
The Impact of BDNF, NTRK2, NGFR, CREB1, GSK3B, AKT, MAPK1, MTOR, PTEN, ARC, and SYN1 Genetic Polymorphisms in Antidepressant Treatment Response Phenotypes

 ,
, 
 ,
,  , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Relapse Phenotype
2.2. Treatment Resistant Depression Phenotype
2.3. Gene Functional Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA Extraction and SNP Analysis
4.3. Functional Enrichment Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castrén, E.; Võikar, V.; Rantamäki, T. Role of neurotrophic factors in depression. Curr. Opin. Pharmacol. 2007, 7, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Castrén, E. Neurotrophic effects of antidepressant drugs. Curr. Opin. Pharmacol. 2004, 4, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Castren, E.; Rantamäki, T. The Role of BDNF and Its Receptors in Depression and Antidepressant Drug Action: Reactivation of Developmental Plasticity. Dev. Neurobiol. 2010, 70, 289–297. [Google Scholar] [CrossRef]
- Duman, R.S.; Heninger, G.R.; Nestler, E.J. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 1997, 54, 597–606. [Google Scholar] [CrossRef]
- Monteggia, L.M.; Barrot, M.; Powell, C.M.; Berton, O.; Galanis, V.; Gemelli, T.; Meuth, S.; Nagy, A.; Greene, R.W.; Nestler, E.J. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc. Natl. Acad. Sci. USA 2004, 101, 10827–10832. [Google Scholar] [CrossRef]
- Ribeiro, L.; Busnello, J.V.; Cantor, R.M.; Whelan, F.; Whittaker, P.; Deloukas, P.; Wong, M.-L.; Licinio, J. The brain-derived neurotrophic factor rs6265 (Val66Met) polymorphism and depression in Mexican-Americans. Neuroreport 2007, 18, 1291–1293. [Google Scholar] [CrossRef]
- Hosang, G.M.; Shiles, C.; Tansey, K.E.; McGuffin, P.; Uher, R. Interaction between stress and the BDNF Val66Met polymorphism in depression: A systematic review and meta-analysis. BMC Med. 2014, 12, 7. [Google Scholar] [CrossRef]
- Ozan, E.; Okur, H.; Eker, C.; Eker, O.D.; Gonul, A.S.; Akarsu, N. The effect of depression, BDNF gene val66met polymorphism and gender on serum BDNF levels. Brain Res. Bull. 2010, 81, 61–65. [Google Scholar] [CrossRef]
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269. [Google Scholar] [CrossRef]
- Frodl, T.; Moller, H.-J.; Meisenzahl, E. Neuroimaging genetics: New perspectives in research on major depression? Acta Psychiatry Scand. 2008, 118, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Wang, Z.; Chen, J.; Fan, J.; Guan, Y.; Zhang, C.; Yuan, C.; Hong, W.; Wang, Y.; et al. The role of BDNF, NTRK2 gene and their interaction in development of treatment-resistant depression: Data from multicenter, prospective, longitudinal clinic practice. J. Psychiatry Res. 2013, 47, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wong, M.-L.; Licinio, J. Sequence variations of ABCB1, SLC6A2, SLC6A3, SLC6A4, CREB1, CRHR1 and NTRK2: Association with major depression and antidepressant response in Mexican-Americans. Mol. Psychiatry 2009, 14, 1105–1118. [Google Scholar] [CrossRef] [PubMed]
- Zagrebelsky, M.; Holz, A.; Dechant, G.; Barde, Y.-A.; Bonhoeffer, T.; Korte, M. The p75 neurotrophin receptor negatively modulates dendrite complexity and spine density in hippocampal neurons. J. Neurosci. 2005, 25, 9989–9999. [Google Scholar] [CrossRef]
- Pilar-Cuéllar, F.; Vidal, R.; Díaz, A.; Castro, E.; dos Anjos, S.; Pascual-Brazo, J.; Linge, R.; Vargas, V.; Blanco, H.; Martínez-Villayandre, B.; et al. Neural plasticity and proliferation in the generation of antidepressant effects: Hippocampal implication. Neural Plast. 2013, 2013, 537265. [Google Scholar] [CrossRef]
- Cowen, D.S. Serotonin and neuronal growth factors—A convergence of signaling pathways. J. Neurochem. 2007, 101, 1161–1171. [Google Scholar] [CrossRef]
- Peltier, J.; O’Neill, A.; Schaffer, D.V. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev. Neurobiol. 2007, 67, 1348–1361. [Google Scholar] [CrossRef]
- Mostany, R.; Valdizán, E.M.; Pazos, A. A role for nuclear β-catenin in SNRI antidepressant-induced hippocampal cell proliferation. Neuropharmacology 2008, 55, 18–26. [Google Scholar] [CrossRef]
- Zubenko, G.S.; Maher, B.; Hughes, H.B.; Zubenko, W.N.; Stiffler, J.S.; Kaplan, B.B.; Marazita, M.L. Genome-wide linkage survey for genetic loci that influence the development of depressive disorders in families with recurrent, early-onset, major depression. Am. J. Med. Genet. B Neuropsychiatr Genet. 2003, 123B, 1–18. [Google Scholar] [CrossRef]
- Calati, R.; Crisafulli, C.; Balestri, M.; Serretti, A.; Spina, E.; Calabrò, M.; Sidoti, A.; Albani, D.; Massat, I.; Höfer, P.; et al. Evaluation of the role of MAPK1 and CREB1 polymorphisms on treatment resistance, response and remission in mood disorder patients. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 44, 271–278. [Google Scholar] [CrossRef]
- Liu, S.; Sun, N.; Xu, Y.; Yang, C.; Ren, Y.; Liu, Z.; Cao, X.; Sun, Y.; Xu, Q.; Zhang, K.; et al. Possible association of the GSK3β gene with the anxiety symptoms of major depressive disorder and P300 waveform. Genet. Test. Mol. Biomarkers 2012, 16, 1382–1389. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, W.; Roh, M.-S.; Friedman, A.B.; Rosborough, K.; Jope, R.S. In vivo regulation of glycogen synthase kinase-3β (GSK3β) by serotonergic activity in mouse brain. Neuropsychopharmacology 2004, 29, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-J.; Liou, Y.-J.; Hong, C.-J.; Yu, Y.W.-Y.; Chen, T.-J. Glycogen synthase kinase-3β gene is associated with antidepressant treatment response in Chinese major depressive disorder. Pharmacogenom. J. 2008, 8, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lee, B.; Liu, R.-J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.-Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; Park, C.S.; Abbassi, N.R.; Tang, S.-J. Roles of glutamate receptors and the mammalian target of rapamycin (mTOR) signaling pathway in activity-dependent dendritic protein synthesis in hippocampal neurons. J. Biol. Chem. 2006, 281, 18802–18815. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.F. Quest for biomarkers of treatment-resistant depression: Shifting the paradigm toward risk. Front. Psychiatry 2013, 4, 57. [Google Scholar] [CrossRef]
- Serafini, G.; Howland, R.H.; Rovedi, F.; Girardi, P.; Amore, M. The role of ketamine in treatment-resistant depression: A systematic review. Curr. Neuropharmacol. 2014, 12, 444–461. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, N.; Yang, C.; Li, X.-M.; Zhou, Z.-Q.; Yang, J.-J. Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur. Psychiatry 2014, 29, 419–423. [Google Scholar] [CrossRef]
- Hashimoto, K. Role of the mTOR signaling pathway in the rapid antidepressant action of ketamine. Expert Rev. Neurother. 2011, 11, 33–36. [Google Scholar] [CrossRef]
- Welberg, L. Psychiatric disorders: Ketamine modifies mood through mTOR. Nat. Rev. Neurosci. 2010, 11, 666. [Google Scholar] [CrossRef]
- Lemos, J.C.; Roth, C.A.; Chavkin, C. Signaling events initiated by kappa opioid receptor activation: Quantification and immunocolocalization using phospho-selective KOR, p38 MAPK, and K(IR) 3.1 antibodies. Methods Mol. Biol 2011, 717, 197–219. [Google Scholar] [CrossRef] [PubMed]
- Gourley, S.L.; Wu, F.J.; Kiraly, D.D.; PLoSki, J.E.; Kedves, A.T.; Duman, R.S.; Taylor, J.R. Regionally specific regulation of ERK MAP kinase in a model of antidepressant-sensitive chronic depression. Biol. Psychiatry 2008, 63, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Mercier, G.; Lennon, A.M.; Renouf, B.; Dessouroux, A.; Ramaugé, M.; Courtin, F.; Pierre, M. MAP kinase activation by fluoxetine and its relation to gene expression in cultured rat astrocytes. J. Mol. Neurosci. 2004, 24, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, Y.; Rizavi, H.S.; Zhang, H.; Roberts, R.C.; Conley, R.R.; Pandey, G.N. Aberrant extracellular signal-regulated kinase (ERK)1/2 signalling in suicide brain: Role of ERK kinase 1 (MEK1). Int. J. Neuropsychopharmacol. 2009, 12, 1337–1354. [Google Scholar] [CrossRef]
- Kwok, J.B.J.; Hallupp, M.; Loy, C.T.; Chan, D.K.Y.; Woo, J.; Mellick, G.D.; Buchanan, D.D.; Silburn, P.A.; Halliday, G.M.; Schofield, P.R. GSK3B polymorphisms alter transcription and splicing in Parkinson’s disease. Ann. Neurol. 2005, 58, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Inkster, B.; Nichols, T.E.; Saemann, P.G.; Auer, D.P.; Holsboer, F.; Muglia, P.; Matthews, P.M. ASsociation of gsk3β polymorphisms with brain structural changes in major depressive disorder. Arch. Gen. Psychiatry 2009, 66, 721–728. [Google Scholar] [CrossRef]
- Liu, Z.; Guo, H.; Cao, X.; Cheng, C.; Yang, C.; Xu, C.; Zhang, A.; Sun, N.; Li, X.; Zhang, K. A combined study of GSK3β polymorphisms and brain network topological metrics in major depressive disorder. Psychiatry Res. 2014, 223, 210–217. [Google Scholar] [CrossRef]
- Yu, G.I.; Kim, S.K.; Park, H.J.; Kim, J.W.; Chung, J.-H.; Shin, D.H. The C allele of synonymous SNP (rs1142636, Asn170Asn) in SYN1 is a risk factor for the susceptibility of Korean female schizophrenia. Synapse 2012, 66, 979–983. [Google Scholar] [CrossRef]
- Garcia, C.C.; Blair, H.J.; Seager, M.; Coulthard, A.; Tennant, S.; Buddles, M.; Curtis, A.; Goodship, J.A. Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J. Med. Genet. 2004, 41, 183–186. [Google Scholar] [CrossRef]
- Ferreira, A.; Chin, L.-S.; Li, L.; Lanier, L.M.; Kosik, K.S.; Greengard, P. Distinct roles of synapsin I and synapsin II during neuronal development. Mol. Med. 1998, 4, 22–28. [Google Scholar] [CrossRef]
- Rosahl, T.W.; Geppert, M.; Spillane, D.; Herz, J.; Hammer, R.E.; Malenka, R.C.; Sudhof, T.C. Short-term synaptic plasticity is altered in mice lacking synapsin I. Cell 1993, 75, 661–670. [Google Scholar] [CrossRef]
- Le-Niculescu, H.; Levey, D.F.; Ayalew, M.; Palmer, L.; Gavrin, L.M.; Jain, N.; Winiger, E.; Bhosrekar, S.; Shankar, G.; Radel, M.; et al. Discovery and validation of blood biomarkers for suicidality. Mol. Psychiatry 2013, 18, 1249–1264. [Google Scholar] [CrossRef]
- Mullins, N.; Hodgson, K.; Tansey, K.E.; Perroud, N.; Maier, W.; Mors, O.; Rietschel, M.; Hauser, J.; Henigsberg, N.; Souery, D.; et al. Investigation of blood mRNA biomarkers for suicidality in an independent sample. Transl. Psychiatry 2014, 4, e474. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; McGue, M. The interacting effect of the BDNF Val66Met polymorphism and stressful life events on adolescent depression is not an artifact of gene-environment correlation: Evidence from a longitudinal twin study. J. Child Psychol. Psychiatry 2013, 54, 1066–1073. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; McGue, M. Interacting effect of BDNF Val66Met polymorphism and stressful life events on adolescent depression. Genes Brain Behav. 2012, 11, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.; Weinberger, D.R.; Mattay, V.S.; Cheng, X.; Toga, A.W.; Thompson, P.M.; Powell-Smith, G.; Cohen-Woods, S.; Simmons, A.; McGuffin, P.; et al. No effect of 5HTTLPR or BDNF Val66Met polymorphism on hippocampal morphology in major depression. Genes Brain Behav. 2011, 10, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.-Y.; Tan, L.-J.; Shen, H.; Liu, Y.-J.; Liu, Y.-Z.; Li, J.; Zhu, X.-Z.; Chen, X.-D.; Tian, Q.; Zhao, M.; et al. SNP rs6265 regulates protein phosphorylation and osteoblast differentiation and influences BMD in humans. J. Bone Miner. Res. 2013, 28, 2498–2507. [Google Scholar] [CrossRef] [PubMed]
- Elfving, B.; Buttenschøn, H.N.; Foldager, L.; Poulsen, P.H.; Andersen, J.H.; Grynderup, M.B.; Hansen, A.M.; Kolstad, H.A.; Kaerlev, L.; Mikkelsen, S.; et al. Depression, the Val66Met polymorphism, age, and gender influence the serum BDNF level. J. Psychiatry Res. 2012, 46, 1118–1125. [Google Scholar] [CrossRef]
- Chi, M.H.; Chang, H.H.; Lee, S.Y.; Lee, I.H.; Gean, P.W.; Yang, Y.K.; Lu, R.B.; Chen, P.S. Brain derived neurotrophic factor gene polymorphism (Val66Met) and short-term antidepressant response in major depressive disorder. J. Affect. Disord. 2010, 126, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Domschke, K.; Lawford, B.; Laje, G.; Berger, K.; Young, R.; Morris, P.; Deckert, J.; Arolt, V.; McMahon, F.J.; Baune, B.T. Brain-derived neurotrophic factor ( BDNF) gene: No major impact on antidepressant treatment response. Int. J. Neuropsychopharmacol. 2010, 13, 93–101. [Google Scholar] [CrossRef]
- Choi, M.-J.; Kang, R.-H.; Lim, S.-W.; Oh, K.-S.; Lee, M.-S. Brain-derived neurotrophic factor gene polymorphism (Val66Met) and citalopram response in major depressive disorder. Brain Res. 2006, 1118, 176–182. [Google Scholar] [CrossRef]
- Alexopoulos, G.S.; Glatt, C.E.; Hoptman, M.J.; Kanellopoulos, D.; Murphy, C.F.; Kelly, R.E., Jr.; Morimoto, S.S.; Lim, K.O.; Gunning, F.M. BDNF val66met polymorphism, white matter abnormalities and remission of geriatric depression. J. Affect. Disord. 2010, 125, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.D.; McQuoid, U.R.; Ashley-Koch, A.; MacFall, J.R.; Bridgers, J.; Krishnan, R.R.; Steffens, D.C. BDNF Val66Met genotype and 6-month remission rates in late-life depression. Pharmacogenom. J. 2011, 11, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-J.; Cheng, C.-Y.; Yu, Y.W.-Y.; Chen, T.-J.; Hong, C.-J. Association study of a brain-derived neurotrophic-factor genetic polymorphism and major depressive disorders, symptomatology, and antidepressant response. Am. J. Med. Genet. B Neuropsy. Genet. 2003, 123B, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Higuchi, H.; Kamata, M.; Takahashi, H.; Inoue, K.; Suzuki, T.; Itoh, K.; Ozaki, N. The G196A polymorphism of the brain-derived neurotrophic factor gene and the antidepressant effect of milnacipran and fluvoxamine. J. Psychopharmacol. 2007, 21, 650–656. [Google Scholar] [CrossRef]
- Zou, Y.-F.; Wang, Y.; Liu, P.; Feng, X.-L.; Wang, B.-Y.; Zang, T.-H.; Yu, X.; Wei, J.; Liu, Z.-C.; Liu, Y.; et al. Association of BDNF Val66Met polymorphism with both baseline HRQOL scores and improvement in HRQOL scores in Chinese major depressive patients treated with fluoxetine. Um. Psychopharmacol. Clin. Exp. 2010, 25, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, A.; Gennarelli, M.; Uher, R.; Breen, G.; Farmer, A.; Aitchison, K.J.; Craig, I.W.; Anacker, C.; Zunsztain, P.A.; McGuffin, P.; et al. Candidate genes expression profile associated with antidepressants response in the GENDEP study: Differentiating between baseline ‘predictors’ and longitudinal ‘targets’. Neuropsychopharmacology 2013, 38, 377–385. [Google Scholar] [CrossRef]
- Kang, R.; Chang, H.; Wong, M.-L.; Choi, M.; Park, J.; Lee, H.; Jung, I.; Joe, S.; Kim, L.; Kim, S.; et al. Brain-derived neurotrophic factor gene polymorphisms and mirtazapine responses in Koreans with major depression. J. Psychopharmacol. 2010, 24, 1755–1763. [Google Scholar] [CrossRef]
- Musil, R.; Zill, P.; Seemuller, F.; Bondy, B.; Obermeier, M.; Spellmann, I.; Bender, W.; Adli, M.; Heuser, I.; Zeiler, J.; et al. No influence of brain-derived neurotrophic factor (BDNF) polymorphisms on treatment response in a naturalistic sample of patients with major depression. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 405–412. [Google Scholar] [CrossRef]
- Correia-Melo, F.S.; Leal, G.C.; Vieira, F.; Jesus-Nunes, A.P.; Mello, R.P.; Magnavita, G.; Caliman-Fontes, A.T.; Echegaray, M.V.F.; Bandeira, I.D.; Silva, S.S.; et al. Efficacy and safety of adjunctive therapy using esketamine or racemic ketamine for adult treatment-resistant depression: A randomized, double-blind, non-inferiority study. J. Affect. Disord. 2020, 264, 527–534. [Google Scholar] [CrossRef]
- Mattson, M.P. Glutamate and Neurotrophic Factors in Neuronal Plasticity and Disease. Ann. N. Y. Acad. Sci. 2008, 1144, 97–112. [Google Scholar] [CrossRef]
- Salahudeen, M.S.; Wright, C.M.; Peterson, G.M. Esketamine: New hope for the treatment of treatment-resistant depression? A narrative review. Ther. Adv. Drug Saf. 2020, 11, 2042098620937899. [Google Scholar] [CrossRef]
- Santos, M.; Carvalho, S.; Lima, L.; Mota-Pereira, J.; Pimentel, P.; Maia, D.; Correia, D.; Gomes, S.; Cruz, A.; Medeiros, R. FAS -670A>G genetic polymorphism Is associated with Treatment Resistant Depression. J. Affect. Disord. 2015, 185, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Carvalho, S.; Lima, L.; Mota-Pereira, J.; Pimentel, P.; Maia, D.; Correia, D.; Gomes, S.; Cruz, A.; Medeiros, R. The role of IL18-607C>A and IL18-137G>C promoter polymorphisms in antidepressant treatment phenotypes: A preliminary report. Neurosci. Lett. 2016, 622, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.H.; Rush, A.J.; Crismon, M.L.; Kashner, T.M.; Toprac, M.G.; Carmody, T.J.; Key, T.; Biggs, M.M.; Shores-Wilson, K.; Witte, B.; et al. Clinical results for patients with major depressive disorder in the Texas Medication Algorithm Project. Arch. Gen. Psychiatry 2004, 61, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.; Ziaugra, L.; Tabbaa, D. SNP Genotyping Using the Sequenom MassARRAY iPLEX Platform. In Current Protocols in Human Genetics; John Wiley & Sons, Inc.: New York, NY, USA, 2001. [Google Scholar] [CrossRef]
- Lachmann, A.; Schilder, B.M.; Wojciechowicz, M.L.; Torre, D.; Kuleshov, M.V.; Keenan, A.B.; Ma’ayan, A. Geneshot: Search engine for ranking genes from arbitrary text queries. Nucleic Acids Res. 2019, 47, W571–W577. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Relapsed | Resistant (TRD) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | Yes | OR | CI 95% | p-Value | No | Yes | OR | CI 95% | p-Value | ||||||
| N | % | N | % | N | % | N | % | ||||||||
| BDNF rs6265 | CC | 23 | 51.1 | 9 | 60.0 | 1.0 | Referent | - | 32 | 53.3 | 5 | 29.4 | 1.0 | Referent | - |
| CT | 17 | 37.8 | 5 | 33.3 | 0.752 | [0.213–2.650] | 0.657 | 22 | 36.7 | 11 | 64.7 | 3.200 | [0.975–10.501] | 0.049 | |
| TT | 5 | 11.1 | 1 | 6.7 | 0.511 | [0.052–5.003] | 1.000 * | 6 | 10.0 | 1 | 5.9 | 1.067 | [0.105–10.825] | 1.000 * | |
| T carrier | 22 | 48.9 | 6 | 40.0 | 0.697 | [0.213–2.284] | 0.550 | 28 | 46.7 | 12 | 70.6 | 2.743 | [0.860–8.750] | 0.081 | |
| PTEN rs12569998 | TT | 41 | 91.1 | 14 | 87.5 | 1.0 | Referent | - | 55 | 90.2 | 13 | 68.4 | 1.0 | Referent | - |
| TG | 4 | 8.9 | 2 | 12.5 | 1.464 | [0.241–8.881] | 0.648 * | 6 | 9.8 | 6 | 31.6 | 4.231 | [1.173–15.261] | 0.020 | |
| GG | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| G carrier | 4 | 8.9 | 2 | 12.5 | 1.464 | [0.241–8.881] | 0.648 * | 6 | 9.8 | 6 | 31.6 | 4.231 | [1.173–15.261] | 0.020 | |
| SYN1 rs1142636 | AA | 28 | 62.2 | 8 | 50.0 | 1.0 | Referent | - | 35 | 59.0 | 6 | 31.6 | 1.0 | Referent | - |
| AG | 12 | 26.7 | 6 | 37.5 | 1.750 | [0.498–6.145] | 0.380 | 18 | 29.5 | 6 | 31.6 | 2.000 | [0.564–7.087] | 0.278 | |
| GG | 5 | 11.1 | 2 | 12.5 | 1.400 | [0.227–8.626] | 0.656 * | 7 | 11.5 | 7 | 36.8 | 6.000 | [1.543–23.333] | 0.006 | |
| G carrier | 17 | 37.8 | 8 | 50.0 | 1.647 | [0.521–5.204] | 0.393 | 25 | 41.0 | 13 | 68.4 | 3.120 | [1.045–9.314] | 0.037 | |
| GSK3B rs6438552 | AA | 20 | 44.4 | 2 | 12.5 | 1.0 | Referent | - | 22 | 36.1 | 9 | 47.4 | 1.0 | Referent | - |
| AG | 16 | 35.6 | 8 | 50.0 | 5.000 | [0.929–26.913] | 0.074 * | 24 | 39.3 | 8 | 42.1 | 0.815 | [0.267–2.483] | 0.718 | |
| GG | 9 | 20.0 | 6 | 37.5 | 6.667 | [1.121–39.660] | 0.042 * | 15 | 24.6 | 2 | 10.5 | 0.326 | [0.062–1.726] | 0.284 * | |
| G carrier | 27 | 55.6 | 14 | 87.5 | 5.600 | [1.137–27.571] | 0.033 * | 39 | 63.9 | 10 | 52.6 | 0.627 | [0.221–1.775] | 0.377 | |
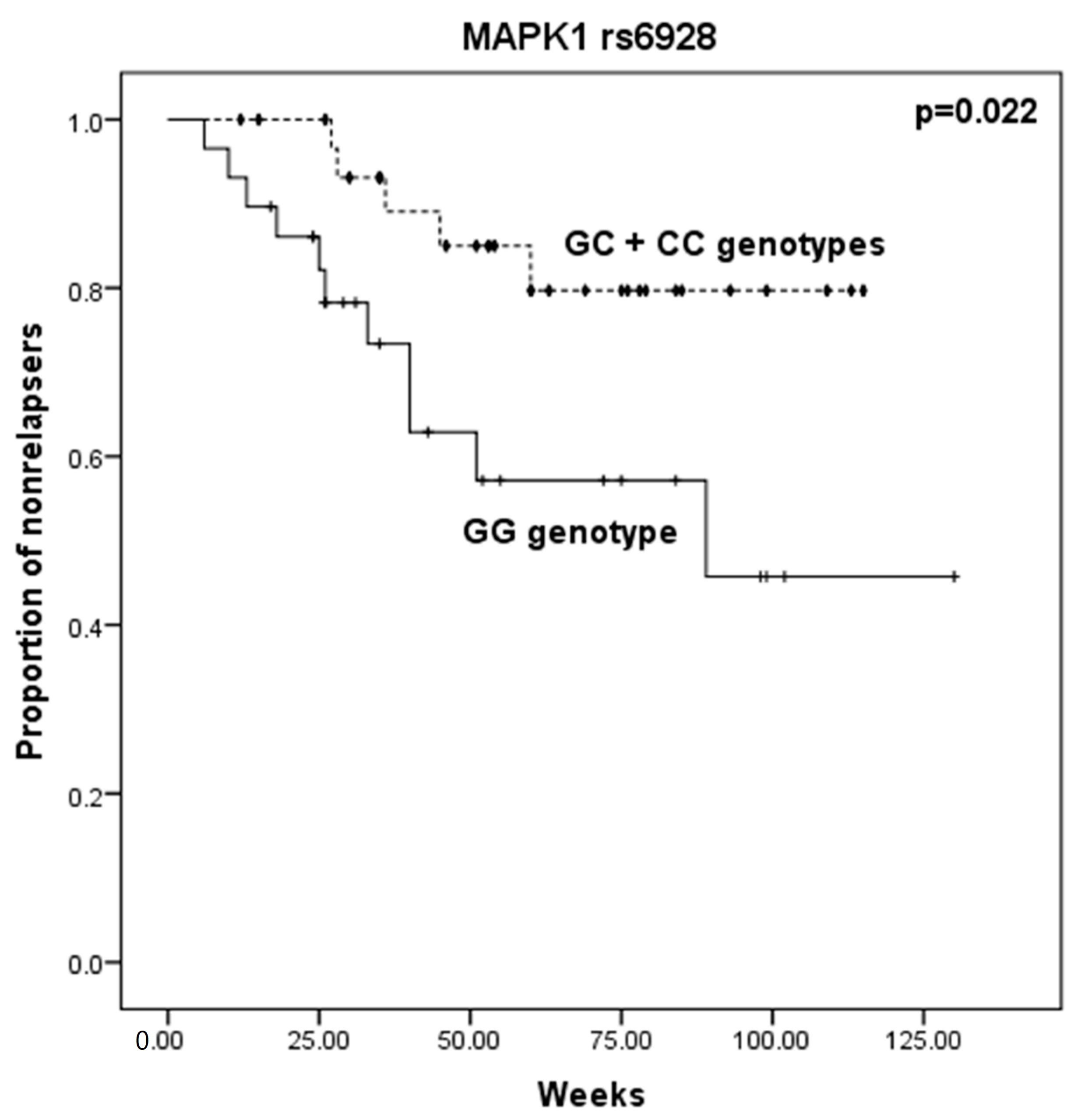
| MAPK1 rs6928 | GG | 18 | 40.0 | 11 | 68.8 | 1.0 | Referent | - | 29 | 47.5 | 5 | 26.3 | 1.0 | Referent | - |
| GC | 19 | 42.2 | 3 | 18.8 | 0.258 | [0.062–1.080] | 0.066 * | 22 | 36.1 | 11 | 57.9 | 2.900 | [0.879–9.567] | 0.074 | |
| CC | 8 | 17.8 | 2 | 12.4 | 0.409 | [0.073–2.288] | 0.445 * | 10 | 16.4 | 3 | 15.8 | 1.740 | [0.351–8.633] | 0.666 * | |
| C carrier | 27 | 60.0 | 5 | 31.2 | 0.303 | [0.090–1.020] | 0.048 * | 32 | 53.5 | 14 | 73.7 | 2.538 | [0.813–7.919] | 0.102 | |
| Index | Name | p-Value | Adjusted p-Value | Odds Ratio | Combined Score |
|---|---|---|---|---|---|
| GO Biological Process | |||||
| 1 | chemical synaptic transmission (GO:0007268) | 5.186 × 10−29 | 1.086 × 10−25 | 22.00 | 1432.77 |
| 2 | anterograde trans-synaptic signaling (GO:0098916) | 3.609 × 10−25 | 3.778 × 10−22 | 22.43 | 1262.23 |
| 3 | modulation of chemical synaptic transmission (GO:0050804) | 3.001 × 10−20 | 2.095 × 10−17 | 33.15 | 1489.97 |
| 4 | glutamate receptor signaling pathway (GO:0007215) | 7.002 × 10−16 | 3.666 × 10−13 | 67.14 | 2343.00 |
| 5 | cellular response to cytokine stimulus (GO:0071345) | 2.163 × 10−15 | 9.060 × 10−13 | 9.56 | 322.96 |
| 6 | regulation of NMDA receptor activity (GO:2000310) | 8.579 × 10−15 | 2.994 × 10−12 | 71.58 | 2318.44 |
| 7 | positive regulation of gene expression (GO:0010628) | 2.058 × 10−14 | 5.422 × 10−12 | 9.08 | 286.14 |
| 8 | positive regulation of multicellular organismal process (GO:0051240) | 2.071 × 10−14 | 5.422 × 10−12 | 11.01 | 346.99 |
| 9 | calcium ion transmembrane import into cytosol (GO:0097553) | 1.848 × 10−13 | 4.224 × 10−11 | 49.19 | 1442.14 |
| 10 | negative regulation of apoptotic process (GO:0043066) | 2.117 × 10−13 | 4.224 × 10−11 | 8.55 | 249.46 |
| GO Molecular Function | |||||
| 1 | receptor ligand activity (GO:0048018) | 7.579 × 10−19 | 2.062 × 10−16 | 14.83 | 618.62 |
| 2 | glutamate receptor activity (GO:0008066) | 2.072 × 10−15 | 2.818 × 10−13 | 140.70 | 4757.03 |
| 3 | hormone activity (GO:0005179) | 5.605 × 10−15 | 5.081 × 10−13 | 32.19 | 1056.44 |
| 4 | ionotropic glutamate receptor activity (GO:0004970) | 8.568 × 10−14 | 5.826 × 10−12 | 137.88 | 4148.62 |
| 5 | glutamate-gated calcium ion channel activity (GO:0022849) | 1.350 × 10−11 | 7.344 × 10−10 | 99,320.00 | 2,485,812.34 |
| 6 | ligand-gated channel activity (GO:0022834) | 1.916 × 10−11 | 8.684 × 10−10 | 56.37 | 1391.10 |
| 7 | ligand-gated ion channel activity (GO:0015276) | 2.567 × 10−11 | 9.976 × 10−10 | 53.92 | 1314.77 |
| 8 | cytokine activity (GO:0005125) | 1.854 × 10−10 | 6.302 × 10−9 | 13.02 | 291.67 |
| 9 | NMDA glutamate receptor activity (GO:0004972) | 7.438 × 10−10 | 2.248 × 10−8 | 252.68 | 5311.25 |
| 10 | G protein-coupled glutamate receptor activity (GO:0001640) | 1.664 × 10−9 | 4.116 × 10−8 | 189.50 | 3830.60 |
| GO Cellular Component | |||||
| 1 | neuron projection (GO:0043005) | 3.351 × 10−24 | 4.290 × 10−22 | 12.87 | 695.42 |
| 2 | dendrite (GO:0030425) | 1.012 × 10−17 | 6.476 × 10−16 | 15.26 | 597.32 |
| 3 | ionotropic glutamate receptor complex (GO:0008328) | 5.840 × 10−13 | 2.170 × 10−11 | 61.13 | 1722.04 |
| 4 | axon (GO:0030424) | 6.781 × 10−13 | 2.170 × 10−11 | 13.95 | 391.00 |
| 5 | integral component of plasma membrane (GO:0005887) | 2.656 × 10−11 | 6.799 × 10−10 | 4.50 | 109.69 |
| 6 | postsynaptic density membrane (GO:0098839) | 4.142 × 10−11 | 8.836 × 10−10 | 82.86 | 1980.97 |
| 7 | postsynaptic specialization membrane (GO:0099634) | 6.178 × 10−11 | 1.130 × 10−9 | 76.94 | 1808.62 |
| 8 | NMDA selective glutamate receptor complex (GO:0017146) | 7.438 × 10−10 | 1.190 × 10−8 | 252.68 | 5311.25 |
| 9 | postsynaptic density (GO:0014069) | 2.637 × 10−9 | 3.750 × 10−8 | 13.68 | 270.15 |
| 10 | cation channel complex (GO:0034703) | 3.444 × 10−8 | 4.408 × 10−7 | 19.04 | 327.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, M.; Lima, L.; Carvalho, S.; Mota-Pereira, J.; Pimentel, P.; Maia, D.; Correia, D.; Barroso, M.F.; Gomes, S.; Cruz, A.; et al. The Impact of BDNF, NTRK2, NGFR, CREB1, GSK3B, AKT, MAPK1, MTOR, PTEN, ARC, and SYN1 Genetic Polymorphisms in Antidepressant Treatment Response Phenotypes. Int. J. Mol. Sci. 2023, 24, 6758. https://doi.org/10.3390/ijms24076758
Santos M, Lima L, Carvalho S, Mota-Pereira J, Pimentel P, Maia D, Correia D, Barroso MF, Gomes S, Cruz A, et al. The Impact of BDNF, NTRK2, NGFR, CREB1, GSK3B, AKT, MAPK1, MTOR, PTEN, ARC, and SYN1 Genetic Polymorphisms in Antidepressant Treatment Response Phenotypes. International Journal of Molecular Sciences. 2023; 24(7):6758. https://doi.org/10.3390/ijms24076758
Chicago/Turabian StyleSantos, Marlene, Luis Lima, Serafim Carvalho, Jorge Mota-Pereira, Paulo Pimentel, Dulce Maia, Diana Correia, M. Fátima Barroso, Sofia Gomes, Agostinho Cruz, and et al. 2023. "The Impact of BDNF, NTRK2, NGFR, CREB1, GSK3B, AKT, MAPK1, MTOR, PTEN, ARC, and SYN1 Genetic Polymorphisms in Antidepressant Treatment Response Phenotypes" International Journal of Molecular Sciences 24, no. 7: 6758. https://doi.org/10.3390/ijms24076758
APA StyleSantos, M., Lima, L., Carvalho, S., Mota-Pereira, J., Pimentel, P., Maia, D., Correia, D., Barroso, M. F., Gomes, S., Cruz, A., & Medeiros, R. (2023). The Impact of BDNF, NTRK2, NGFR, CREB1, GSK3B, AKT, MAPK1, MTOR, PTEN, ARC, and SYN1 Genetic Polymorphisms in Antidepressant Treatment Response Phenotypes. International Journal of Molecular Sciences, 24(7), 6758. https://doi.org/10.3390/ijms24076758