Abstract
Gastroesophageal reflux disease (GERD) significantly impacts patient quality of life and is a major risk factor for the development of Barrett’s esophagus (BE) and esophageal adenocarcinoma (EAC). Proton pump inhibitors (PPIs) are the standard-of-care for GERD and are among the most prescribed drugs in the world, but do not protect against nonacid components of reflux such as pepsin, or prevent reflux-associated carcinogenesis. We recently identified an HIV protease inhibitor amprenavir that inhibits pepsin and demonstrated the antireflux therapeutic potential of its prodrug fosamprenavir in a mouse model of laryngopharyngeal reflux. In this study, we assessed the capacity of amprenavir to protect against esophageal epithelial barrier disruption in vitro and related molecular events, E-cadherin cleavage, and matrix metalloproteinase induction, which are associated with GERD severity and esophageal cancer. Herein, weakly acidified pepsin (though not acid alone) caused cell dissociation accompanied by regulated intramembrane proteolysis of E-cadherin. Soluble E-cadherin responsive matrix metalloproteinases (MMPs) were transcriptionally upregulated 24 h post-treatment. Amprenavir, at serum concentrations achievable given the manufacturer-recommended dose of fosamprenavir, protected against pepsin-induced cell dissociation, E-cadherin cleavage, and MMP induction. These results support a potential therapeutic role for amprenavir in GERD recalcitrant to PPI therapy and for preventing GERD-associated neoplastic changes.
1. Introduction
Gastroesophageal reflux disease (GERD) affects up to 25% of adults in high-income countries and has significant adverse effects on patient quality of life [1,2,3]. Typical symptoms of GERD include heartburn and regurgitation, and concomitant extraesophageal symptoms such as cough and hoarseness are common [1,4,5]. Patients with GERD are at increased risk for developing complications such as esophageal strictures and Barrett’s esophagus (BE; columnar metaplasia of the esophageal epithelium) and have a 43.5-fold greater risk of developing esophageal adenocarcinoma (EAC) [1,6,7,8].
Proton pump inhibitors (PPIs) are the first-line therapy against GERD and are among the most prescribed drugs in the world. Studies conducted in developed nations have reported PPI use by up to 30% of all adults and greater than 50% of those over 65 with inappropriate prescriptions, and over-the-counter availability contributing to excessive use [9,10,11,12,13,14,15]. The global economic burden of PPIs exceeds over $25 billion annually [16,17]. Despite the widespread use of PPIs, the global incidence of EAC has rapidly risen in recent decades and at current rates will reach 141,300 new cases in 2040 [18,19,20,21]. A recent meta-analysis to evaluate the chemopreventive benefit of PPIs found no significant reduction in EAC risk [22]. Instead, high-quality observational trials have demonstrated that long-term high adherence to PPIs may increase EAC risk [23,24]. The association of PPI use with EAC may be due to symptom masking or the carcinogenic potential of the drugs themselves as supported by the association of their use with increased incidence of digestive tract cancers generally, and pancreatic, gastric, and liver cancers specifically [25]. Physicians and patients have grown increasingly concerned regarding the health risks of long-term PPI therapy prompting the investigation of alternatives [26,27].
PPIs reduce the secretion of acid by parietal cells in the stomach, thereby reducing the acidity of the refluxate. PPIs do not reduce the number of reflux events [28,29] and do not address damage by nonacid reflux components such as pepsin and bile salts [30,31]. Instead, studies indicate that PPIs may increase the concentration and toxicity of nonacid constituents of refluxate [30,32]. In the context of weakly to nonacidic refluxate such as that of patients taking PPIs or in extraesophageal reflux, the gastric enzyme pepsin is active up to pH6.5 and stable up to pH8 and can be endocytosed, leading to inflammatory and carcinogenic changes in the aerodigestive tract mucosa, including in the esophagus [8,33,34,35,36].
Our group recently identified the HIV protease inhibitor, amprenavir, as a potential pepsin-targeting therapeutic for laryngopharyngeal reflux (LPR). Amprenavir inhibited pepsin at low micromolar concentrations and its prodrug fosamprenavir prevented histologic changes in an LPR mouse model [37]. In parallel work, we found that pepsin was required for esophageal and laryngeal epithelial barrier disruption at pH4 and that amprenavir preserved laryngeal epithelial integrity and prevented damage to a critical cell adhesion molecule elicited by pepsin at pH4 [38,39]. Barrier disruption is a key mechanism of GERD pathogenesis, contributing to symptom origination, sensitivity to reflux insult, immune cell infiltration, and prolonged recovery [40,41,42,43,44]. Adhesion molecules such as E-cadherin confer epithelial integrity and regulate important cellular processes such as cell proliferation, migration, and epithelial mesenchymal transition. E-cadherin damage or misexpression is associated with epithelial barrier dysfunction during GERD, progression of BE to EAC, and EAC prognosis [45,46,47,48]. This study aimed to investigate the therapeutic potential of amprenavir for GERD by investigating its protection against pepsin-induced epithelial barrier disruption and related cancer-associated molecular changes in an esophageal cell-culture model.
2. Results
2.1. Amprenavir Rescues Pepsin-Mediated Cell Dissociation
Near-confluent hTERT-immortalized Barrett’s esophageal cells (BAR-T) cells were mock-treated or treated with pH4 acid only, 1 mg/mL pepsin in pH4 acid, or pepsin acid plus amprenavir at 1 or 10 µM for one hour (Figure 1, Supplemental Table S1). Acidified pepsin caused a near-complete cell dissociation (p < 0.0001, control versus pepsin pH4), whereas treatment with acid alone did not result in cell dissociation (p = 0.80, control versus pH4). Treatment with 1 µM amprenavir partially rescued pepsin-mediated cell dissociation to a mean of 34.9% confluence (p = 0.0008, pepsin pH4 versus +1 µM APR). The full rescue was achieved by 10 µM amprenavir treatment (p < 0.0001, pepsin pH4 versus +10 µM APR).
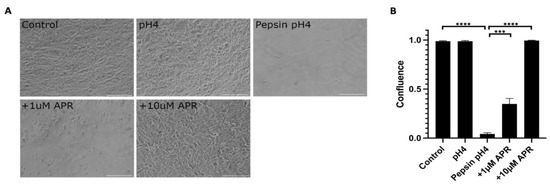
Figure 1.
Amprenavir (10 µM) rescued cell dissociation caused by 1 mg/mL pepsin at pH4. The partial rescue was achieved by 1 µM APR. Acid alone did not cause cell dissociation. (A) Representative images of experiments performed in triplicate. Scale bars equal 100 µm. (B) Confluence as calculated using the FIJI plug-in PHANTAST (see Methods for details). Where significant, the following comparisons are shown: control vs. pH4, control vs. pepsin pH4, pepsin pH4 vs. +1 µM APR, and pepsin pH4 vs. +10 µM APR. *** = p < 0.001, **** = p < 0.0001.
2.2. Amprenavir Partially Rescues Pepsin-Mediated E-Cadherin Cleavage
We next utilized a Western blot to investigate the ability of amprenavir to rescue E-cadherin regulated intramembrane proteolysis (RIP). Treatment of BAR-T cells with acidified pepsin, though not acid alone, resulted in the depletion of full-length E-cadherin and the production of C-terminal intracellular fragments of molecular weights consistent with E-cadherin RIP (Figure 2). Treatment with 10 µM amprenavir significantly rescued E-cadherin RIP, whereas 1 µM amprenavir resulted in a slight rescue of full-length E-cadherin cleavage though it had no statistically significant effect on C-terminal intracellular fragments (Figure 2, Supplemental Table S2).
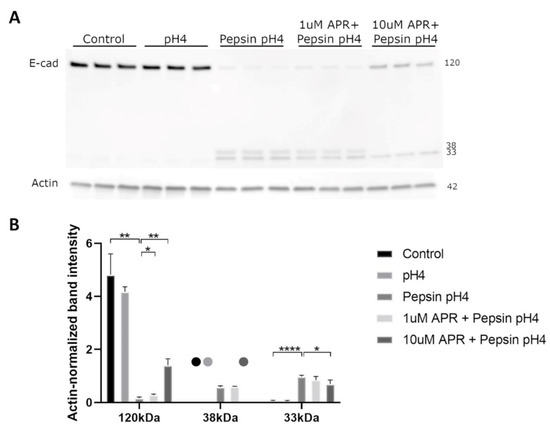
Figure 2.
Western blot (A) analyzed by densitometry (B) showed that acidified pepsin elicited E-cadherin cleavage via regulated intramembrane proteolysis (RIP), producing C-terminal intracellular fragments of 33 and 38 kDa. Acid alone did not cause E-cadherin RIP. 10 µM amprenavir partially rescued E-cadherin RIP, with increased full-length E-cadherin and decreased 33 and 38 kDa fragments compared to acidified pepsin, whereas 1 µM amprenavir resulted in a slight increase in full-length E-cadherin only. Dots represent samples for which no bands were detected. Where significant and where bands were detectable for analysis, the following comparisons are shown: control vs. pH4, control vs. pepsin pH4, pepsin pH4 vs. +1 µM APR, and pepsin pH4 vs. +10 µM APR. * = p < 0.05, ** = p < 0.01, **** = p < 0.0001.
2.3. Amprenavir Protected against Pepsin-Mediated Upregulation of Matrix Metalloproteinases
We next investigated the ability of amprenavir to rescue pepsin-mediated upregulation of matrix metalloproteinases (MMPs) that have been shown to be upregulated by E-cadherin fragments following RIP and/or that are known to be associated with GERD pathology. BAR-T cells were treated for 15 min, washed, and then cultured for 24 h in normal growth media prior to RNA harvest. Significant upregulation compared to an untreated control was observed following treatment with acidified pepsin for MMP1 (p = 0.0004), MMP2 (p = 0.033), MMP7 (p = 0.041), MMP9 (p = 0.0052), and MMP14 (p = 0.0089), whereas only MMP9 (p = 0.0048) and MMP14 (p = 0.0056) showed statistically significant upregulation by acid alone (Figure 3, Supplemental Table S3) Treatment with 10 µM amprenavir led to statistically significant reductions in pH4 pepsin-mediated induction of MMP1 (p = 0.0006), MMP7 (p = 0.043), MMP9 (p = 0.0066), and MMP14 (p = 0.034), as well as a reduction in MMP2 expression that did not reach statistical significance (p = 0.077). Treatment with 1 µM amprenavir did not significantly reduce the peptic induction of any MMP investigated.
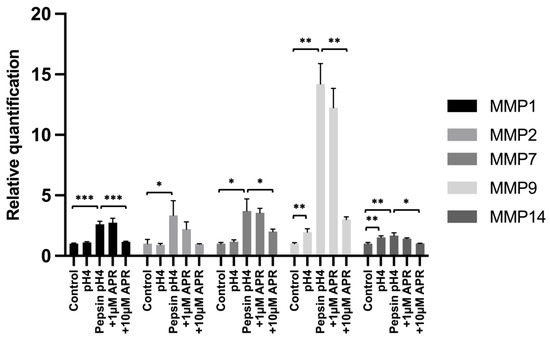
Figure 3.
Expression of MMPs following mock treatment, treatment with acidified pepsin ±1 or 10 µM amprenavir, or acid alone. Where significant, the following comparisons are shown: control vs. pH4, control vs. pepsin pH4, pepsin pH4 vs. +1 µM APR, and pepsin pH4 vs. +10 µM APR. * = p < 0.05, ** = p < 0.01, *** = p < 0.001.
3. Discussion
The development of the PPI class of drugs represented a breakthrough in the treatment of GERD. While PPIs have provided symptom relief for millions of patients, important therapeutic challenges remain in the management of GERD. PPIs reduce the acidity of refluxate but do not target nonacid components such as pepsin and bile salts and in fact may increase their toxicity and concentration [30,31]. Ten Kate et al. found that 60 mg daily omeprazole increased pepsin in refluxate from 0.8 ± 0.1 to 2.7 ± 0.4 mg/mL (unmedicated versus omeprazole cohorts) by virtue of reducing gastric acid secretion, and thereby, volume [30]. The concentration of pepsin by PPIs may have a pathologic consequence, as indicated by experiments in an airway epithelial cell culture model which found greater inflammatory cytokine secretion, barrier disruption, and neutrophil transepithelial migration in cells exposed to gastric juice from acid-suppressed patients relative to those not taking PPIs [44]. The dose-dependent effects of pepsin have similarly been illustrated in animal and culture models of the esophagus [8,49,50].
Pepsin is found in all refluxate, and a substantial body of literature supports its contribution to reflux-attributed pathology irrespective of acid. While much of this work focused on anatomical locations proximal to the esophagus [34,51,52,53,54,55,56,57,58,59] evidence suggests that pepsin, independent of acid, elicits damage to the esophagus via similar molecular mechanisms. Pepsin harbors its greatest enzymatic activity at pH2, retains 70% of its activity up to pH6.5, and is stable up to pH8 [36]. In a nonacidic environment, pepsin is endocytosed by aerodigestive tract mucosa, including that of the larynx and esophagus. This results in cell and molecular biological changes associated with inflammation, stress, toxicity, and carcinogenesis, presumably triggered by enzymatic reactivation of pepsin in endocytic vesicles of low pH where pepsin is stored for up to 36 h following endocytosis or pepsin interaction with a receptor on the cell surface [33,34,53,59,60,61]. In alignment with an emerging model of GERD pathogenesis, mediated by inflammatory insult rather than caustic acid-induced injury [62], interleukin-8 (IL-8), a neutrophil chemoattractant and nexus of cell signaling networks regulating proliferation, apoptosis, epithelial-mesenchymal transition, and migration, appears to play a key role in the peptic injury of the larynx and esophagus [8,44,53,63,64]. IL-8 is elevated in GERD patients, highest in patients with BE and EAC, and reduced in BE patients following antireflux surgery [8,64]. In esophageal cells, chronic stimulation with nonacid pepsin elicited secretion of IL-8 and induced transition from a noncancer (KRT10 high, KRT8 low) to BE-associated cytokeratin profile (KRT10 low and KRT8 high) [8,65]. Weeks of acid pulses alone (pH 3.5 for 1 h, 3 days per week) did not incur such changes [66] in accord with poor chemoprevention by acid suppression therapy. Further, brief (5 min) exposure to pepsin with 4 or 20-h exposure to pepsin at pH7 doubled the expression of PTGS2 (gene encoding COX-2), which is associated with chronic inflammation and cancer, and is a prognostic indicator of EAC [67,68]. COX-2 inhibitors have been shown to abrogate cell proliferation in EAC cells in vitro [69], reduce tumor incidence in animal models of EAC [70], and slow BE cell proliferation in a clinical trial [71]. Interestingly, an epidemiologic study demonstrated that long-term PPI therapy has no advantageous effect on COX-2 expression [72], suggesting that nonacid constituents of refluxate may be responsible for continued dysregulation of COX-2 in patients taking PPIs. Stable ectopic coexpression of pepsinogen and the gastric proton pump in a BE cell line also led to upregulated expression of transcripts associated with BE, EAC, and carcinogenesis including TGFB1 and ERBB2 [35]; pathway analysis of the global transcriptomic changes identified cancer as the top associated disease, regulation of epithelial–mesenchymal transition by growth factors as a top associated canonical pathway and cell cycle regulation, cell growth/proliferation/death/survival, DNA replication and repair, and lipid metabolism as top associated networks [35]. In contrast to the hyperproliferative effects of pepsin, acid (pH4) alone has been shown to exert an antiproliferative effect in esophageal cells [73,74].
The refluxate of untreated GERD patients is commonly acidic (63% pH < 4) with which >80% of symptom episodes are associated [75]. That of acid-suppressed GERD patients is commonly weakly to nonacidic (80% episodes pH ≥ 4) giving rise to 72% of symptom episodes [76]. In the context of acidic or weakly acidic reflux, pepsin is enzymatically active and is required for some forms of mucosal injury. For example, Tobey et al. showed that 1 mg/mL pepsin at pH1.7 caused histological damage to esophageal mucosa and irreversible loss of epithelial barrier integrity (electrical resistance) in rabbit esophagi ex vivo, whereas histological damage was absent and barrier disruption milder and reversible, provided an acid pH1.7 alone [49]. Nagahama et al. demonstrated that treatment with pepstatin, an inhibitor of pepsin, prevented esophageal lesions in a surgical rat model of reflux without altering the acidity of reflux [77]. Goldberg et al. demonstrated that acid pH > 1.3 alone caused minimal esophagitis in an in vivo cat model and that pepsin dramatically exacerbated esophagitis and histological epithelial damage at pH 1.6–2.0 in a dose-dependent manner [50]. Similarly, we recently demonstrated that weakly acidified pepsin, though not acid (pH4) alone, caused barrier dysfunction and cell detachment in a human esophageal cell culture model and that a pepsin inhibitor, sodium alginate, could prevent such damage [39].
Barrier disruption by pepsin is thought to be caused by damage to the protein constituents of apical junction complexes which facilitate cell–cell junctions, maintain epithelial integrity, and regulate cell proliferation and migration. Accordingly, damage to intercellular junctions was shown to precede pepsin-induced erosive lesions in the rabbit esophageal epithelium by Tobey et al. [49], and cleavage of the major protein constituent of adherens junctions, E-cadherin, is observed in GERD and LPR biopsies [45,78,79,80]. Jovov et al. demonstrated that the deletion of E-cadherin in the adult mouse esophagus was sufficient to cause barrier dysfunction, as indicated by macromolecular flux [45]. In agreement with these findings, we recently demonstrated that E-cadherin was cleaved by weakly acidic pepsin (though not acid pH4 alone) in human esophageal and laryngeal cells in vitro and that E-cadherin cleavage was rescued by pepsin inhibitors (sodium alginate in esophageal cells and amprenavir in laryngeal cells) [38,81]. The E-cadherin cleavage fragments produced by human esophageal and laryngeal epithelial cells in response to pepsin were consistent with those observed in GERD and LPR biopsies and indicative of RIP. RIP is a highly regulated and evolutionarily conserved process by which a protease (“sheddase”) cleaves an intramembrane protein near the cell surface, thereby initiating subsequent cleavage near the cytoplasmic surface by intramembrane cleavage proteases and release of cleavage fragments which harbor biological activity [82]. In the case of E-cadherin RIP, the extracellularly released fragment promotes cancer-associated changes including increased signaling through growth factor receptors (epithelial growth factor receptor and insulin-dependent growth factor receptor one), inhibition of hippo signaling and related apoptosis, immune evasion, cell migration/invasion, and transcriptional upregulation of MMP-2, 9, and 14; the intracellular domain has been implicated in Wnt/β-catenin signaling, which in turn regulates cell proliferation and migration as well as the transcription of specific MMPs [83,84,85]. MMPs are a family of enzymes that weaken epithelial integrity by degrading extracellular matrix components. Their upregulation by E-cadherin RIP fragments is especially notable given that several MMPs are associated with GERD, its severity, and EAC, and have known roles in carcinogenesis [86,87,88,89].
Herein, the treatment of esophageal cells with acidified pepsin, though not acid alone (pH4), caused near complete E-cadherin RIP within 30 min and cell dissociation in one hour. Prior work has indicated that E-cadherin RIP fragments elicit transcription of MMP-2, 9, and 14 24 h poststimulation in lung cells [83]. Herein, MMP-2, 9, and 14 were elevated at 24 h postexposure to pepsin acid. This suggests that E-cadherin RIP fragments produced by esophageal cells in response to pepsin-acid stimulation are biologically active and exert carcinogenic changes consistent with their function in other cell types. Importantly, the pepsin inhibitor, amprenavir, attenuated or rescued these effects and provided concentrations found in the serum of patients taking the manufacturer-recommended dose of fosamprenavir for HIV [90]. These data are in alignment with prior work demonstrating that the pepsin inhibitor, pepstatin, prevents esophageal lesions in a surgical rat model of GERD [77] and that the pepsin inhibitor, sodium alginate, prevents esophageal and laryngeal epithelial barrier disruption by pepsin pH4 [39]. Pepsin-acid-induced E-cadherin RIP and protection by amprenavir appear to be conserved across cell types. In a recently submitted work, we found that amprenavir abrogates epithelial cell disruption, E-cadherin cleavage, and MMP dysregulation by pepsin pH4 in laryngeal cells, supporting the potential utility of amprenavir for both GERD and LPR [38]. Given that amprenavir protects epithelial barrier integrity, which is, in turn, a facet of GERD symptom origination, the data herein suggest that amprenavir may provide relief for the 20–40% of GERD patients with symptoms recalcitrant to PPI therapy [91,92,93]. Further, the capacity of amprenavir to rescue a pepsin-induced proinvasive cell phenotype characterized by E-cadherin RIP and MMP induction supports its chemopreventive potential. This is particularly intriguing in light of the failure of PPIs to demonstrate chemopreventive benefits for patients with GERD as well as burgeoning evidence that pepsin promotes carcinogenic changes in the esophagus [23,24,30,31,35,53,57,61]. Future in vitro work is warranted to address amprenavir protection against additional pepsin-induced molecular changes harboring a strong correlation with GERD severity and/or EAC, such as IL-8 and PTGS2/COX-2, and cancer-related cellular processes known to be impacted by pepsin such as cell-cycle regulation. Corroboration of the chemopreventive benefits of amprenavir is warranted in in vivo models of BE and EAC.
In summary, our results demonstrate that amprenavir, at serum concentrations achievable using the manufacturer-recommended dose of fosamprenavir, protects against esophageal epithelial barrier disruption and contributes to molecular mechanisms in a cell culture model of weakly acid reflux. Further, these data suggest that amprenavir may provide chemopreventive benefits. This work has important implications for the treatment of PPI-recalcitrant GERD, which affects a large number of patients, and for the prevention of EAC which is increasing at an alarming rate and is poorly addressed by current first-line therapy for GERD. This work supports the potential benefit of pepsin-targeting therapeutics as adjunctive therapies for GERD and an upcoming randomized clinical trial planned to assess the efficacy of amprenavir for the prevention of signs and symptoms of GERD.
4. Materials and Methods
4.1. Cell Culture and Treatment
hTERT-immortalized Barrett’s esophageal cells BAR-T [94] (the kind gift of Rhonda F. Souza) were cultured as previously described [39] to 90% confluence for examining protein expression and 50% for gene expression.
The IC50 of amprenavir for porcine pepsin (3.56 uM) was determined in a prior study [37]. The dose for study herein was selected based upon the serum concentration observed in patients taking the manufacturer-recommended dose for the treatment of HIV [90].
In triplicate, unless noted, cultures were treated in HBSS or HBSS pH4 ±1 mg/mL porcine pepsin (Sigma-Aldrich, St Louis, MO, USA) ±1–10 uM amprenavir (APR; Sigma Aldrich) at 37 °C/5% CO2 for 30 min (Western blot). To measure cell dissociation, cells were treated for 75 min and washed in HBSS. For MMP qPCR, cells were treated for 15 min, washed twice in HBSS, and incubated in normal growth media at 37 °C/5% CO2 for 24 h prior to harvest.
4.2. Cell Dissociation
Images were obtained on an inverted microscope (Nikon, Tokyo, Japan and Metamorph Inc., Nashville, TN, USA). The percent cell-free area of a single image from each well (n = 3) was quantified using the PHANTAST plug-in for FIJI [95,96].
4.3. Western Blot
Treatment solutions were collected and 2 uM NaOH and protease inhibitor cocktail (ThermoFisher Scientific, Waltham, MA, USA) added. Cells were harvested in cold RIPA lysis buffer (1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 50 mM Tris-Cl, pH7.4) containing protease inhibitor. Proteins were separated (4–20% TGX, Bio Rad Laboratories, Hercules, CA, USA) and transferred to polyvinylidene difluoride. Membranes were blocked (5% milk, 0.1% Tween-20, PBS) and probed with: E-cadherin C-terminal (4A2C7; ThermoFisher), actin (CP01, Sigma-Aldrich), and HRP-conjugated secondary (Agilent Technologies, Santa Clara, CA, USA). Densitometry was performed using Image J (version 1.52a, National Institutes of Health, Bethesda, MD, USA).
4.4. Real Time qPCR
RNA was extracted via TRIZOL (ThermoFisher Scientific) and the purity and concentration were assessed by UV spectroscopy. RNA was reverse transcribed using Superscript IV VILO (ThermoFisher Scientific). The qPCR was performed in quadruplicate reactions using Taqman gene expression assays in a Viia7 instrument (ThermoFisher Scientific) per the manufacturer’s instructions. Threshold cycle (Ct) values < 36 were used for analysis, and gene expression was normalized to the housekeeping gene HPRT1.
4.5. Statistical Analysis
Student’s t-test was used to compare groups. For qPCR, RQs were calculated via the delta-delta method, and p < 0.05 was considered significant.
Supplementary Materials
The supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24076765/s1.
Author Contributions
Conceptualization, T.L.S. and N.J.; methodology, S.B.-S., T.L.S. and N.J.; validation, S.B.-S., T.L.S. and N.J.; formal analysis, K.Y.; investigation, S.B.-S. and T.L.S.; resources, T.L.S. and N.J.; data curation, S.B.-S. and T.L.S.; writing—original draft preparation, S.B.-S. and T.L.S.; writing—review and editing, S.B.-S., T.L.S., K.Y. and N.J.; visualization, S.B.-S. and T.L.S.; supervision, N.J.; project administration, N.J.; funding acquisition, N.J. All authors have read and agreed to the published version of the manuscript.
Funding
Work was funded by the Medical College of Wisconsin Department of Otolaryngology and Communication Sciences and kind donations of Jamie Koufman and Eric Becker and his family.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data presented in this study are available herein and in the Supplementary Materials.
Acknowledgments
We thank Rhonda F. Souza for the generous gift of BAR-T cells used in this study.
Conflicts of Interest
N.J. is a co-founder, Chief Scientific Officer, and an investor in N-Zyme Biomedical. N.J. is an inventor on: International Patent Application PCT/US2021/027758, Aerosolized formulations of HIV protease inhibitors for the treatment of airway reflux, filed 16 April 2021, and U.S. Patent Application 63/392,929, Sustained-release oral fosamprenavir for the treatment of reflux, filed 28 July 2022. T.S. is an investor in N-Zyme Biomedical. Other authors have no financial relationships or conflicts of interest to disclose.
References
- Maret-Ouda, J.; Markar, S.R.; Lagergren, J. Gastroesophageal Reflux Disease: A Review. JAMA 2020, 324, 2536. [Google Scholar] [CrossRef] [PubMed]
- Eusebi, L.H.; Ratnakumaran, R.; Yuan, Y.; Solaymani-Dodaran, M.; Bazzoli, F.; Ford, A.C. Global Prevalence of, and Risk Factors for, Gastro-Oesophageal Reflux Symptoms: A Meta-Analysis. Gut 2018, 67, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.E.; Rubenstein, J.H. Presentation and Epidemiology of Gastroesophageal Reflux Disease. Gastroenterology 2018, 154, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.M.; Patel, D.; Vaezi, M.F. Laryngopharyngeal Reflux and Atypical Gastroesophageal Reflux Disease. Gastrointest. Endosc. Clin. N. Am. 2020, 30, 361–376. [Google Scholar] [CrossRef]
- Lechien, J.R.; Saussez, S.; Karkos, P.D. Laryngopharyngeal Reflux Disease: Clinical Presentation, Diagnosis and Therapeutic Challenges in 2018. Curr. Opin. Otolaryngol. Head Neck Surg. 2018, 26, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J.; Bergström, R.; Lindgren, A.; Nyrén, O. Symptomatic Gastroesophageal Reflux as a Risk Factor for Esophageal Adenocarcinoma. N. Engl. J. Med. 1999, 340, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Ruigómez, A.; García Rodríguez, L.A.; Wallander, M.-A.; Johansson, S.; Eklund, S. Esophageal Stricture: Incidence, Treatment Patterns, and Recurrence Rate. Am. J. Gastroenterol. 2006, 101, 2685–2692. [Google Scholar] [CrossRef]
- Samuels, T.L.; Altman, K.W.; Gould, J.C.; Kindel, T.; Bosler, M.; MacKinnon, A.; Hagen, C.E.; Johnston, N. Esophageal Pepsin and Proton Pump Synthesis in Barrett’s Esophagus and Esophageal Adenocarcinoma. Laryngoscope 2019, 129, 2687–2695. [Google Scholar] [CrossRef]
- Rückert-Eheberg, I.-M.; Nolde, M.; Ahn, N.; Tauscher, M.; Gerlach, R.; Güntner, F.; Günter, A.; Meisinger, C.; Linseisen, J.; Amann, U.; et al. Who Gets Prescriptions for Proton Pump Inhibitors and Why? A Drug-Utilization Study with Claims Data in Bavaria, Germany, 2010-2018. Eur. J. Clin. Pharm. 2022, 78, 657–667. [Google Scholar] [CrossRef]
- Bustillos, H.; Leer, K.; Kitten, A.; Reveles, K.R. A Cross-Sectional Study of National Outpatient Gastric Acid Suppressant Prescribing in the United States between 2009 and 2015. PLoS ONE 2018, 13, e0208461. [Google Scholar] [CrossRef]
- Torres-Bondia, F.; de Batlle, J.; Galván, L.; Buti, M.; Barbé, F.; Piñol-Ripoll, G. Evolution of the Consumption Trend of Proton Pump Inhibitors in the Lleida Health Region between 2002 and 2015. BMC Public Health 2022, 22, 818. [Google Scholar] [CrossRef] [PubMed]
- Lassalle, M.; Le Tri, T.; Bardou, M.; Biour, M.; Kirchgesner, J.; Rouby, F.; Dumarcet, N.; Zureik, M.; Dray-Spira, R. Use of Proton Pump Inhibitors in Adults in France: A Nationwide Drug Utilization Study. Eur. J. Clin. Pharm. 2020, 76, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Othman, F.; Card, T.R.; Crooks, C.J. Proton Pump Inhibitor Prescribing Patterns in the UK: A Primary Care Database Study. Pharmacoepidemiol. Drug Saf. 2016, 25, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Muheim, L.; Signorell, A.; Markun, S.; Chmiel, C.; Neuner-Jehle, S.; Blozik, E.; Ursprung, P.; Rosemann, T.; Senn, O. Potentially Inappropriate Proton-Pump Inhibitor Prescription in the General Population: A Claims-Based Retrospective Time Trend Analysis. Ther. Adv. Gastroenterol. 2021, 14, 1756284821998928. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Katz, P.O.; Armstrong, D.; Cohen, H.; Delaney, B.C.; Howden, C.W.; Katelaris, P.; Tutuian, R.I.; Castell, D.O. The Safety of Appropriate Use of Over-the-Counter Proton Pump Inhibitors: An Evidence-Based Review and Delphi Consensus. Drugs 2017, 77, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Jaynes, M.; Kumar, A.B. The Risks of Long-Term Use of Proton Pump Inhibitors: A Critical Review. Ther. Adv. Drug Saf. 2019, 10, 2042098618809927. [Google Scholar] [CrossRef]
- Shaheen, N.J.; Hansen, R.A.; Morgan, D.R.; Gangarosa, L.M.; Ringel, Y.; Thiny, M.T.; Russo, M.W.; Sandler, R.S. The Burden of Gastrointestinal and Liver Diseases, 2006. Am. J. Gastroenterol. 2006, 101, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, M.H.; Arnold, M.; Brewster, D.H.; Going, J.J.; Mitchell, D.R.; Forman, D.; McColl, K.E.L. Worldwide Inverse Association between Gastric Cancer and Esophageal Adenocarcinoma Suggesting a Common Environmental Factor Exerting Opposing Effects. Am. J. Gastroenterol. 2016, 111, 228–239. [Google Scholar] [CrossRef] [PubMed]
- McColl, K.E.L. What Is Causing the Rising Incidence of Esophageal Adenocarcinoma in the West and Will It Also Happen in the East? J. Gastroenterol. 2019, 54, 669–673. [Google Scholar] [CrossRef]
- Blot, W.J.; Devesa, S.S.; Kneller, R.W.; Fraumeni, J.F. Rising Incidence of Adenocarcinoma of the Esophagus and Gastric Cardia. JAMA 1991, 265, 1287–1289. [Google Scholar] [CrossRef]
- Morgan, E.; Soerjomataram, I.; Rumgay, H.; Coleman, H.G.; Thrift, A.P.; Vignat, J.; Laversanne, M.; Ferlay, J.; Arnold, M. The Global Landscape of Esophageal Squamous Cell Carcinoma and Esophageal Adenocarcinoma Incidence and Mortality in 2020 and Projections to 2040: New Estimates From GLOBOCAN 2020. Gastroenterology 2022, 163, 649–658.e2. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Sun, T.-T.; Hong, J.; Fang, J.-Y.; Xiong, H.; Meltzer, S.J. Proton Pump Inhibitors Do Not Reduce the Risk of Esophageal Adenocarcinoma in Patients with Barrett’s Esophagus: A Systematic Review and Meta-Analysis. PLoS ONE 2017, 12, e0169691. [Google Scholar] [CrossRef] [PubMed]
- Hvid-Jensen, F.; Pedersen, L.; Funch-Jensen, P.; Drewes, A.M. Proton Pump Inhibitor Use May Not Prevent High-Grade Dysplasia and Oesophageal Adenocarcinoma in Barrett’s Oesophagus: A Nationwide Study of 9883 Patients. Aliment. Pharmacol. Ther. 2014, 39, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Masclee, G.M.C.; Coloma, P.M.; Spaander, M.C.W.; Kuipers, E.J.; Sturkenboom, M.C.J.M. NSAIDs, Statins, Low-Dose Aspirin and PPIs, and the Risk of Oesophageal Adenocarcinoma among Patients with Barrett’s Oesophagus: A Population-Based Case-Control Study. BMJ Open 2015, 5, e006640. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Cheng, Y.; Luo, D.; Wang, J.; Yang, J.; Jiang, L.; Zhuo, Z.; Guo, K.; Wu, H.; Leung, F.W.; et al. Comprehensive Analysis of Proton Pump Inhibitors and Risk of Digestive Tract Cancers. Eur. J. Cancer 2021, 156, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Yibirin, M.; De Oliveira, D.; Valera, R.; Plitt, A.E.; Lutgen, S. Adverse Effects Associated with Proton Pump Inhibitor Use. Cureus 2021, 13, e12759. [Google Scholar] [CrossRef] [PubMed]
- Scarpignato, C.; Hunt, R.H. Acid Suppressant Therapy: A Step Forward with Potassium-Competitive Acid Blockers. Curr. Treat. Options Gastroenterol. 2021, 19, 94–132. [Google Scholar] [CrossRef]
- Tamhankar, A.P.; Peters, J.H.; Portale, G.; Hsieh, C.-C.; Hagen, J.A.; Bremner, C.G.; DeMeester, T.R. Omeprazole Does Not Reduce Gastroesophageal Reflux: New Insights Using Multichannel Intraluminal Impedance Technology. J. Gastrointest. Surg. 2004, 8, 890–897, discussion 897–898. [Google Scholar] [CrossRef] [PubMed]
- Vela, M.F.; Camacho-Lobato, L.; Srinivasan, R.; Tutuian, R.; Katz, P.O.; Castell, D.O. Simultaneous Intraesophageal Impedance and PH Measurement of Acid and Nonacid Gastroesophageal Reflux: Effect of Omeprazole. Gastroenterology 2001, 120, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Ten Kate, R.W.; Tuynman, H.A.; Festen, H.P.; Pals, G.; Meuwissen, S.G. Effect of High Dose Omeprazole on Gastric Pepsin Secretion and Serum Pepsinogen Levels in Man. Eur. J. Clin. Pharmacol. 1988, 35, 173–176. [Google Scholar] [CrossRef]
- Alsalahi, O.; Dobrian, A.D. Proton Pump Inhibitors: The Culprit for Barrett’s Esophagus? Front. Oncol. 2014, 4, 373. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, K.D.; Strugala, V.; Dettmar, P.W. Reflux Revisited: Advancing the Role of Pepsin. Int. J. Otolaryngol. 2012, 2012, 646901. [Google Scholar] [CrossRef] [PubMed]
- Johnston, N.; Wells, C.W.; Blumin, J.H.; Toohill, R.J.; Merati, A.L. Receptor-Mediated Uptake of Pepsin by Laryngeal Epithelial Cells. Ann. Otol. Rhinol. Laryngol. 2007, 116, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Samuels, T.L.; Johnston, N. Pepsin in Gastroesophageal and Extraesophageal Reflux: Molecular Pathophysiology and Diagnostic Utility. Curr. Opin. Otolaryngol. Head Neck Surg. 2020, 28, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Stabenau, K.A.; Samuels, T.L.; Lam, T.K.; Mathison, A.J.; Wells, C.; Altman, K.W.; Battle, M.A.; Johnston, N. Pepsinogen/Proton Pump Co-Expression in Barrett’s Esophageal Cells Induces Cancer-Associated Changes. Laryngoscope 2023, 133, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Johnston, N.; Dettmar, P.W.; Bishwokarma, B.; Lively, M.O.; Koufman, J.A. Activity/Stability of Human Pepsin: Implications for Reflux Attributed Laryngeal Disease. Laryngoscope 2007, 117, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Johnston, N.; Samuels, T.L.; Goetz, C.J.; Arnold, L.A.; Smith, B.C.; Seabloom, D.; Wuertz, B.; Ondrey, F.; Wiedmann, T.S.; Vuksanovic, N.; et al. Oral and Inhaled Fosamprenavir Reverses Pepsin-Induced Damage in a Laryngopharyngeal Reflux Mouse Model. Laryngoscope 2023, 133 (Suppl. S1), S1–S11. [Google Scholar] [CrossRef] [PubMed]
- Samuels, T.L.; Blaine-Sauer, S.; Shafiee, S.; Lesnick, A.; Yan, K.; Joshi, A.; Johnston, N. Amprenavir Protects against Pepsin-Mediated Laryngeal E-Cadherin Cleavage and Matrix Metalloprotease Induction in Vitro. Laryngoscope 2023, Submitted. [Google Scholar]
- Samuels, T.L.; Yan, K.; Patel, N.; Plehhova, K.; Coyle, C.; Hurley, B.P.; Johnston, N. Alginates for Protection Against Pepsin-Acid Induced Aerodigestive Epithelial Barrier Disruption. Laryngoscope 2022, 132, 2327–2334. [Google Scholar] [CrossRef]
- Weijenborg, P.W.; Smout, A.J.P.M.; Verseijden, C.; van Veen, H.A.; Verheij, J.; de Jonge, W.J.; Bredenoord, A.J. Hypersensitivity to Acid Is Associated with Impaired Esophageal Mucosal Integrity in Patients with Gastroesophageal Reflux Disease with and without Esophagitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G323–G329. [Google Scholar] [CrossRef] [PubMed]
- Woodland, P.; Al-Zinaty, M.; Yazaki, E.; Sifrim, D. In Vivo Evaluation of Acid-Induced Changes in Oesophageal Mucosa Integrity and Sensitivity in Non-Erosive Reflux Disease. Gut 2013, 62, 1256–1261. [Google Scholar] [CrossRef]
- Woodland, P.; Lee, C.; Duraisamy, Y.; Farré, R.; Dettmar, P.; Sifrim, D. Assessment and Protection of Esophageal Mucosal Integrity in Patients with Heartburn without Esophagitis. Am. J. Gastroenterol. 2013, 108, 535–543. [Google Scholar] [CrossRef]
- Xie, C.; Sifrim, D.; Li, Y.; Chen, M.; Xiao, Y. Esophageal Baseline Impedance Reflects Mucosal Integrity and Predicts Symptomatic Outcome With Proton Pump Inhibitor Treatment. J. Neurogastroenterol. Motil. 2018, 24, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Hurley, B.P.; Jugo, R.H.; Snow, R.F.; Samuels, T.L.; Yonker, L.M.; Mou, H.; Johnston, N.; Rosen, R. Pepsin Triggers Neutrophil Migration Across Acid Damaged Lung Epithelium. Sci. Rep. 2019, 9, 13778. [Google Scholar] [CrossRef]
- Jovov, B.; Que, J.; Tobey, N.A.; Djukic, Z.; Hogan, B.L.M.; Orlando, R.C. Role of E-Cadherin in the Pathogenesis of Gastroesophageal Reflux Disease. Am. J. Gastroenterol. 2011, 106, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Tischoff, I.; Tannapfel, A. Barrett’s Esophagus: Can Biomarkers Predict Progression to Malignancy? Expert Rev. Gastroenterol. Hepatol. 2008, 2, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Feith, M.; Stein, H.J.; Mueller, J.; Siewert, J.R. Malignant Degeneration of Barrett’s Esophagus: The Role of the Ki-67 Proliferation Fraction, Expression of E-Cadherin and P53. Dis. Esophagus 2004, 17, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Charalabopoulos, A.; Golias, C. E-Cadherin Expression in Barrett’s Esophagus and Esophageal Carcinoma. Esophagus 2014, 11, 153–161. [Google Scholar] [CrossRef]
- Tobey, N.A.; Hosseini, S.S.; Caymaz-Bor, C.; Wyatt, H.R.; Orlando, G.S.; Orlando, R.C. The Role of Pepsin in Acid Injury to Esophageal Epithelium. Am. J. Gastroenterol. 2001, 96, 3062–3070. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, H.I.; Dodds, W.J.; Gee, S.; Montgomery, C.; Zboralske, F.F. Role of Acid and Pepsin in Acute Experimental Esophagitis. Gastroenterology 1969, 56, 223–230. [Google Scholar] [CrossRef]
- Crapko, M.; Kerschner, J.E.; Syring, M.; Johnston, N. Role of Extra-Esophageal Reflux in Chronic Otitis Media with Effusion. Laryngoscope 2007, 117, 1419–1423. [Google Scholar] [CrossRef] [PubMed]
- Gill, G.A.; Johnston, N.; Buda, A.; Pignatelli, M.; Pearson, J.; Dettmar, P.W.; Koufman, J. Laryngeal Epithelial Defenses against Laryngopharyngeal Reflux: Investigations of E-Cadherin, Carbonic Anhydrase Isoenzyme III, and Pepsin. Ann. Otol. Rhinol. Laryngol. 2005, 114, 913–921. [Google Scholar] [CrossRef]
- Johnston, N.; Yan, J.C.; Hoekzema, C.R.; Samuels, T.L.; Stoner, G.D.; Blumin, J.H.; Bock, J.M. Pepsin Promotes Proliferation of Laryngeal and Pharyngeal Epithelial Cells. Laryngoscope 2012, 122, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Luebke, K.; Samuels, T.L.; Chelius, T.H.; Sulman, C.G.; McCormick, M.E.; Kerschner, J.E.; Johnston, N.; Chun, R.H. Pepsin as a Biomarker for Laryngopharyngeal Reflux in Children with Laryngomalacia. Laryngoscope 2017, 127, 2413–2417. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, R.C.; Soundar, S.; Tonb, D.; Bolling, L.; Yoo, E.; Nadal, T.; Grindle, C.; Field, E.; He, Z. The Role of Gastric Pepsin in the Inflammatory Cascade of Pediatric Otitis Media. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 350–357. [Google Scholar] [CrossRef]
- Samuels, T.L.; Johnston, N. Pepsin as a Causal Agent of Inflammation during Nonacidic Reflux. Otolaryngol. Head Neck Surg. 2009, 141, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Sereg-Bahar, M.; Jerin, A.; Hocevar-Boltezar, I. Higher Levels of Total Pepsin and Bile Acids in the Saliva as a Possible Risk Factor for Early Laryngeal Cancer. Radiol. Oncol. 2015, 49, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Southwood, J.E.; Hoekzema, C.R.; Samuels, T.L.; Wells, C.; Poetker, D.M.; Johnston, N.; Loehrl, T.A. The Impact of Pepsin on Human Nasal Epithelial Cells In Vitro: A Potential Mechanism for Extraesophageal Reflux Induced Chronic Rhinosinusitis. Ann. Otol. Rhinol. Laryngol. 2015, 124, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, Z.; Ren, J.; Xu, Y.; Zhang, Y.; Lei, L.; Zheng, Y.; Huang, L.; He, Z. Effects of Pepsin A on Heat Shock Protein 70 Response in Laryngopharyngeal Reflux Patients with Chronic Rhinosinusitis. Acta Otolaryngol. 2017, 137, 1253–1259. [Google Scholar] [CrossRef]
- Johnston, N.; Wells, C.W.; Samuels, T.L.; Blumin, J.H. Rationale for Targeting Pepsin in the Treatment of Reflux Disease. Ann. Otol. Rhinol. Laryngol. 2010, 119, 547–558. [Google Scholar] [CrossRef]
- Kelly, E.A.; Samuels, T.L.; Johnston, N. Chronic Pepsin Exposure Promotes Anchorage-Independent Growth and Migration of a Hypopharyngeal Squamous Cell Line. Otolaryngol. Head Neck Surg. 2014, 150, 618–624. [Google Scholar] [CrossRef]
- Huo, X.; Souza, R.F. Acid Burn or Cytokine Sizzle in the Pathogenesis of Heartburn? J. Gastroenterol. Hepatol. 2013, 28, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.-J.; Wang, L.; Mo, T.-T.; Wang, J.; Wang, M.-G.; Li, X.-P. Pepsin Promotes IL-8 Signaling-Induced Epithelial-Mesenchymal Transition in Laryngeal Carcinoma. Cancer Cell Int. 2019, 19, 64. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.S.; DeMeester, S.R.; Vallbohmer, D.; Mori, R.; Kuramochi, H.; Hagen, J.A.; Lipham, J.; Danenberg, K.D.; Danenberg, P.V.; Chandrasoma, P.; et al. Reduction of Interleukin 8 Gene Expression in Reflux Esophagitis and Barrett’s Esophagus with Antireflux Surgery. Arch. Surg. 2007, 142, 554–559, discussion 559–560. [Google Scholar] [CrossRef] [PubMed]
- van Baal, J.W.P.M.; Bozikas, A.; Pronk, R.; Ten Kate, F.J.W.; Milano, F.; Rygiel, A.M.; Rosmolen, W.D.; Peppelenbosch, M.P.; Bergman, J.J.G.H.M.; Krishnadath, K.K. Cytokeratin and CDX-2 Expression in Barrett’s Esophagus. Scand. J. Gastroenterol. 2008, 43, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Kosoff, R.E.; Gardiner, K.L.; Merlo, L.M.F.; Pavlov, K.; Rustgi, A.K.; Maley, C.C. Development and Characterization of an Organotypic Model of Barrett’s Esophagus. J. Cell. Physiol. 2012, 227, 2654–2659. [Google Scholar] [CrossRef] [PubMed]
- Samuels, T.; Hoekzema, C.; Gould, J.; Goldblatt, M.; Frelich, M.; Bosler, M.; Lee, S.-H.; Johnston, N. Local Synthesis of Pepsin in Barrett’s Esophagus and the Role of Pepsin in Esophageal Adenocarcinoma. Ann. Otol. Rhinol. Laryngol. 2015, 124, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Huang, J.; Zhu, Z.; Zhang, J.; Li, K. Systematic Review and Meta-Analysis of Tumor Biomarkers in Predicting Prognosis in Esophageal Cancer. BMC Cancer 2013, 13, 539. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.F.; Shewmake, K.; Beer, D.G.; Cryer, B.; Spechler, S.J. Selective Inhibition of Cyclooxygenase-2 Suppresses Growth and Induces Apoptosis in Human Esophageal Adenocarcinoma Cells. Cancer Res. 2000, 60, 5767–5772. [Google Scholar]
- Oyama, K.; Fujimura, T.; Ninomiya, I.; Miyashita, T.; Kinami, S.; Fushida, S.; Ohta, T.; Koichi, M. A COX-2 Inhibitor Prevents the Esophageal Inflammation-Metaplasia-Adenocarcinoma Sequence in Rats. Carcinogenesis 2005, 26, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.S.; Khamnehei, N.; Iravani, M.; Namburu, S.S.; Lin, O.; Triadafilopoulos, G. Rofecoxib Inhibits Cyclooxygenase 2 Expression and Activity and Reduces Cell Proliferation in Barrett’s Esophagus. Gastroenterology 2002, 123, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Lao-Sirieix, P.; Roy, A.; Worrall, C.; Vowler, S.L.; Gardiner, S.; Fitzgerald, R.C. Effect of Acid Suppression on Molecular Predictors for Esophageal Cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 288–293. [Google Scholar] [CrossRef]
- Feagins, L.A.; Zhang, H.-Y.; Hormi-Carver, K.; Quinones, M.H.; Thomas, D.; Zhang, X.; Terada, L.S.; Spechler, S.J.; Ramirez, R.D.; Souza, R.F. Acid Has Antiproliferative Effects in Nonneoplastic Barrett’s Epithelial Cells. Am. J. Gastroenterol. 2007, 102, 10–20. [Google Scholar] [CrossRef]
- Zhang, H.-Y.; Zhang, X.; Hormi-Carver, K.; Feagins, L.A.; Spechler, S.J.; Souza, R.F. In Non-Neoplastic Barrett’s Epithelial Cells, Acid Exerts Early Antiproliferative Effects through Activation of the Chk2 Pathway. Cancer Res. 2007, 67, 8580–8587. [Google Scholar] [CrossRef] [PubMed]
- Boeckxstaens, G.E.; Smout, A. Systematic Review: Role of Acid, Weakly Acidic and Weakly Alkaline Reflux in Gastro-Oesophageal Reflux Disease. Aliment. Pharmacol. Ther. 2010, 32, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Zerbib, F.; Roman, S.; Ropert, A.; des Varannes, S.B.; Pouderoux, P.; Chaput, U.; Mion, F.; Vérin, E.; Galmiche, J.-P.; Sifrim, D. Esophageal PH-Impedance Monitoring and Symptom Analysis in GERD: A Study in Patients off and on Therapy. Am. J. Gastroenterol. 2006, 101, 1956–1963. [Google Scholar] [CrossRef]
- Nagahama, K.; Yamato, M.; Nishio, H.; Takeuchi, K. Essential Role of Pepsin in Pathogenesis of Acid Reflux Esophagitis in Rats. Dig. Dis. Sci. 2006, 51, 303–309. [Google Scholar] [CrossRef]
- Im, N.-R.; Lee, D.Y.; Kim, B.; Kim, J.; Jung, K.-Y.; Kim, T.H.; Baek, S.-K. Role of Matrix Metalloproteinases 7 in the Pathogenesis of Laryngopharyngeal Reflux: Decreased E-Cadherin in Acid Exposed Primary Human Pharyngeal Epithelial Cells. Int. J. Mol. Sci. 2019, 20, 5276. [Google Scholar] [CrossRef]
- Im, N.-R.; Kim, B.; Jung, K.-Y.; Baek, S.-K. Matrix Metalloproteinase-7 Induces E-Cadherin Cleavage in Acid-Exposed Primary Human Pharyngeal Epithelial Cells via the ROS/ERK/c-Jun Pathway. J. Mol. Med. 2022, 100, 313–322. [Google Scholar] [CrossRef]
- Reichel, O.; Mayr, D.; Durst, F.; Berghaus, A. E-Cadherin but Not Beta-Catenin Expression Is Decreased in Laryngeal Biopsies from Patients with Laryngopharyngeal Reflux. Eur. Arch. Otorhinolaryngol. 2008, 265, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Samuels, T.L.; Blaine-Sauer, S.; Yan, K.; Plehhova, K.; Coyle, C.; Johnston, N. Topical Alginate Protection against Pepsin-Mediated Esophageal Damage: E-Cadherin Proteolysis and Matrix Metalloproteinase Induction. Int. J. Mol. Sci. 2023, Submitted. [Google Scholar]
- Lal, M.; Caplan, M. Regulated Intramembrane Proteolysis: Signaling Pathways and Biological Functions. Physiology 2011, 26, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki-Raby, B.; Gilles, C.; Polette, M.; Bruyneel, E.; Laronze, J.-Y.; Bonnet, N.; Foidart, J.-M.; Mareel, M.; Birembaut, P. Upregulation of MMPs by Soluble E-Cadherin in Human Lung Tumor Cells. Int. J. Cancer 2003, 105, 790–795. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Rajasekaran, A.K. Dishonorable Discharge: The Oncogenic Roles of Cleaved E-Cadherin Fragments. Cancer Res. 2012, 72, 2917–2923. [Google Scholar] [CrossRef]
- Hu, Q.-P.; Kuang, J.-Y.; Yang, Q.-K.; Bian, X.-W.; Yu, S.-C. Beyond a Tumor Suppressor: Soluble E-Cadherin Promotes the Progression of Cancer. Int. J. Cancer 2016, 138, 2804–2812. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Jarquín, Á.J.; de Lara-Cisneros, L.G.V.; López-Colombo, A.; Solís-Mendoza, H.A.; Palmer-Márquez, M.L.; Romero-Figueroa, M.S. Expression of Metalloproteinase-9 in Patients with Mild and Severe Forms of Gastroesophageal Reflux Disease. Cir. Cir. 2019, 87, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Davelaar, A.L.; Straub, D.; Buttar, N.S.; Fockens, P.; Krishnadath, K.K. Active Matrix Metalloproteases Are Expressed Early on and Are High during the Barrett’s Esophagus Malignancy Sequence. Scand. J. Gastroenterol. 2015, 50, 321–332. [Google Scholar] [CrossRef]
- Cheung, W.Y.; Zhai, R.; Bradbury, P.; Hopkins, J.; Kulke, M.H.; Heist, R.S.; Asomaning, K.; Ma, C.; Xu, W.; Wang, Z.; et al. Single Nucleotide Polymorphisms in the Matrix Metalloproteinase Gene Family and the Frequency and Duration of Gastroesophageal Reflux Disease Influence the Risk of Esophageal Adenocarcinoma. Int. J. Cancer 2012, 131, 2478–2486. [Google Scholar] [CrossRef]
- Grimm, M.; Lazariotou, M.; Kircher, S.; Stuermer, L.; Reiber, C.; Höfelmayr, A.; Gattenlöhner, S.; Otto, C.; Germer, C.T.; von Rahden, B.H.A. MMP-1 Is a (Pre-)Invasive Factor in Barrett-Associated Esophageal Adenocarcinomas and Is Associated with Positive Lymph Node Status. J. Transl. Med. 2010, 8, 99. [Google Scholar] [CrossRef]
- GlaskoSmithKline Lexiva Prescribing Information 2009. Available online: https://gskpro.com/content/dam/global/hcpportal/en_US/Prescribing_Information/Lexiva/pdf/LEXIVA-PI-PIL.PDF (accessed on 21 February 2023).
- Hussain, Z.H.; Henderson, E.E.; Maradey-Romerao, C.; George, N.; Fass, R.; Lacy, B.E. The Proton Pump Inhibitor Non-Responder: A Clinical Conundrum. Clin. Transl. Gastroenterol. 2015, 6, e106. [Google Scholar] [CrossRef]
- Delshad, S.D.; Almario, C.V.; Chey, W.D.; Spiegel, B.M.R. Prevalence of Gastroesophageal Reflux Disease and Proton Pump Inhibitor-Refractory Symptoms. Gastroenterology 2020, 158, 1250–1261.e2. [Google Scholar] [CrossRef] [PubMed]
- Mainie, I.; Tutuian, R.; Shay, S.; Vela, M.; Zhang, X.; Sifrim, D.; Castell, D.O. Acid and Non-Acid Reflux in Patients with Persistent Symptoms despite Acid Suppressive Therapy: A Multicentre Study Using Combined Ambulatory Impedance-PH Monitoring. Gut 2006, 55, 1398–1402. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, K.R.; Morales, C.P.; Feagins, L.A.; Gandia, K.G.; Zhang, X.; Zhang, H.-Y.; Hormi-Carver, K.; Shen, Y.; Elder, F.; Ramirez, R.D.; et al. Characterization of Telomerase-Immortalized, Non-Neoplastic, Human Barrett’s Cell Line (BAR-T). Dis. Esophagus 2007, 20, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Jaccard, N.; Griffin, L.D.; Keser, A.; Macown, R.J.; Super, A.; Veraitch, F.S.; Szita, N. Automated Method for the Rapid and Precise Estimation of Adherent Cell Culture Characteristics from Phase Contrast Microscopy Images. Biotechnol. Bioeng. 2014, 111, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).