
Transcriptomic Changes Predict Metabolic Alterations in LC3 Associated Phagocytosis in Aged Mice


Abstract
:1. Introduction
2. Results
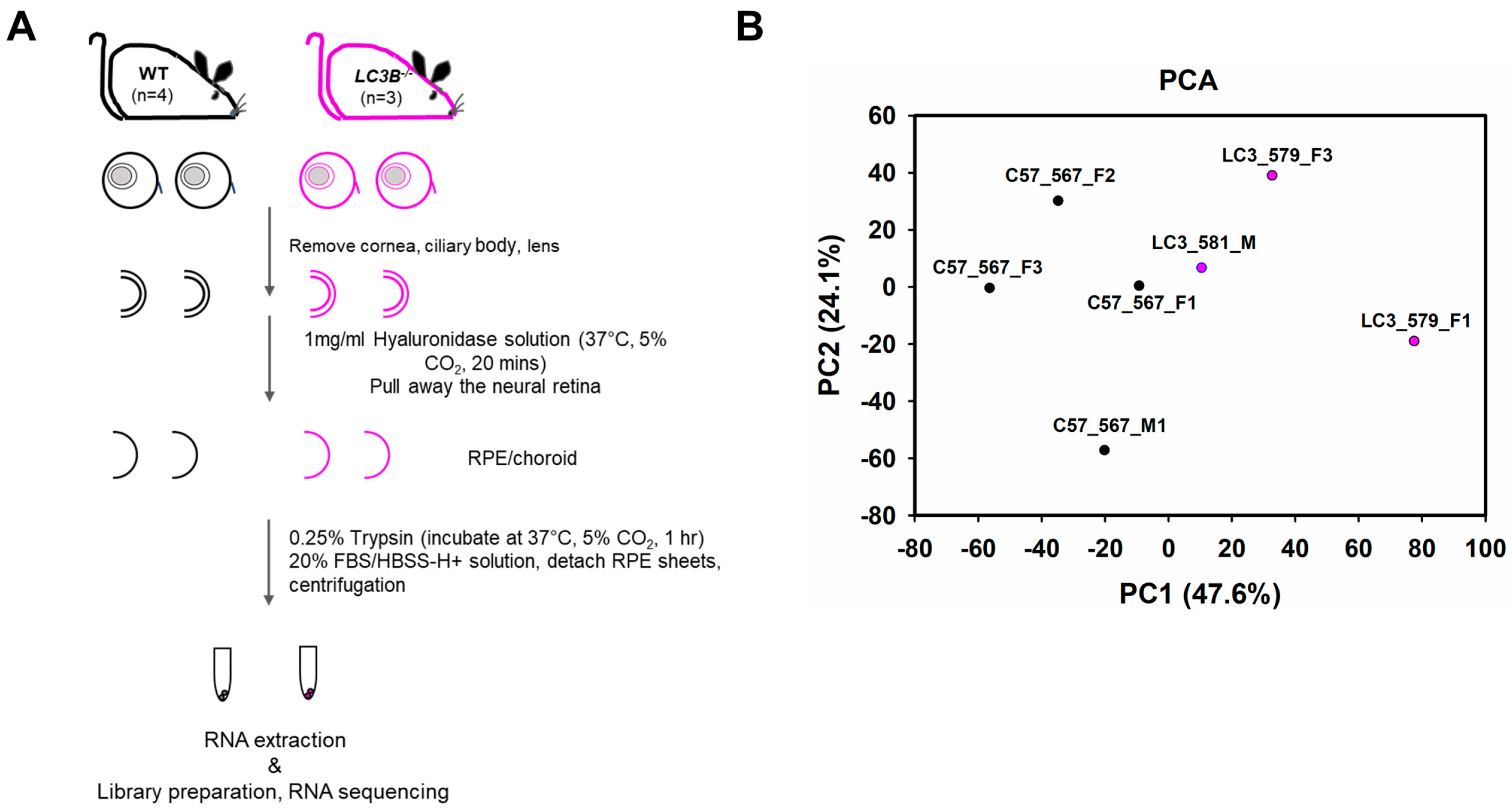
2.1. RNAseq Data Source and Quality (High Throughput RNA Sequencing Data Set Preparation for Analysis)
2.2. RNA-Sequencing Analysis to Identify Differentially Expressed Genes (DEGs)
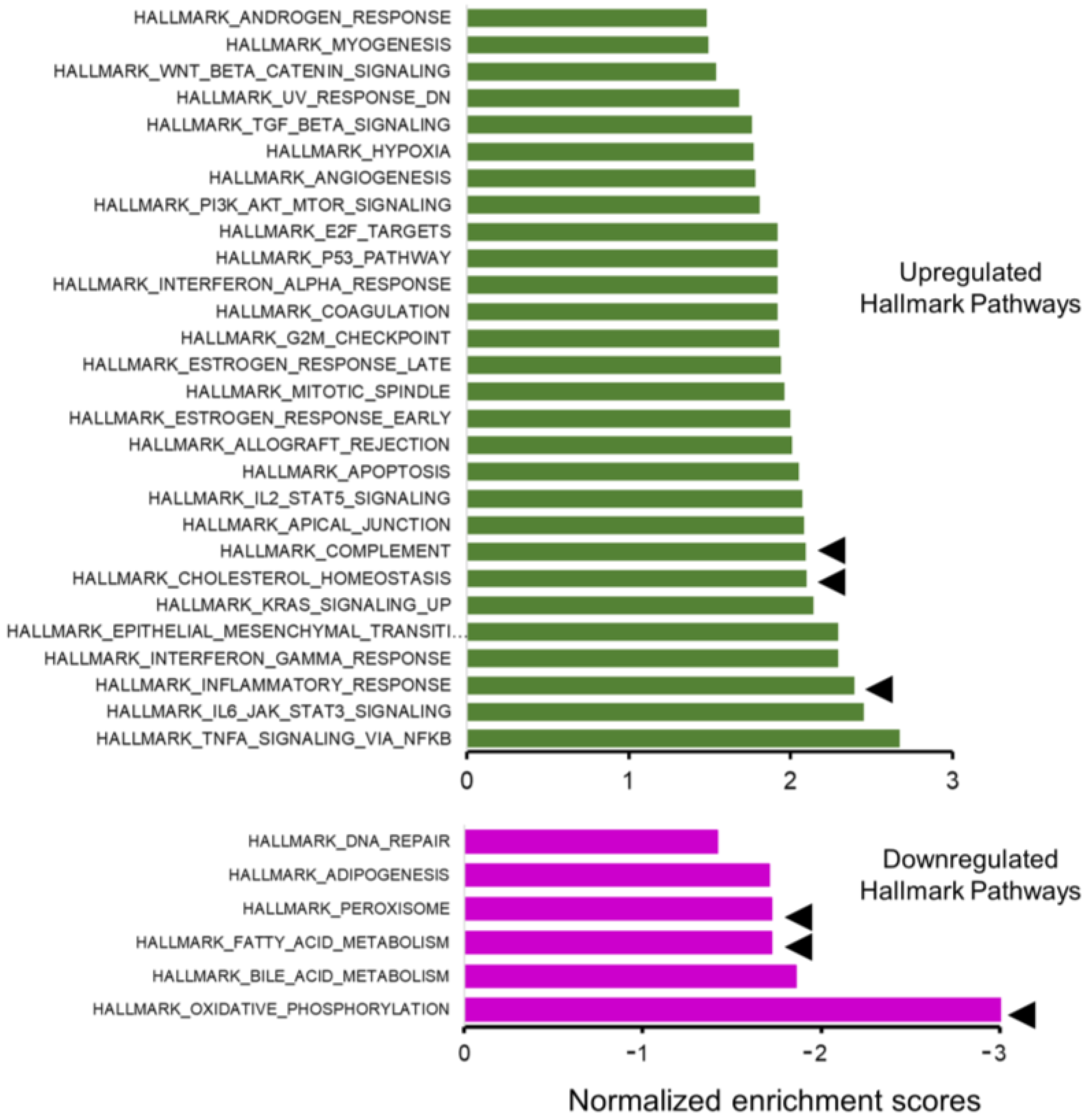
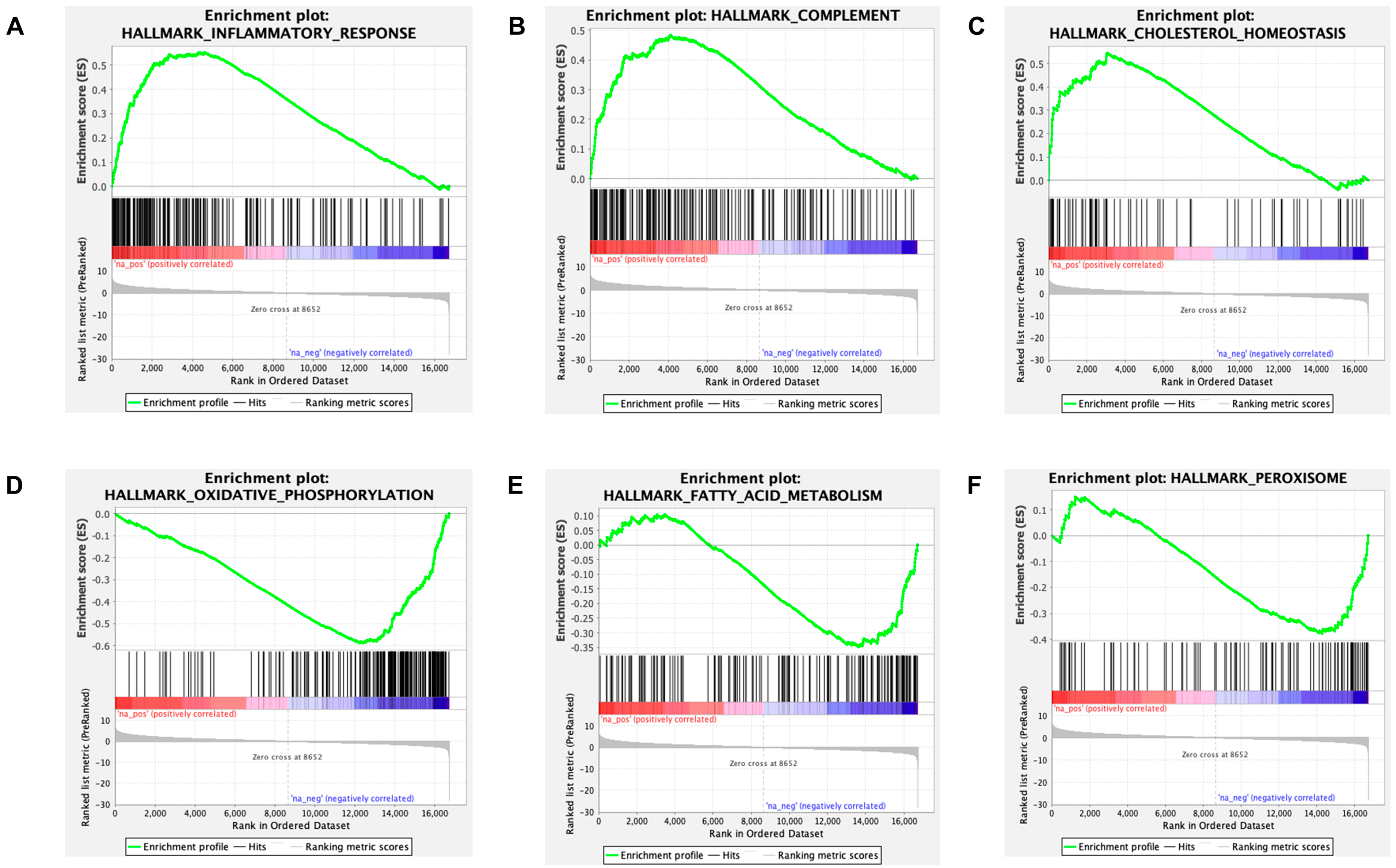
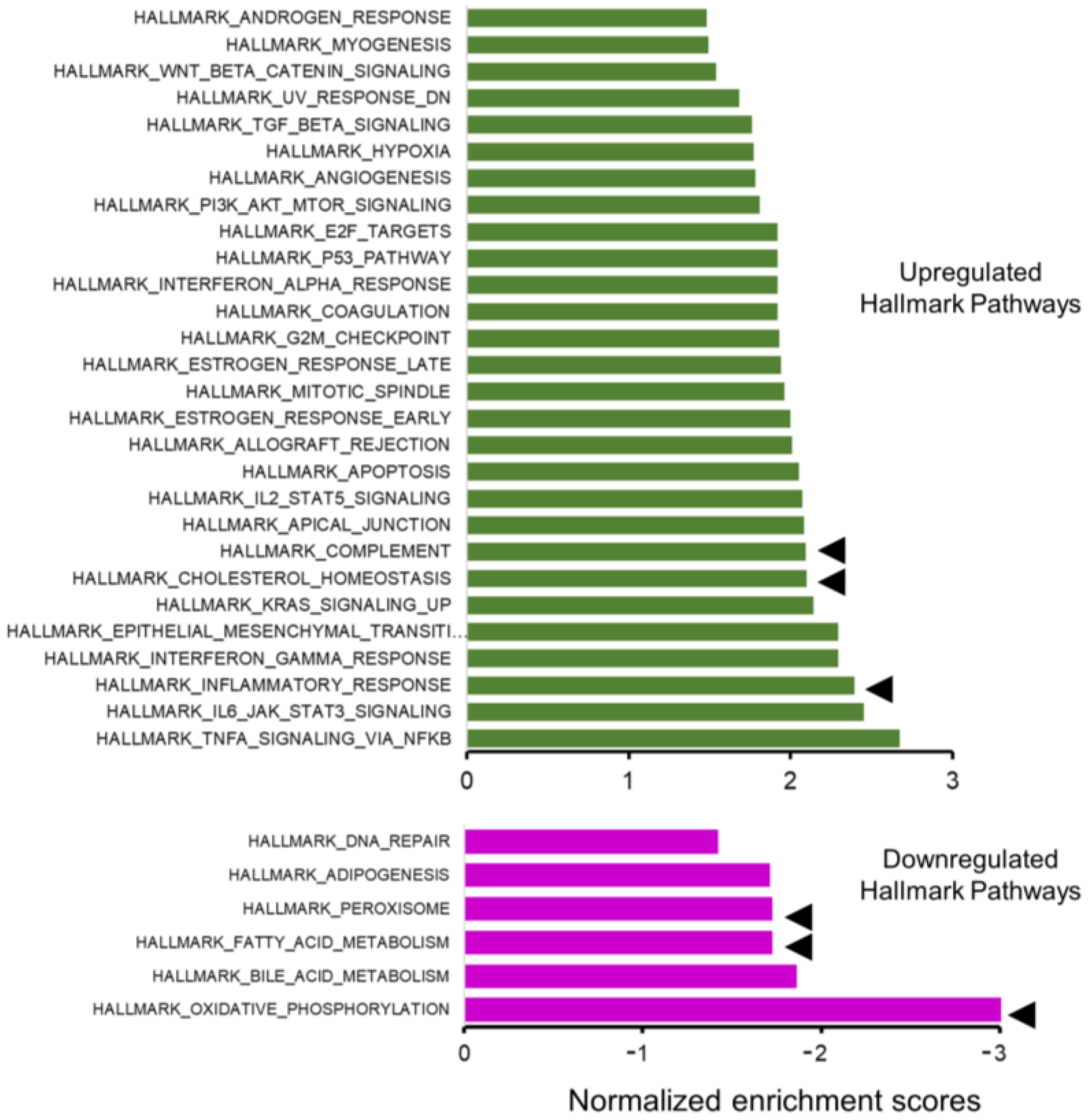
2.3. RNA-Sequencing Reveals Pathways Related to Inflammation and Metabolism
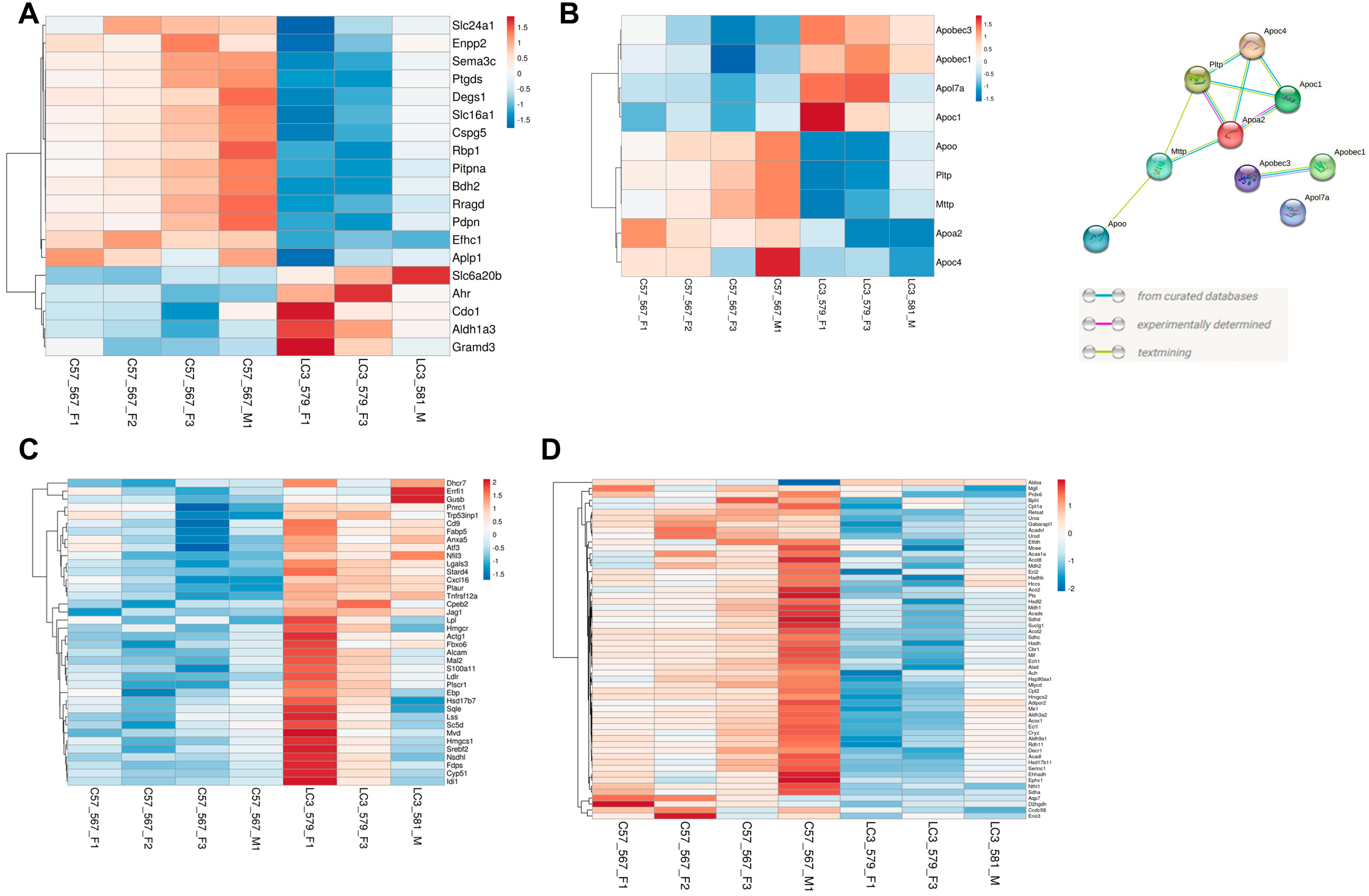
2.4. Several Solute Carrier Family Genes Are Differentially Expressed
2.5. Loss of LC3b Is Associated with Differential Expression of Select RPE Signature and Lipid Metabolism Related Genes
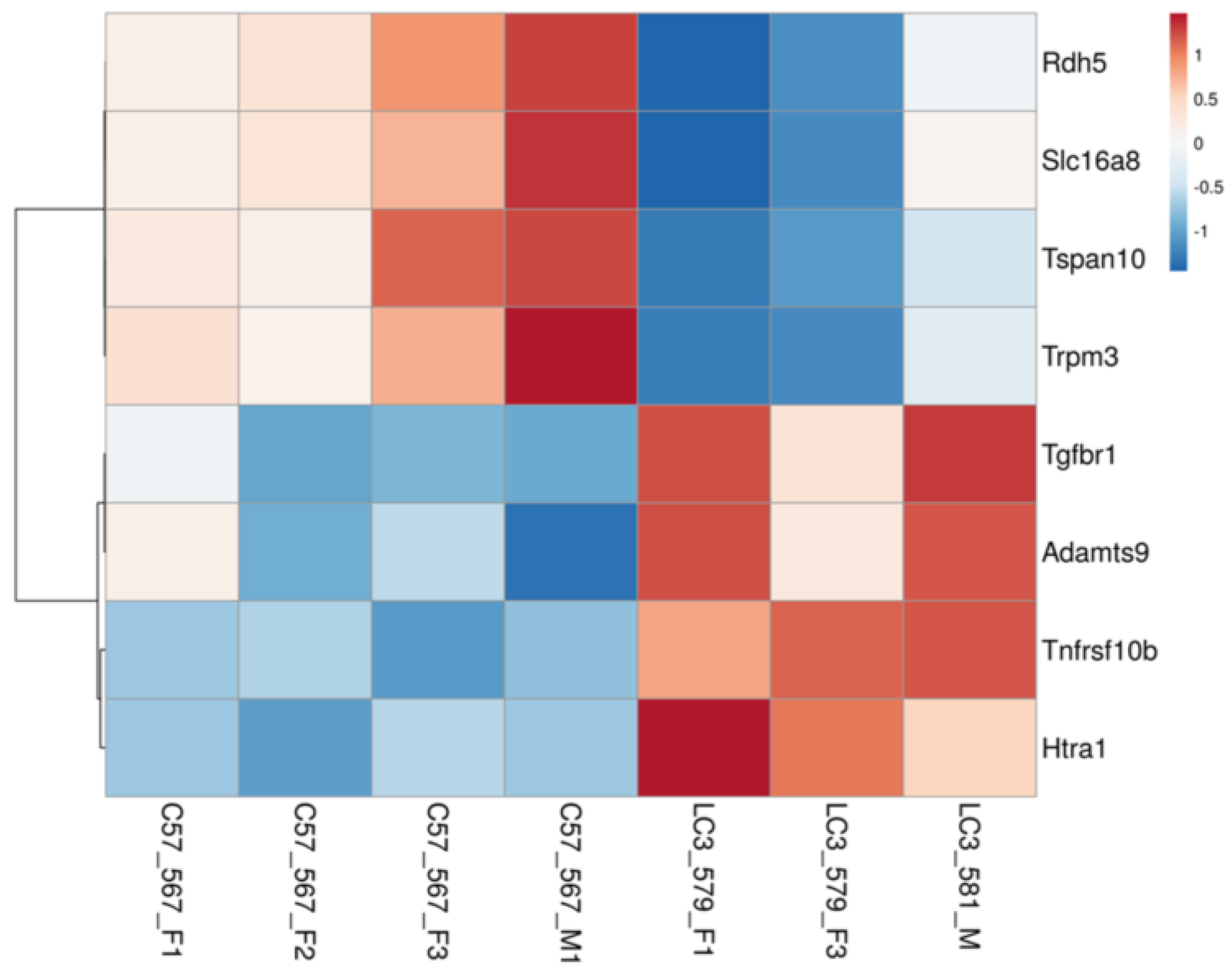
2.6. DEGs with Potential Role in Age-Related Retinal Diseases
3. Discussion
3.1. LC3b and Lipid Metabolic Dysregulation
3.2. Defective LAP, Inflammation, and Complement System
3.3. Lapses in LAP and AMD Associated Phenotype
4. Materials and Methods
4.1. Animals
4.2. Antibodies
4.3. RPE Cell Isolation
4.4. RNA Isolation and Library Preparation
4.5. RNAseq Data Analysis
4.6. Enrichment Analysis
4.7. Gene Expression Visualization
4.8. Protein–Protein Interaction Network Analysis
4.9. Immunoblotting
4.10. Immunostaining
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Cemma, M.; Brumell, J.H. Interactions of pathogenic bacteria with autophagy systems. Curr. Biol. 2012, 22, R540–R545. [Google Scholar] [CrossRef] [Green Version]
- Peña-Martinez, C.; Rickman, A.D.; Heckmann, B.L. Beyond autophagy: LC3-associated phagocytosis and endocytosis. Sci. Adv. 2022, 8, eabn1702. [Google Scholar] [CrossRef]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Liton, P.B.; Boesze-Battaglia, K.; Boulton, M.E.; Boya, P.; Ferguson, T.A.; Ganley, I.G.; Kauppinnen, A.; Laurie, G.W.; Mizushima, N.; Morishita, H.; et al. Autophagy in the Eye: From Physiology to Pathophysology. Autophagy Rep. 2023, 2, 2178996. [Google Scholar] [CrossRef]
- Xie, Y.; Li, J.; Kang, R.; Tang, D. Interplay Between Lipid Metabolism and Autophagy. Front. Cell Dev. Biol. 2020, 8, 431. [Google Scholar] [CrossRef]
- Heckmann, B.L.; Green, D.R. LC3-associated phagocytosis at a glance. J. Cell Sci. 2019, 132, jcs222984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grijmans, B.J.M.; van der Kooij, S.B.; Varela, M.; Meijer, A.H. LAPped in Proof: LC3-Associated Phagocytosis and the Arms Race Against Bacterial Pathogens. Front. Cell. Infect. Microbiol. 2021, 11, 809121. [Google Scholar] [CrossRef] [PubMed]
- Asare, P.F.; Tran, H.B.; Hurtado, P.R.; Perkins, G.B.; Nguyen, P.; Jersmann, H.; Roscioli, E.; Hodge, S. Inhibition of LC3-associated phagocytosis in COPD and in response to cigarette smoke. Adv. Respir. Dis. 2021, 15, 17534666211039769. [Google Scholar] [CrossRef]
- Kim, J.Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.C.; Zhou, Z.; Green, D.R.; et al. Noncanonical autophagy promotes the visual cycle. Cell 2013, 154, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Frost, L.S.; Lopes, V.S.; Bragin, A.; Reyes-Reveles, J.; Brancato, J.; Cohen, A.; Mitchell, C.H.; Williams, D.S.; Boesze-Battaglia, K. The Contribution of Melanoregulin to Microtubule-Associated Protein 1 Light Chain 3 (LC3) Associated Phagocytosis in Retinal Pigment Epithelium. Mol. Neurobiol. 2015, 52, 1135–1151. [Google Scholar] [CrossRef] [Green Version]
- Yefimova, M.G.; Ravel, C.; Rolland, A.D.; Bourmeyster, N.; Jégou, B. MERTK-Mediated LC3-Associated Phagocytosis (LAP) of Apoptotic Substrates in Blood-Separated Tissues: Retina, Testis, Ovarian Follicles. Cells 2021, 10, 1443. [Google Scholar] [CrossRef] [PubMed]
- Panneerdoss, S.; Viswanadhapalli, S.; Abdelfattah, N.; Onyeagucha, B.C.; Timilsina, S.; Mohammad, T.A.; Chen, Y.; Drake, M.; Vuori, K.; Kumar, T.R.; et al. Cross-talk between miR-471-5p and autophagy component proteins regulates LC3-associated phagocytosis (LAP) of apoptotic germ cells. Nat. Commun. 2017, 8, 598. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [Green Version]
- Minami, S.; Nakamura, S.; Yoshimori, T. Rubicon in Metabolic Diseases and Ageing. Front. Cell Dev. Biol. 2022, 9, 3798. [Google Scholar] [CrossRef]
- Martinez, J.; Malireddi, R.K.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Lam, H.C.; Chen, Z.H.; Choi, A.M. Deadly triplex: Smoke, autophagy and apoptosis. Autophagy 2011, 7, 436–437. [Google Scholar] [CrossRef] [Green Version]
- Kesireddy, V.S.; Chillappagari, S.; Ahuja, S.; Knudsen, L.; Henneke, I.; Graumann, J.; Meiners, S.; Ochs, M.; Ruppert, C.; Korfei, M.; et al. Susceptibility of microtubule-associated protein 1 light chain 3beta (MAP1LC3B/LC3B) knockout mice to lung injury and fibrosis. FASEB J. 2019, 33, 12392–12408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asare, P.F.; Roscioli, E.; Hurtado, P.R.; Tran, H.B.; Mah, C.Y.; Hodge, S. LC3-Associated Phagocytosis (LAP): A Potentially Influential Mediator of Efferocytosis-Related Tumor Progression and Aggressiveness. Front. Oncol. 2020, 10, 1298. [Google Scholar] [CrossRef]
- Cunha, L.D.; Yang, M.; Carter, R.; Guy, C.; Harris, L.; Crawford, J.C.; Quarato, G.; Boada-Romero, E.; Kalkavan, H.; Johnson, M.D.L.; et al. LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 2018, 175, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J. LAP it up, fuzz ball: A short history of LC3-associated phagocytosis. Curr. Opin. Immunol. 2018, 55, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, P.; Xiao, W. β-hydroxybutyrate as an Anti-Aging Metabolite. Nutrients 2021, 13, 3420. [Google Scholar] [CrossRef]
- Yang, S.; Zhou, J.; Li, D. Functions and Diseases of the Retinal Pigment Epithelium. Front. Pharmacol. 2021, 12, 727870. [Google Scholar] [CrossRef] [PubMed]
- Kevany, B.M.; Palczewski, K. Phagocytosis of retinal rod and cone photoreceptors. Physiology 2010, 25, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volland, S.; Esteve-Rudd, J.; Hoo, J.; Yee, C.; Williams, D.S. A comparison of some organizational characteristics of the mouse central retina and the human macula. PLoS ONE 2015, 10, e0125631. [Google Scholar] [CrossRef] [Green Version]
- Dhingra, A.; Bell, B.A.; Peachey, N.S.; Daniele, L.L.; Reyes-Reveles, J.; Sharp, R.C.; Jun, B.; Bazan, N.G.; Sparrow, J.R.; Kim, H.J.; et al. Microtubule-Associated Protein 1 Light Chain 3B, (LC3B) Is Necessary to Maintain Lipid-Mediated Homeostasis in the Retinal Pigment Epithelium. Front. Cell. Neurosci. 2018, 12, 351. [Google Scholar] [CrossRef] [Green Version]
- Strauss, O. The Retinal Pigment Epithelium in Visual Function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cross, S.D.; Stanton, J.B.; Marmorstein, A.D.; Le, Y.Z.; Marmorstein, L.Y. Early AMD-like defects in the RPE and retinal degeneration in aged mice with RPE-specific deletion of Atg5 or Atg7. Mol. Vis. 2017, 23, 228–241. [Google Scholar] [PubMed]
- Curcio, C.A.; Johnson, M.; Rudolf, M.; Huang, J.D. The oil spill in ageing Bruch membrane. Br. J. Ophthalmol. 2011, 95, 1638–1645. [Google Scholar] [CrossRef]
- Malek, G.; Li, C.M.; Guidry, C.; Medeiros, N.E.; Curcio, C.A. Apolipoprotein B in cholesterol-containing drusen and basal deposits of human eyes with age-related maculopathy. Am. J. Pathol. 2003, 162, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curcio, C.A.; Presley, J.B.; Millican, C.L.; Medeiros, N.E. Basal deposits and drusen in eyes with age-related maculopathy: Evidence for solid lipid particles. Exp. Eye Res. 2005, 80, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Messinger, J.D.; Zhang, Y.; Spaide, R.F.; Freund, K.B.; Curcio, C.A. SUBRETINAL DRUSENOID DEPOSIT IN AGE-RELATED MACULAR DEGENERATION: Histologic Insights Into Initiation, Progression to Atrophy, and Imaging. Retina 2020, 40, 618–631. [Google Scholar] [CrossRef]
- Reyes-Reveles, J.; Dhingra, A.; Alexander, D.; Bragin, A.; Philp, N.J.; Boesze-Battaglia, K. Phagocytosis-dependent ketogenesis in retinal pigment epithelium. J. Biol. Chem. 2017, 292, 8038–8047. [Google Scholar] [CrossRef] [Green Version]
- Adijanto, J.; Du, J.; Moffat, C.; Seifert, E.L.; Hurle, J.B.; Philp, N.J. The retinal pigment epithelium utilizes fatty acids for ketogenesis. J. Biol. Chem. 2014, 289, 20570–20582. [Google Scholar] [CrossRef] [Green Version]
- Cotter, D.G.; Ercal, B.; Huang, X.; Leid, J.M.; d’Avignon, D.A.; Graham, M.J.; Dietzen, D.J.; Brunt, E.M.; Patti, G.J.; Crawford, P.A. Ketogenesis prevents diet-induced fatty liver injury and hyperglycemia. J. Clin. Investig. 2014, 124, 5175–5190. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.K.; Lee, J.A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpilka, T.; Weidberg, H.; Pietrokovski, S.; Elazar, Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biol. 2011, 12, 226. [Google Scholar] [CrossRef]
- Dhingra, A.; Alexander, D.; Reyes-Reveles, J.; Sharp, R.; Boesze-Battaglia, K. Microtubule-Associated Protein 1 Light Chain 3 (LC3) Isoforms in RPE and Retina. Adv. Exp. Med. Biol. 2018, 1074, 609–616. [Google Scholar] [CrossRef]
- Daniele, L.L.; Caughey, J.; Volland, S.; Sharp, R.C.; Dhingra, A.; Williams, D.S.; Philp, N.J.; Boesze-Battaglia, K. Peroxisome turnover and diurnal modulation of antioxidant activity in retinal pigment epithelia utilizes microtubule-associated protein 1 light chain 3B (LC3B). Am. J. Physiol. Cell Physiol. 2019, 317, C1194–C1204. [Google Scholar] [CrossRef]
- Lakkaraju, A.; Umapathy, A.; Tan, L.X.; Daniele, L.; Philp, N.J.; Boesze-Battaglia, K.; Williams, D.S. The cell biology of the retinal pigment epithelium. Prog. Retin. Eye Res. 2020, 78, 100846. [Google Scholar] [CrossRef] [PubMed]
- Adijanto, J.; Philp, N.J. The SLC16A family of monocarboxylate transporters (MCTs)--physiology and function in cellular metabolism, pH homeostasis, and fluid transport. Curr. Top. Membr. 2012, 70, 275–311. [Google Scholar] [CrossRef] [PubMed]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef] [PubMed]
- Philp, N.J.; Ochrietor, J.D.; Rudoy, C.; Muramatsu, T.; Linser, P.J. Loss of MCT1, MCT3, and MCT4 expression in the retinal pigment epithelium and neural retina of the 5A11/basigin-null mouse. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1305–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.Y.S.; Kinoshita, J.; Bisetto, S.; Bell, B.A.; Nowak, R.A.; Peachey, N.S.; Philp, N.J. Role of monocarboxylate transporters in regulating metabolic homeostasis in the outer retina: Insight gained from cell-specific Bsg deletion. FASEB J. 2020, 34, 5401–5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strunnikova, N.V.; Maminishkis, A.; Barb, J.J.; Wang, F.; Zhi, C.; Sergeev, Y.; Chen, W.; Edwards, A.O.; Stambolian, D.; Abecasis, G.; et al. Transcriptome analysis and molecular signature of human retinal pigment epithelium. Hum. Mol. Genet. 2010, 19, 2468–2486. [Google Scholar] [CrossRef]
- Borooah, S.; Phillips, M.J.; Bilican, B.; Wright, A.F.; Wilmut, I.; Chandran, S.; Gamm, D.; Dhillon, B. Using human induced pluripotent stem cells to treat retinal disease. Prog. Retin. Eye Res. 2013, 37, 163–181. [Google Scholar] [CrossRef] [Green Version]
- Calderon-Dominguez, M.; Gil, G.; Medina, M.A.; Pandak, W.M.; Rodriguez-Agudo, D. The StarD4 subfamily of steroidogenic acute regulatory-related lipid transfer (START) domain proteins: New players in cholesterol metabolism. Int. J. Biochem. Cell Biol. 2014, 49, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Watts, G.F.; Ding, P.Y.A.; George, P.; Hagger, M.S.; Hu, M.; Lin, J.; Khoo, K.L.; Marais, A.D.; Miida, T.; Nawawi, H.M.; et al. Translational Research for Improving the Care of Familial Hypercholesterolemia: The “Ten Countries Study” and Beyond. J. Atheroscler. Thromb. 2016, 23, 891–900. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.K.; Ramli, F.F.; Ali, A.; Ibrahim, N. Genetics of Cholesterol-Related Genes in Metabolic Syndrome: A Review of Current Evidence. Biomedicines 2022, 10, 3239. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Costello, J.; Godinho, L.F.; Islinger, M. Peroxisome-mitochondria interplay and disease. J. Inherit. Metab. Dis. 2015, 38, 681–702. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhoven, P.P. Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 2010, 51, 2863–2895. [Google Scholar] [CrossRef] [Green Version]
- Handa, J.T.; Cano, M.; Wang, L.; Datta, S.; Liu, T. Lipids, oxidized lipids, oxidation-specific epitopes, and Age-related Macular Degeneration. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2017, 1862, 430–440. [Google Scholar] [CrossRef] [Green Version]
- van Leeuwen, E.M.; Emri, E.; Merle, B.M.J.; Colijn, J.M.; Kersten, E.; Cougnard-Gregoire, A.; Dammeier, S.; Meester-Smoor, M.; Pool, F.M.; de Jong, E.K.; et al. A new perspective on lipid research in age-related macular degeneration. Prog. Retin. Eye Res. 2018, 67, 56–86. [Google Scholar] [CrossRef]
- Landowski, M.; Bowes Rickman, C. Targeting Lipid Metabolism for the Treatment of Age-Related Macular Degeneration: Insights from Preclinical Mouse Models. J. Ocul. Pharmacol. Ther. 2022, 38, 3–32. [Google Scholar] [CrossRef]
- Tan, L.X.; Germer, C.J.; La Cunza, N.; Lakkaraju, A. Complement activation, lipid metabolism, and mitochondrial injury: Converging pathways in age-related macular degeneration. Redox Biol. 2020, 37, 101781. [Google Scholar] [CrossRef]
- Cherepanoff, S.; McMenamin, P.; Gillies, M.C.; Kettle, E.; Sarks, S.H. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br. J. Ophthalmol. 2010, 94, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef] [Green Version]
- Strunz, T.; Kiel, C.; Sauerbeck, B.L.; Weber, B.H.F. Learning from Fifteen Years of Genome-Wide Association Studies in Age-Related Macular Degeneration. Cells 2020, 9, 2267. [Google Scholar] [CrossRef]
- Daniele, L.L.; Sauer, B.; Gallagher, S.M.; Pugh, E.N., Jr.; Philp, N.J. Altered visual function in monocarboxylate transporter 3 (Slc16a8) knockout mice. Am. J. Physiol. Cell Physiol. 2008, 295, C451–C457. [Google Scholar] [CrossRef] [Green Version]
- Occelli, L.M.; Daruwalla, A.; De Silva, S.R.; Winkler, P.A.; Sun, K.; Pasmanter, N.; Minella, A.; Querubin, J.; Lyons, L.A.; Consortium, L.; et al. A large animal model of RDH5-associated retinopathy recapitulates important features of the human phenotype. Hum. Mol. Genet. 2021, 31, 1263–1277. [Google Scholar] [CrossRef]
- Liu, B.; Calton, M.A.; Abell, N.S.; Benchorin, G.; Gloudemans, M.J.; Chen, M.; Hu, J.; Li, X.; Balliu, B.; Bok, D.; et al. Genetic analyses of human fetal retinal pigment epithelium gene expression suggest ocular disease mechanisms. Commun. Biol. 2019, 2, 186. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.K.; Yang, J.; Hsu, C.W.; Gore, A.; Bassuk, A.G.; Brown, L.M.; Colligan, R.; Sengillo, J.D.; Mahajan, V.B.; Tsang, S.H. HTRA1, an age-related macular degeneration protease, processes extracellular matrix proteins EFEMP1 and TSP1. Aging Cell 2018, 17, e12710. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Sigrist, S. Autophagy and proteostasis in the control of synapse aging and disease. Curr. Opin. Neurobiol. 2018, 48, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Iker Etchegaray, J.; Kelley, S.; Penberthy, K.; Karvelyte, L.; Nagasaka, Y.; Gasperino, S.; Paul, S.; Seshadri, V.; Raymond, M.; Marco, A.R.; et al. Phagocytosis in the retina promotes local insulin production in the eye. Nat. Metab. 2023, 5, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Cann, G.M.; Guignabert, C.; Ying, L.; Deshpande, N.; Bekker, J.M.; Wang, L.; Zhou, B.; Rabinovitch, M. Developmental expression of LC3alpha and beta: Absence of fibronectin or autophagy phenotype in LC3beta knockout mice. Dev. Dyn. 2008, 237, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Esteve-Rudd, J.; Lopes, V.S.; Diemer, T.; Lillo, C.; Rump, A.; Williams, D.S. Microtubule motors transport phagosomes in the RPE, and lack of KLC1 leads to AMD-like pathogenesis. J. Cell Biol. 2015, 210, 595–611. [Google Scholar] [CrossRef] [Green Version]
- Fliesler, A.J.; Anderson, R.E. Chemistry and metabolism of lipids in the vertebrate retina. Prog. Lipid Res. 1983, 22, 79–131. [Google Scholar] [CrossRef]
- Beier, K.; Völkl, A.; Fahimi, H.D. The impact of aging on enzyme proteins of rat liver peroxisomes: Quantitative analysis by immunoblotting and immunoelectron microscopy. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1993, 63, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Feher, J.; Kovacs, I.; Artico, M.; Cavallotti, C.; Papale, A.; Balacco Gabrieli, C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 2006, 27, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Liles, M.R.; Newsome, D.A.; Oliver, P.D. Antioxidant enzymes in the aging human retinal pigment epithelium. Arch. Ophthalmol. 1991, 109, 1285–1288. [Google Scholar] [CrossRef] [PubMed]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Grenell, A.; Zhong, F.; Yam, M.; Hauer, A.; Gregor, E.; Zhu, S.; Lohner, D.; Zhu, J.; Du, J. Metabolic signature of the aging eye in mice. Neurobiol. Aging 2018, 71, 223–233. [Google Scholar] [CrossRef] [PubMed]
- d’Avignon, D.A.; Puchalska, P.; Ercal, B.; Chang, Y.; Martin, S.E.; Graham, M.J.; Patti, G.J.; Han, X.; Crawford, P.A. Hepatic ketogenic insufficiency reprograms hepatic glycogen metabolism and the lipidome. J. Clin. Investig. 2018, 3, e99762. [Google Scholar] [CrossRef] [Green Version]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.R.; Ferrington, D.A. Perspective on AMD Pathobiology: A Bioenergetic Crisis in the RPE. Investig. Opthalmol. Vis. Sci. 2018, 59, AMD41–AMD47. [Google Scholar] [CrossRef] [Green Version]
- Asif, S.; Kim, R.Y.; Fatica, T.; Sim, J.; Zhao, X.; Oh, Y.; Denoncourt, A.; Cheung, A.C.; Downey, M.; Mulvihill, E.E.; et al. Hmgcs2-mediated ketogenesis modulates high-fat diet-induced hepatosteatosis. Mol. Metab. 2022, 61, 101494. [Google Scholar] [CrossRef]
- Pang, X.Y.; Wang, S.; Jurczak, M.J.; Shulman, G.I.; Moise, A.R. Retinol saturase modulates lipid metabolism and the production of reactive oxygen species. Arch. Biochem. Biophys. 2017, 633, 93–102. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [Green Version]
- Moise, A.R.; Lobo, G.P.; Erokwu, B.; Wilson, D.L.; Peck, D.; Alvarez, S.; Domínguez, M.; Alvarez, R.; Flask, C.A.; de Lera, A.R.; et al. Increased adiposity in the retinol saturase-knockout mouse. FASEB J. 2010, 24, 1261–1270. [Google Scholar] [CrossRef] [Green Version]
- Pikuleva, I.A.; Curcio, C.A. Cholesterol in the retina: The best is yet to come. Prog. Retin. Eye Res. 2014, 41, 64–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhaescu, I.; Izzedine, H. Mevalonate pathway: A review of clinical and therapeutical implications. Clin. Biochem. 2007, 40, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Fliesler, S.J.; Bretillon, L. The ins and outs of cholesterol in the vertebrate retina. J. Lipid Res. 2010, 51, 3399–3413. [Google Scholar] [CrossRef] [Green Version]
- Solsona-Vilarrasa, E.; Fucho, R.; Torres, S.; Nuñez, S.; Nuño-Lámbarri, N.; Enrich, C.; García-Ruiz, C.; Fernández-Checa, J.C. Cholesterol enrichment in liver mitochondria impairs oxidative phosphorylation and disrupts the assembly of respiratory supercomplexes. Redox Biol. 2019, 24, 101214. [Google Scholar] [CrossRef]
- Schirris, T.J.J.; Rossell, S.; de Haas, R.; Frambach, S.; Hoogstraten, C.A.; Renkema, G.H.; Beyrath, J.D.; Willems, P.; Huynen, M.A.; Smeitink, J.A.M.; et al. Stimulation of cholesterol biosynthesis in mitochondrial complex I-deficiency lowers reductive stress and improves motor function and survival in mice. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 166062. [Google Scholar] [CrossRef]
- Dhingra, A.; Sharp, R.C.; Kim, T.; Popov, A.V.; Ying, G.S.; Pietrofesa, R.A.; Park, K.; Christofidou-Solomidou, M.; Boesze-Battaglia, K. Assessment of a Small Molecule Synthetic Lignan in Enhancing Oxidative Balance and Decreasing Lipid Accumulation in Human Retinal Pigment Epithelia. Int. J. Mol. Sci. 2021, 22, 5764. [Google Scholar] [CrossRef]
- Wang, L.; Clark, M.E.; Crossman, D.K.; Kojima, K.; Messinger, J.D.; Mobley, J.A.; Curcio, C.A. Abundant lipid and protein components of drusen. PLoS ONE 2010, 5, e10329. [Google Scholar] [CrossRef] [PubMed]
- Li, C.M.; Chung, B.H.; Presley, J.B.; Malek, G.; Zhang, X.; Dashti, N.; Li, L.; Chen, J.; Bradley, K.; Kruth, H.S.; et al. Lipoprotein-like particles and cholesteryl esters in human Bruch’s membrane: Initial characterization. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2576–2586. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, C.M.; Rudolf, M.; Belyaeva, O.V.; Chung, B.H.; Messinger, J.D.; Kedishvili, N.Y.; Curcio, C.A. Lipoprotein particles of intraocular origin in human Bruch membrane: An unusual lipid profile. Investig. Opthalmology Vis. Sci. 2009, 50, 870–877. [Google Scholar] [CrossRef]
- Sirwi, A.; Hussain, M.M. Lipid transfer proteins in the assembly of apoB-containing lipoproteins. J. Lipid Res. 2018, 59, 1094–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Léveillard, T.; Philp, N.J.; Sennlaub, F. Is Retinal Metabolic Dysfunction at the Center of the Pathogenesis of Age-related Macular Degeneration? Int. J. Mol. Sci. 2019, 20, 762. [Google Scholar] [CrossRef] [Green Version]
- Philp, N.J.; Wang, D.; Yoon, H.; Hjelmeland, L.M. Polarized expression of monocarboxylate transporters in human retinal pigment epithelium and ARPE-19 cells. Investig. Opthalmol. Vis. Sci. 2003, 44, 1716–1721. [Google Scholar] [CrossRef] [Green Version]
- Adijanto, J.; Philp, N.J. Cultured primary human fetal retinal pigment epithelium (hfRPE) as a model for evaluating RPE metabolism. Exp. Eye Res. 2014, 126, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Vogt, S.D.; Curcio, C.A.; Wang, L.; Li, C.M.; McGwin, G., Jr.; Medeiros, N.E.; Philp, N.J.; Kimble, J.A.; Read, R.W. Retinal pigment epithelial expression of complement regulator CD46 is altered early in the course of geographic atrophy. Exp. Eye Res. 2011, 93, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439, 439e431–439e432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Yasumura, D.; Li, X.; Matthes, M.; Lloyd, M.; Nielsen, G.; Ahern, K.; Snyder, M.; Bok, D.; Dunaief, J.L.; et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J. Clin. Investig. 2011, 121, 369–383. [Google Scholar] [CrossRef] [Green Version]
- Tam, J.M.; Mansour, M.K.; Khan, N.S.; Seward, M.; Puranam, S.; Tanne, A.; Sokolovska, A.; Becker, C.E.; Acharya, M.; Baird, M.A.; et al. Dectin-1–Dependent LC3 Recruitment to Phagosomes Enhances Fungicidal Activity in Macrophages. J. Infect. Dis. 2014, 210, 1844–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Becker, C.; Lowell, C.A.; Underhill, D.M. Dectin-1-triggered Recruitment of Light Chain 3 Protein to Phagosomes Facilitates Major Histocompatibility Complex Class II Presentation of Fungal-derived Antigens. J. Biol. Chem. 2012, 287, 34149–34156. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.-Y.; Cringle, S.J. Retinal degeneration and local oxygen metabolism. Exp. Eye Res. 2005, 80, 745–751. [Google Scholar] [CrossRef]
- Trakkides, T.O.; Schäfer, N.; Reichenthaler, M.; Kühn, K.; Brandwijk, R.; Toonen, E.J.M.; Urban, F.; Wegener, J.; Enzmann, V.; Pauly, D. Oxidative Stress Increases Endogenous Complement-Dependent Inflammatory and Angiogenic Responses in Retinal Pigment Epithelial Cells Independently of Exogenous Complement Sources. Antioxidants 2019, 8, 548. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Montenegro, D.; Zhao, J.; Sparrow, J.R. Bisretinoids of the Retina: Photo-Oxidation, Iron-Catalyzed Oxidation, and Disease Consequences. Antioxidants 2021, 10, 1382. [Google Scholar] [CrossRef]
- Asatryan, A.; Bazan, N.G. Molecular mechanisms of signaling via the docosanoid neuroprotectin D1 for cellular homeostasis and neuroprotection. J. Biol. Chem. 2017, 292, 12390–12397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Radu, R.A.; Hu, J.; Yuan, Q.; Welch, D.L.; Makshanoff, J.; Lloyd, M.; McMullen, S.; Travis, G.H.; Bok, D. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J. Biol. Chem. 2011, 286, 18593–18601. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, A.R.; Yshii, L.M.; Viel, T.A.; Buck, H.S.; Mattson, M.P.; Scavone, C.; Kawamoto, E.M. Intermittent fasting attenuates lipopolysaccharide-induced neuroinflammation and memory impairment. J. Neuroinflamm. 2014, 11, 85. [Google Scholar] [CrossRef] [Green Version]
- Youm, Y.-H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujol-Lereis, L.M.; Schäfer, N.; Kuhn, L.B.; Rohrer, B.; Pauly, D. Interrelation Between Oxidative Stress and Complement Activation in Models of Age-Related Macular Degeneration. Retin. Degener. Dis. Mech. Exp. Ther. 2016, 854, 87–93. [Google Scholar]
- Ding, J.D.; Kelly, U.; Groelle, M.; Christenbury, J.G.; Zhang, W.; Bowes Rickman, C. The role of complement dysregulation in AMD mouse models. Adv. Exp. Med. Biol. 2014, 801, 213–219. [Google Scholar] [CrossRef]
- Toomey, C.B.; Johnson, L.V.; Bowes Rickman, C. Complement factor H in AMD: Bridging genetic associations and pathobiology. Prog. Retin. Eye Res. 2018, 62, 38–57. [Google Scholar] [CrossRef]
- Hadziahmetovic, M.; Kumar, U.; Song, Y.; Grieco, S.; Song, D.; Li, Y.; Tobias, J.W.; Dunaief, J.L. Microarray analysis of murine retinal light damage reveals changes in iron regulatory, complement, and antioxidant genes in the neurosensory retina and isolated RPE. Investig. Opthalmol. Vis. Sci. 2012, 53, 5231–5241. [Google Scholar] [CrossRef] [PubMed]
- Rutar, M.; Natoli, R.; Albarracin, R.; Valter, K.; Provis, J. 670-nm light treatment reduces complement propagation following retinal degeneration. J. Neuroinflamm. 2012, 9, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Jiang, N.; Chu, Y.; Postnikova, O.; Varghese, R.; Horvath, A.; Cheema, A.K.; Golestaneh, N. Dysregulated metabolic pathways in age-related macular degeneration. Sci. Rep. 2020, 10, 2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Lin, Y.; Terry, R.; Nelson, K.; Bernstein, P.S. Role of long-chain and very-long-chain polyunsaturated fatty acids in macular degenerations and dystrophies. Clin. Lipidol. 2011, 6, 593–613. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Morishita, R. The roles of lipid and glucose metabolism in modulation of β-amyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front. Aging Neurosci. 2015, 7, 199. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.W. Advances in Understanding of the Role of Lipid Metabolism in Aging. Cells 2021, 10, 880. [Google Scholar] [CrossRef]
- Curcio, C.A.; Presley, J.B.; Malek, G.; Medeiros, N.E.; Avery, D.V.; Kruth, H.S. Esterified and unesterified cholesterol in drusen and basal deposits of eyes with age-related maculopathy. Exp. Eye Res. 2005, 81, 731–741. [Google Scholar] [CrossRef]
- Neale, B.M.; Fagerness, J.; Reynolds, R.; Sobrin, L.; Parker, M.; Raychaudhuri, S.; Tan, P.L.; Oh, E.C.; Merriam, J.E.; Souied, E.; et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc. Natl. Acad. Sci. USA 2010, 107, 7395–7400. [Google Scholar] [CrossRef] [Green Version]
- Vavvas, D.G.; Daniels, A.B.; Kapsala, Z.G.; Goldfarb, J.W.; Ganotakis, E.; Loewenstein, J.I.; Young, L.H.; Gragoudas, E.S.; Eliott, D.; Kim, I.K.; et al. Regression of Some High-risk Features of Age-related Macular Degeneration (AMD) in Patients Receiving Intensive Statin Treatment. EBioMedicine 2016, 5, 198–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Stambolian, D.; Edwards, A.O.; Branham, K.E.; Othman, M.; Jakobsdottir, J.; Tosakulwong, N.; Pericak-Vance, M.A.; Campochiaro, P.A.; Klein, M.L.; et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 7401–7406. [Google Scholar] [CrossRef] [Green Version]
- Golestaneh, N.; Chu, Y.; Xiao, Y.-Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2018, 8, e2537. [Google Scholar] [CrossRef] [Green Version]
- Paterno, J.J.; Koskela, A.; Hyttinen, J.M.T.; Vattulainen, E.; Synowiec, E.; Tuuminen, R.; Watala, C.; Blasiak, J.; Kaarniranta, K. Autophagy Genes for Wet Age-Related Macular Degeneration in a Finnish Case-Control Study. Genes 2020, 11, 1318. [Google Scholar] [CrossRef]
- Orozco, L.D.; Chen, H.H.; Cox, C.; Katschke, K.J., Jr.; Arceo, R.; Espiritu, C.; Caplazi, P.; Nghiem, S.S.; Chen, Y.J.; Modrusan, Z.; et al. Integration of eQTL and a Single-Cell Atlas in the Human Eye Identifies Causal Genes for Age-Related Macular Degeneration. Cell Rep. 2020, 30, 1246–1259.e1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Fu, Y.; Baird, P.N.; Guymer, R.H.; Das, T.; Iwata, T. Exploring the contribution of ARMS2 and HTRA1 genetic risk factors in age-related macular degeneration. Prog. Retin. Eye Res. 2022, 101159. [Google Scholar] [CrossRef]
- Klipfel, L.; Cordonnier, M.; Thiébault, L.; Clérin, E.; Blond, F.; Millet-Puel, G.; Mohand-Saïd, S.; Goureau, O.; Sahel, J.-A.; Nandrot, E.F.; et al. A Splice Variant in SLC16A8 Gene Leads to Lactate Transport Deficit in Human iPS Cell-Derived Retinal Pigment Epithelial Cells. Cells 2021, 10, 179. [Google Scholar] [CrossRef]
- Melo, E.; Oertle, P.; Trepp, C.; Meistermann, H.; Burgoyne, T.; Sborgi, L.; Cabrera, A.C.; Chen, C.Y.; Hoflack, J.C.; Kam-Thong, T.; et al. HtrA1 Mediated Intracellular Effects on Tubulin Using a Polarized RPE Disease Model. EBioMedicine 2018, 27, 258–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Hollander, A.I.; Mullins, R.F.; Orozco, L.D.; Voigt, A.P.; Chen, H.-H.; Strunz, T.; Grassmann, F.; Haines, J.L.; Kuiper, J.J.W.; Tumminia, S.J.; et al. Systems genomics in age-related macular degeneration. Exp. Eye Res. 2022, 225, 109248. [Google Scholar] [CrossRef]
- Yamamoto, H.; Yakushijin, K.; Kusuhara, S.; Escaño, M.F.; Nagai, A.; Negi, A. A novel RDH5 gene mutation in a patient with fundus albipunctatus presenting with macular atrophy and fading white dots. Am. J. Ophthalmol. 2003, 136, 572–574. [Google Scholar] [CrossRef]
- Kutty, R.K.; Samuel, W.; Boyce, K.; Cherukuri, A.; Duncan, T.; Jaworski, C.; Nagineni, C.N.; Redmond, T.M. Proinflammatory cytokines decrease the expression of genes critical for RPE function. Mol. Vis. 2016, 22, 1156. [Google Scholar]
- Behrendt, M. TRPM3 in the eye and in the nervous system—From new findings to novel mechanisms. Biol. Chem. 2022, 403, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Held, K.; Tóth, B.I. TRPM3 in Brain (Patho)Physiology. Front. Cell Dev. Biol. 2021, 9, 635659. [Google Scholar] [CrossRef]
- Shiels, A. TRPM3_miR-204: A complex locus for eye development and disease. Hum. Genom. 2020, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Miyagishima, K.J.; Dong, L.; Rising, A.; Nimmagadda, M.; Liang, G.; Sharma, R.; Dejene, R.; Wang, Y.; Abu-Asab, M.; et al. Regulation of phagolysosomal activity by miR-204 critically influences structure and function of retinal pigment epithelium/retina. Hum. Mol. Genet. 2019, 28, 3355–3368. [Google Scholar] [CrossRef] [Green Version]
- Adijanto, J.; Castorino, J.J.; Wang, Z.X.; Maminishkis, A.; Grunwald, G.B.; Philp, N.J. Microphthalmia-associated transcription factor (MITF) promotes differentiation of human retinal pigment epithelium (RPE) by regulating microRNAs-204/211 expression. J. Biol. Chem. 2012, 287, 20491–20503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naylor, J.; Li, J.; Milligan, C.J.; Zeng, F.; Sukumar, P.; Hou, B.; Sedo, A.; Yuldasheva, N.; Majeed, Y.; Beri, D.; et al. Pregnenolone sulphate- and cholesterol-regulated TRPM3 channels coupled to vascular smooth muscle secretion and contraction. Circ. Res. 2010, 106, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Bamps, D.; Vriens, J.; Hoon, J.d.; Voets, T. TRP Channel Cooperation for Nociception: Therapeutic Opportunities. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 655–677. [Google Scholar] [CrossRef]
- Su, S.; Yudin, Y.; Kim, N.; Tao, Y.-X.; Rohacs, T. TRPM3 Channels Play Roles in Heat Hypersensitivity and Spontaneous Pain after Nerve Injury. J. Neurosci. 2021, 41, 2457–2474. [Google Scholar] [CrossRef]
- Philp, N.J.; Yoon, H.; Grollman, E.F. Monocarboxylate transporter MCT1 is located in the apical membrane and MCT3 in the basal membrane of rat RPE. Am. J. Physiol. 1998, 274, R1824–R1828. [Google Scholar] [CrossRef]
- Fernandez-Godino, R.; Garland, D.L.; Pierce, E.A. Isolation, culture and characterization of primary mouse RPE cells. Nat. Protoc. 2016, 11, 1206–1218. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Frankish, A.; Diekhans, M.; Jungreis, I.; Lagarde, J.; Loveland, J.E.; Mudge, J.M.; Sisu, C.; Wright, J.C.; Armstrong, J.; Barnes, I.; et al. GENCODE 2021. Nucleic Acids Res. 2021, 49, D916–D923. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, S.; Pohl, M.O.; Zhou, Y.; Rodriguez-Frandsen, A.; Wang, G.; Stein, D.A.; Moulton, H.M.; DeJesus, P.; Che, J.; Mulder, L.C.; et al. Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 2015, 18, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020, 10, 20560. [Google Scholar] [CrossRef]
- Silvestri, A.; Fiorilli, V.; Miozzi, L.; Accotto, G.P.; Turina, M.; Lanfranco, L. In silico analysis of fungal small RNA accumulation reveals putative plant mRNA targets in the symbiosis between an arbuscular mycorrhizal fungus and its host plant. BMC Genom. 2019, 20, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | GO | Description | LogP | Enrichment | Z-Score |
|---|---|---|---|---|---|
| GO Molecular Functions | GO:0050839 | cell adhesion molecule binding (↑) | −22 | 3.2 | 12 |
| GO:0045296 | cadherin binding (↑) | −17 | 3.6 | 11 | |
| GO:0019900 | kinase binding (↑) | −14 | 2.4 | 8.9 | |
| GO:0005509 | calcium ion binding (↑) | −13 | 2.4 | 8.7 | |
| GO:0019901 | protein kinase binding (↑) | −12 | 2.4 | 8.2 | |
| GO:0004672 | protein kinase activity (↑) | −11 | 2.5 | 8 | |
| GO:0016773 | phosphotransferase activity, alcohol group as acceptor (↑) | −11 | 2.3 | 7.7 | |
| GO:0005543 | phospholipid binding (↑) | −9.9 | 2.5 | 7.5 | |
| GO:0016301 | kinase activity (↑) | −9.8 | 2.2 | 7.3 | |
| GO:0004175 | endopeptidase activity (↑) | −8.9 | 2.5 | 7.1 | |
| GO Cellular Components | GO:0005911 | cell-cell junction (↑) | −22 | 3.4 | 12 |
| GO:0009925 | basal plasma membrane (↑) (↓) | −18 | 4.2 | 12 | |
| GO:0045178 | basal part of cell (↑) (↓) | −17 | 3.9 | 11 | |
| GO:0045177 | apical part of cell (↑) (↓) | −16 | 3.2 | 10 | |
| GO:0016324 | apical plasma membrane (↑) (↓) | −16 | 3.4 | 10 | |
| GO:0016323 | basolateral plasma membrane (↑) (↓) | −15 | 4 | 10 | |
| GO:0030055 | cell-substrate junction | −14 | 3 | 9.2 | |
| GO:0005925 | focal adhesion | −14 | 3 | 9.2 | |
| GO:0005764 | Lysosome (↑) (↓) | −13 | 2.3 | 8.4 | |
| GO:0000323 | lytic vacuole (↑) (↓) | −13 | 2.3 | 8.4 | |
| GO Biological Processes | GO:0030855 | epithelial cell differentiation (↑) | −18 | 2.9 | 11 |
| GO:0006954 | inflammatory response (↑) | −17 | 3 | 11 | |
| GO:0071345 | cellular response to cytokine stimulus (↑) | −17 | 2.6 | 10 | |
| GO:0098609 | cell-cell adhesion (↑) | −17 | 2.9 | 10 | |
| GO:0009611 | response to wounding (↑) | −16 | 3.1 | 10 | |
| GO:0001775 | cell activation (↑) | −16 | 2.6 | 9.7 | |
| GO:0008544 | epidermis development (↑) | −16 | 3.6 | 10 | |
| GO:0048729 | tissue morphogenesis | −16 | 2.8 | 9.8 | |
| GO:0008610 | lipid biosynthetic process (↓) | −15 | 2.7 | 9.6 | |
| GO:0030162 | regulation of proteolysis (↑) | −15 | 2.5 | 9.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhingra, A.; Tobias, J.W.; Philp, N.J.; Boesze-Battaglia, K. Transcriptomic Changes Predict Metabolic Alterations in LC3 Associated Phagocytosis in Aged Mice. Int. J. Mol. Sci. 2023, 24, 6716. https://doi.org/10.3390/ijms24076716
Dhingra A, Tobias JW, Philp NJ, Boesze-Battaglia K. Transcriptomic Changes Predict Metabolic Alterations in LC3 Associated Phagocytosis in Aged Mice. International Journal of Molecular Sciences. 2023; 24(7):6716. https://doi.org/10.3390/ijms24076716
Chicago/Turabian StyleDhingra, Anuradha, John W. Tobias, Nancy J. Philp, and Kathleen Boesze-Battaglia. 2023. "Transcriptomic Changes Predict Metabolic Alterations in LC3 Associated Phagocytosis in Aged Mice" International Journal of Molecular Sciences 24, no. 7: 6716. https://doi.org/10.3390/ijms24076716