C191) | Mouse hippocampal cultures; acute hippocampal slices; in vivo (Thy1-GCaMP6 mice) | Binding to the γ-Aminobutyric Acid (GABA) receptor and modulation of GABAergic neurotransmission | [124] |
| In vivo (5xFAD MT5-MMP−/− mice); AD brain human samples | Accumulation in the surrounding of amyloid plaques in dystrophic neurites of transgenic mice and AD patients and contribution to cognitive decline | [63,64] | ||
| In vitro assay; SH-SY5Y/APP751 cells; H4/APP751 human neuroglioma cells | Processed by Cathepsin L, although its physiological role has not been investigated | [125] | ||
| Aη-α Aη-β | Murine hippocampal neurons and brain slices; human CSF | Both detected in mouse brain homogenates and human CSF (5-fold higher levels than Aβ); Aη-α, but not Aη-β inhibited LTP, suppressed neuronal activity and resides in the halo of the amyloid plaque. | [64] | |
| Non-canonical—Meprin-β | sAPP1–380/3 sAPP1–124 | - | Unknown physiologic and pathologic properties | - |
| CTF | - | Unknown physiologic and pathologic properties | - | |
| Aβ2–X | In vivo (APPswe-based mouse models); AD patient brain samples | Increased potential to aggregate compared to Aβ1–X peptides | [126] | |
| p11 | Differentiated SH-SY5Y cells; in vivo (wt, APP–/– and APLP2–/– C57BL6J × 129/Sv mice); AD brain human samples | Increased production detected during neuronal differentiation | [127] | |
| Non-canonical— θ-secretase Non-canonical—Caspases | sAPPθ | - | Unknown physiologic and pathologic properties | - |
| θ-CTF (C80) | - | Unknown physiologic and pathologic properties | - | |
| APP-Ncas | - | Unknown physiologic and pathologic properties | - | |
| APP-Ccas (C31) | Neuro-2a cells; AD brain human samples | Synaptic dysfunction, neuronal apoptosis and death; APP-dependent toxicity | [128] | |
| JCasp | Primary neuronal cultures | Induction of neuronal apoptosis by transducing a tyrosine-dependent signalling | [129] | |
| Non-canonical—Cathepsin B | Aβ3–X Aβ11–X (pE-Aβ) | In vitro assay; HEK293, 20E2, 2EB2, HAW and BAW cell lines; human AD brain samples | High aggregation propensity, cellular toxicity, disruption of LTP and proposed as predominant Aβ peptide species in AD brain patients | [38] |
| Other secretases | Aβ4–X Aβ5–X | In vivo (APPswe mouse models); AD patient brain samples | Increased potential to aggregate compared to Aβ1–X peptides | [126] |
| N-APP18–286 | neonatal C57BL/6 × SJL mice hippocampal cultures | Binding an unknown neuronal receptor and increase of phosphatidylinositol phosphate levels | [77] | |
| APP1–119 APP1–121 APP1–122 APP1–123 APP1–126 | Human CSF | ~12 kDa sAPP fragments reported by mass spectrometry analyses with unknown physiologic or pathologic properties | [76] | |
| 17–28 kDa APP N-Terminal Fragments | SH-SY5Y cells with HSV-1-mediated APP expression; rat cortical neuronal cultures | Generated through a developmentally regulated PKC-related APP processing pathway independent of α-secretase or β-secretase 1, with a putative important role in APP physiological function | [127,130] |
| Gene | Accession Number | Location | Encoded Protein | sAPPα-Mediated Regulation |
|---|---|---|---|---|
| TSPAN3 | NM_198902.3 | 15q24.3 | Tetraspanin-3 | Up-regulated |
| ISM2 | NM_199296 | 14q24.3 | Isthmin 2 | Up-regulated |
| MNAT1 | AY165512.1 | 14q23.1 | MNAT1 component of CDK activating kinase | Up-regulated |
| HSPA8 | NM_153201.4 | 11q24.1 | Heat Shock Protein Family A (Hsp70) Member 8 | Up-regulated |
| SOX11 | AB028641.1 | 2p25.2 | SRY-Box Transcription Factor 11 | Up-regulated |
| USE1 | AB097050.1 | 19p13.11 | Unconventional SNARE in the ER 1 | Up-regulated |
| KALRN | NM_007064.5 | 3q21.1-q21.2 | Kalirin RhoGEF kinase | Up-regulated |
| MAP3K5 | NM_005923 | 6q23.3 | Mitogen-Activated Protein Kinase Kinase Kinase 5 | Up-regulated |
| PTGES3 | NM_006601.7 | 12q13.3; 12 | Prostaglandin E Synthase 3 | Up-regulated |
| BACE1 | BC065492.1 | 11q23.3 | β-secretase 1 | Down-regulated |
| VEGFA | AF022375.1 | 6p21.1 | Vascular Endothelial Growth Factor A | Down-regulated |
| RBM8A | NM_005105.5 | 1q21.1 | RNA Binding Motif Protein 8A | Down-regulated |
| SLC27A3 | BC003654.2 | 1q21.3 | Solute Carrier Family 27 Member 3 | Down-regulated |
| METTL21A | BC009462.1 | 2q33.3 | Methyltransferase 21A, HSPA Lysine | Down-regulated |
| RPS28 | NM_001031.5 | 19p13.2 | Ribosomal Protein S28 | Down-regulated |
| RPL24 | NM_000986.4 | 3q12.3 | Ribosomal Protein L24 | Down-regulated |
| UBE3A | NM_130839.5 | 15q11.2 | Ubiquitin Protein Ligase E3A | Down-regulated |
| QDPR | BC000576.2 | 4p15.32 | Quinoid Dihydropteridine Reductase | Down-regulated |
| MTX1 | BC001906.1 | 1q22 | Metaxin 1 | Down-regulated |
| COMMD1 | NM_152516.4 | 2p15 | Copper Metabolism Domain Containing 1 | Down-regulated |
| PSMC6 | NM_002806.5 | 14q22.1 | Proteasome 26S Subunit, ATPase 6 | Down-regulated |
| Gene | Reported Correlation with AD/TBI | Ref. |
|---|---|---|
| HSPA8 | Decreased in AD patients’ samples and possible molecular biomarker for prognosis among HSP70 family in AD; part of the alcohol-sensitive protein networks modulated by AD-associated proteins that exacerbate neural and behavioural pathology upon alcohol drinking; potential drug target for preventing protein misfolding aggregation and cell death in AD and other neurodegenerative pathologies | [146,147,148] |
| SOX11 | Hypothesised to participate in the modulation of neuron plasticity in the dentate gyrus of the hippocampus; involved in the early attempts of axon regeneration and neuronal survival; modulates peripheral nerve regeneration in adult mice and BDNF expression in an exon promoter-specific manner; favours endogenous neurogenesis and locomotor recovery in mice spinal cord injury | [149,150,151,152,153] |
| USE1 | Involved in ER morphology maintenance and in the regulation of ER stress-induced neuronal apoptosis | [154] |
| KALRN | Key role in synaptic plasticity and in dendritic arbours and spines formation; proposed as possible therapeutic target for pharmacological intervention for synapse dysregulation; its deficit contributes to AD-related cognitive decline and memory loss; prevents dendritic spine dysgenesis induced by Aβ oligomers | [155,156,157] |
| MAP3K5 | Neuroprotective action against apoptosis as part of the Notch1/ASK1/p38 MAPK signalling pathway; neuroprotective role against oxidative stress; involved in neuronal death after its IRE1α-mediated recruitment upon ER stress and UPR | [158,159,160] |
| RBM8A | Potential contribution to AD pathophysiological changes by regulating components of key complexes in autophagy | [161] |
| SLC27A3 | Highly expressed in in human neural stem cells and involved in early brain development | [162] |
| RPS28 | Promotion of P-body assembly and hypothesised to be linked with neurodegeneration | [163,164] |
| UBE3A | Involved in synaptic function and plasticity; age-dependently decreased in AD mice models; possible critical player in AD pathogenesis and potential therapeutic target | [165,166] |
| COMMD1 | Hypothesised to be involved in hypoxia-induced AD neurodegeneration and brain injury via its interaction with Hypoxia-Inducible Factor 1 alpha (HIF-1α) | [167] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masi, M.; Biundo, F.; Fiou, A.; Racchi, M.; Pascale, A.; Buoso, E. The Labyrinthine Landscape of APP Processing: State of the Art and Possible Novel Soluble APP-Related Molecular Players in Traumatic Brain Injury and Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 6639. https://doi.org/10.3390/ijms24076639
Masi M, Biundo F, Fiou A, Racchi M, Pascale A, Buoso E. The Labyrinthine Landscape of APP Processing: State of the Art and Possible Novel Soluble APP-Related Molecular Players in Traumatic Brain Injury and Neurodegeneration. International Journal of Molecular Sciences. 2023; 24(7):6639. https://doi.org/10.3390/ijms24076639
Chicago/Turabian StyleMasi, Mirco, Fabrizio Biundo, André Fiou, Marco Racchi, Alessia Pascale, and Erica Buoso. 2023. "The Labyrinthine Landscape of APP Processing: State of the Art and Possible Novel Soluble APP-Related Molecular Players in Traumatic Brain Injury and Neurodegeneration" International Journal of Molecular Sciences 24, no. 7: 6639. https://doi.org/10.3390/ijms24076639