
Molecular Docking and Dynamics Simulation Studies Predict Potential Anti-ADAR2 Inhibitors: Implications for the Treatment of Cancer, Neurological, Immunological and Infectious Diseases

 ,
,  , ,
, ,
Abstract
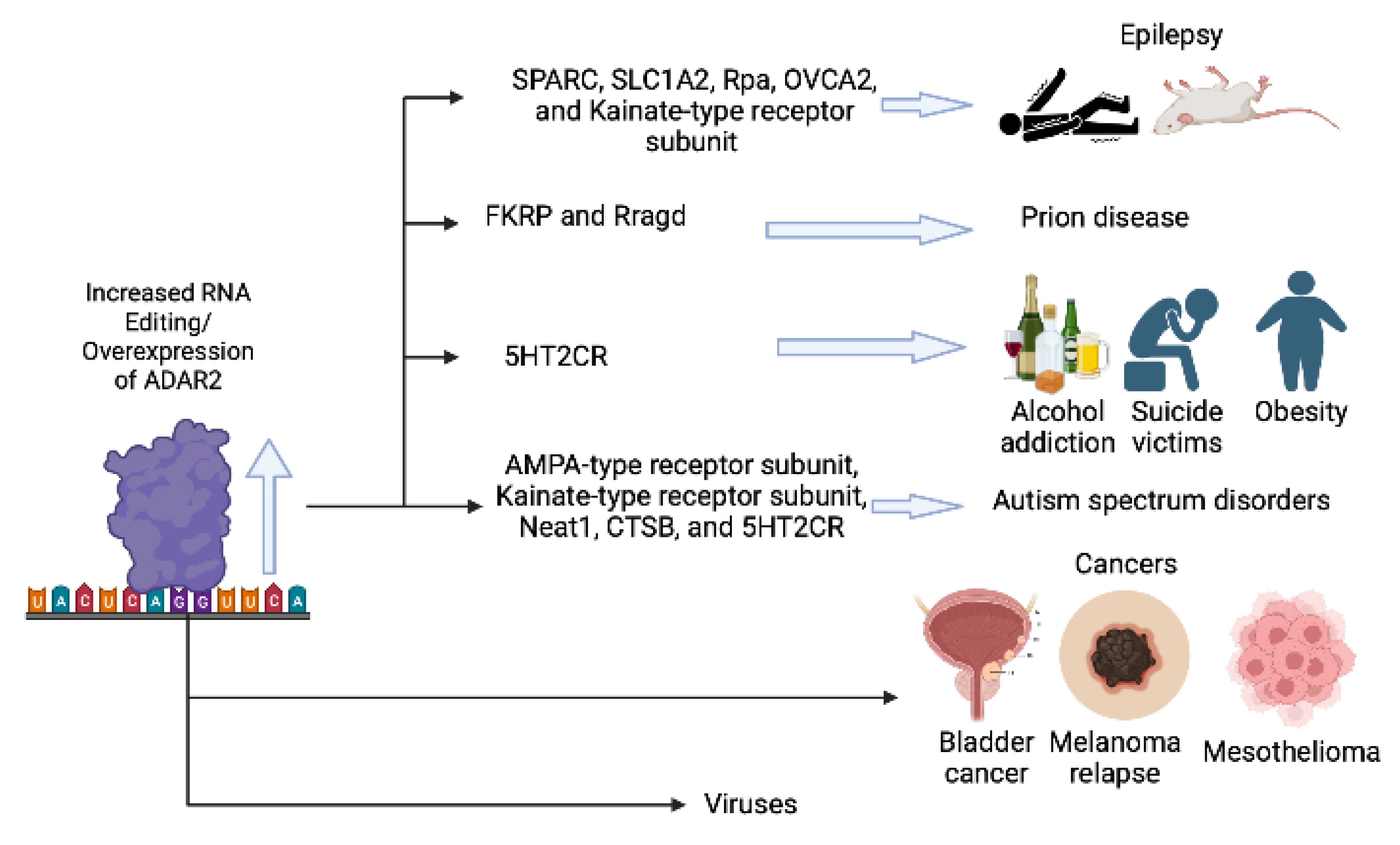
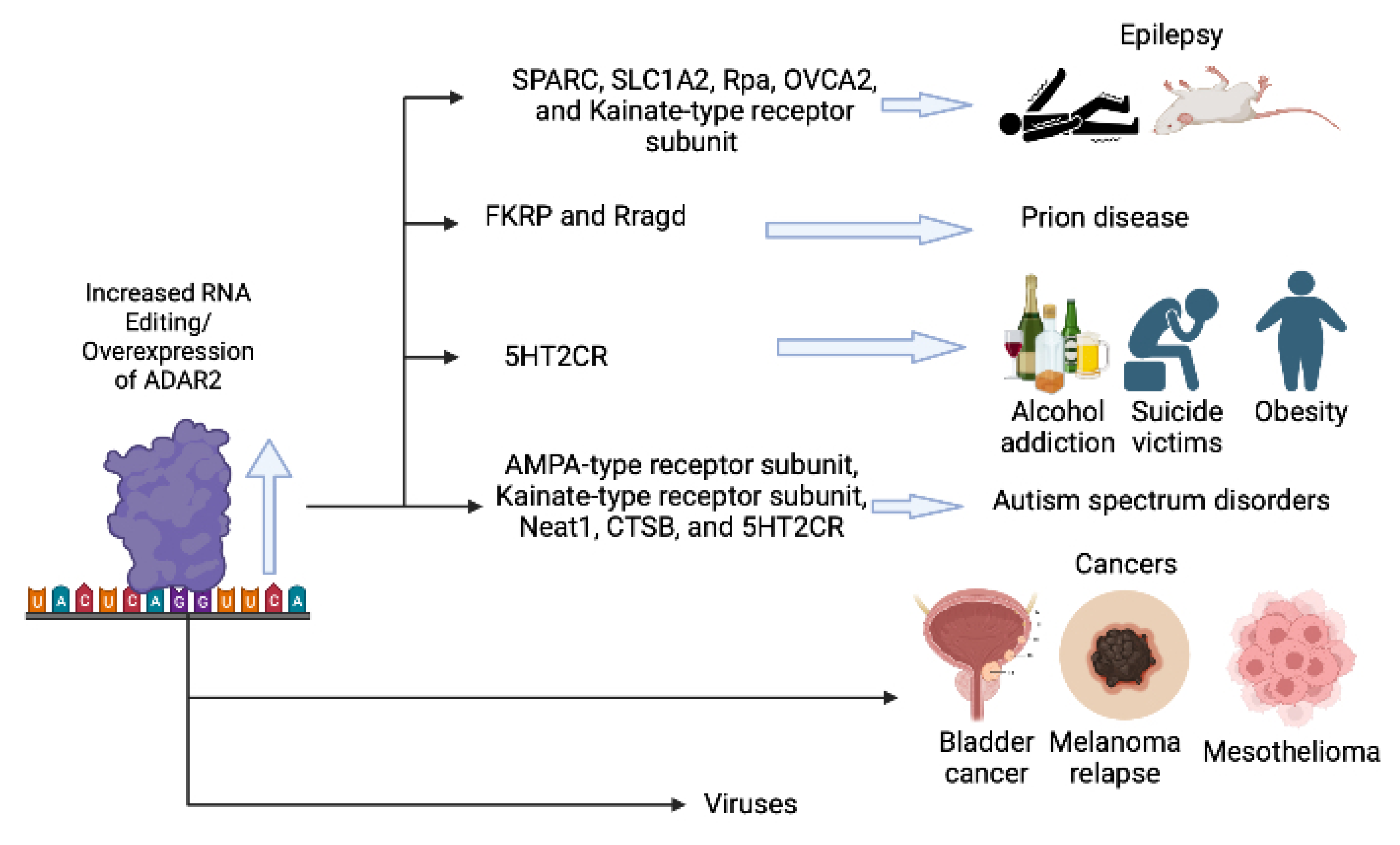
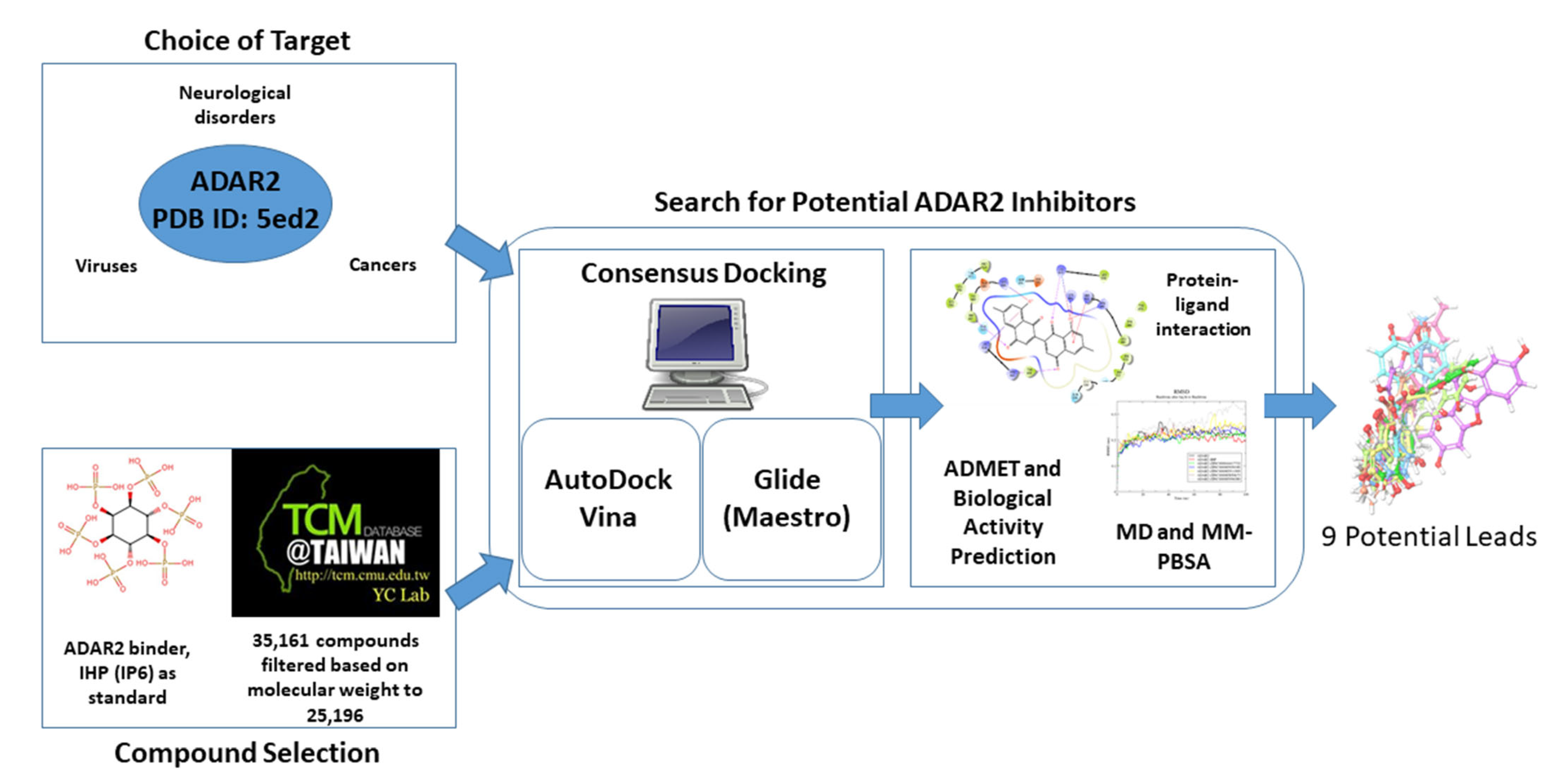
:1. Introduction
2. Results and Discussions
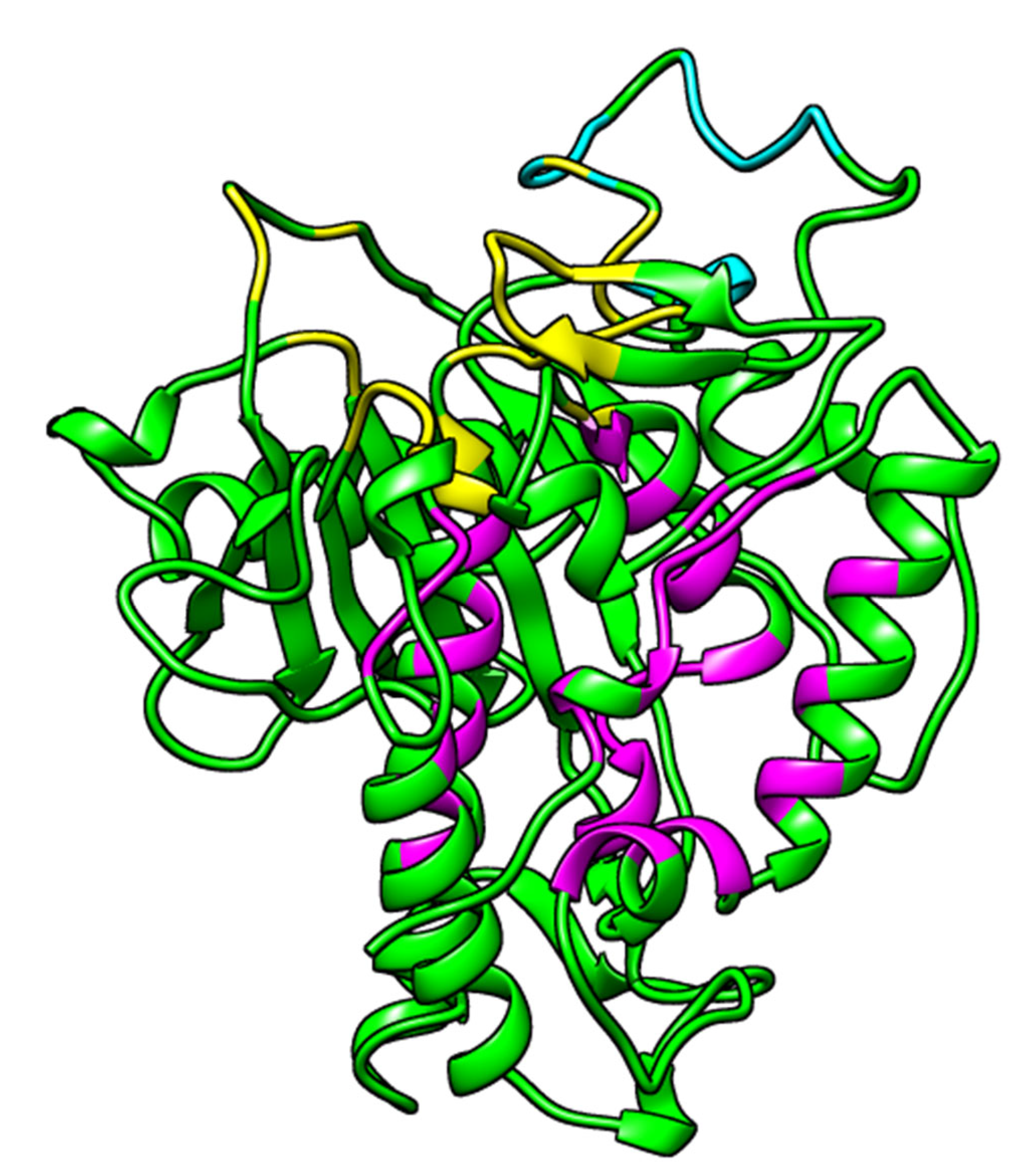
2.1. Prediction of Binding Sites
2.2. Molecular Docking
2.2.1. Validation of Docking Protocols
2.2.2. Molecular Docking via AutoDock Vina
2.2.3. Molecular Docking via Maestro
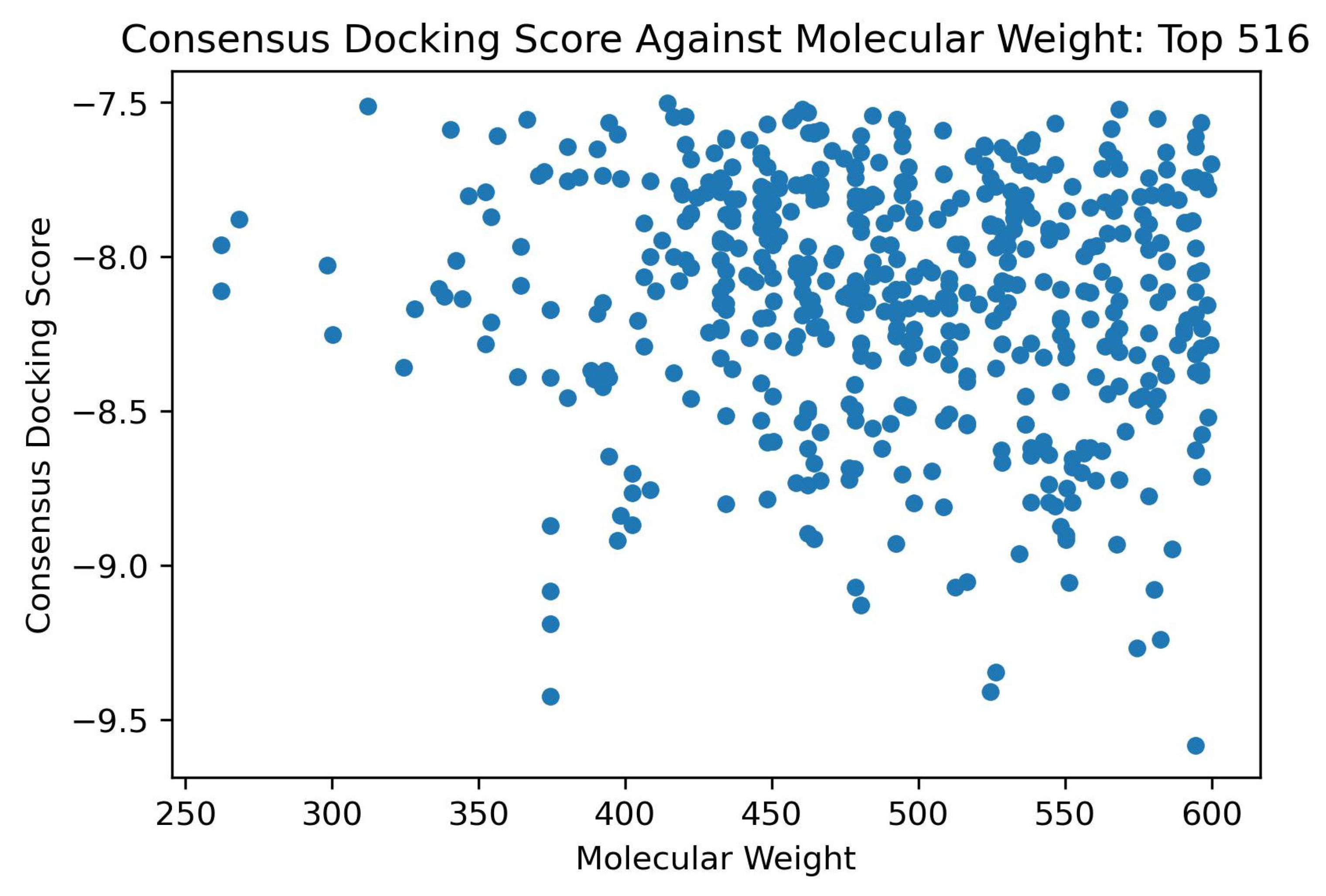
2.2.4. Shortlisting Compounds Based on Consensus Score
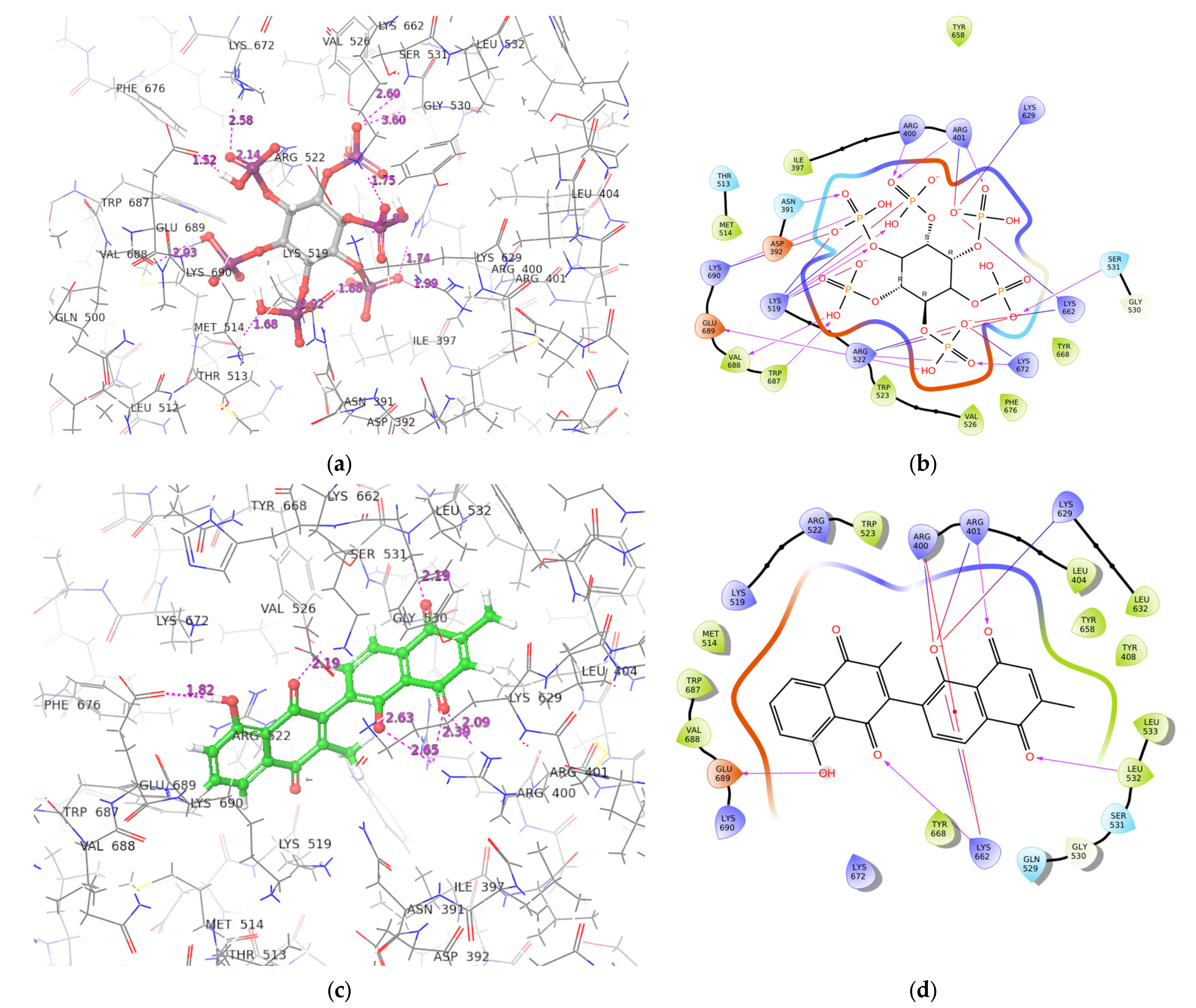
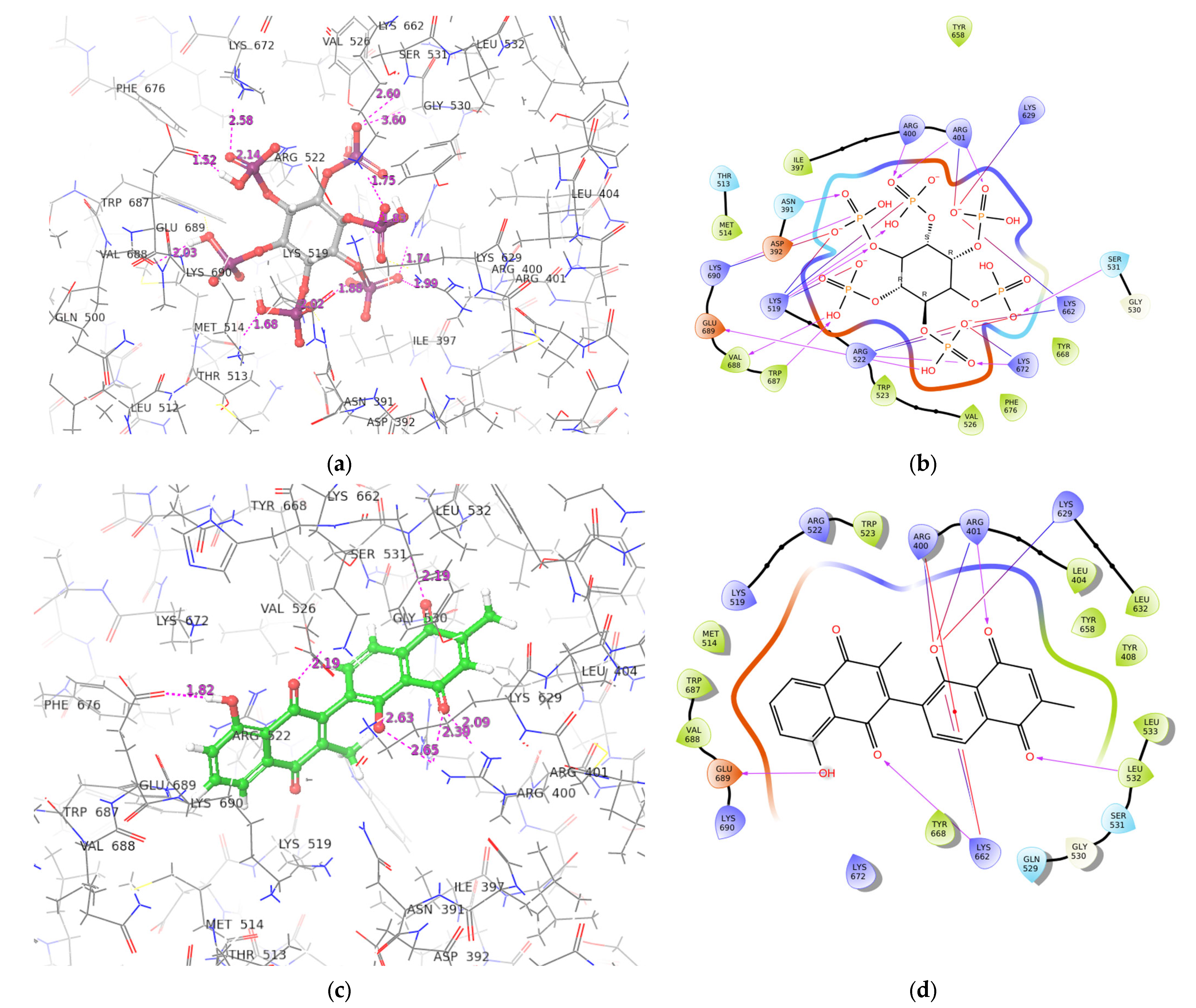
2.3. ADAR2-Ligand Interaction Profiling
2.4. ADMET Prediction
2.5. Structural Similarity Search and Prediction of Biological Activity of Shortlisted Compounds
2.6. Molecular Dynamics Simulations
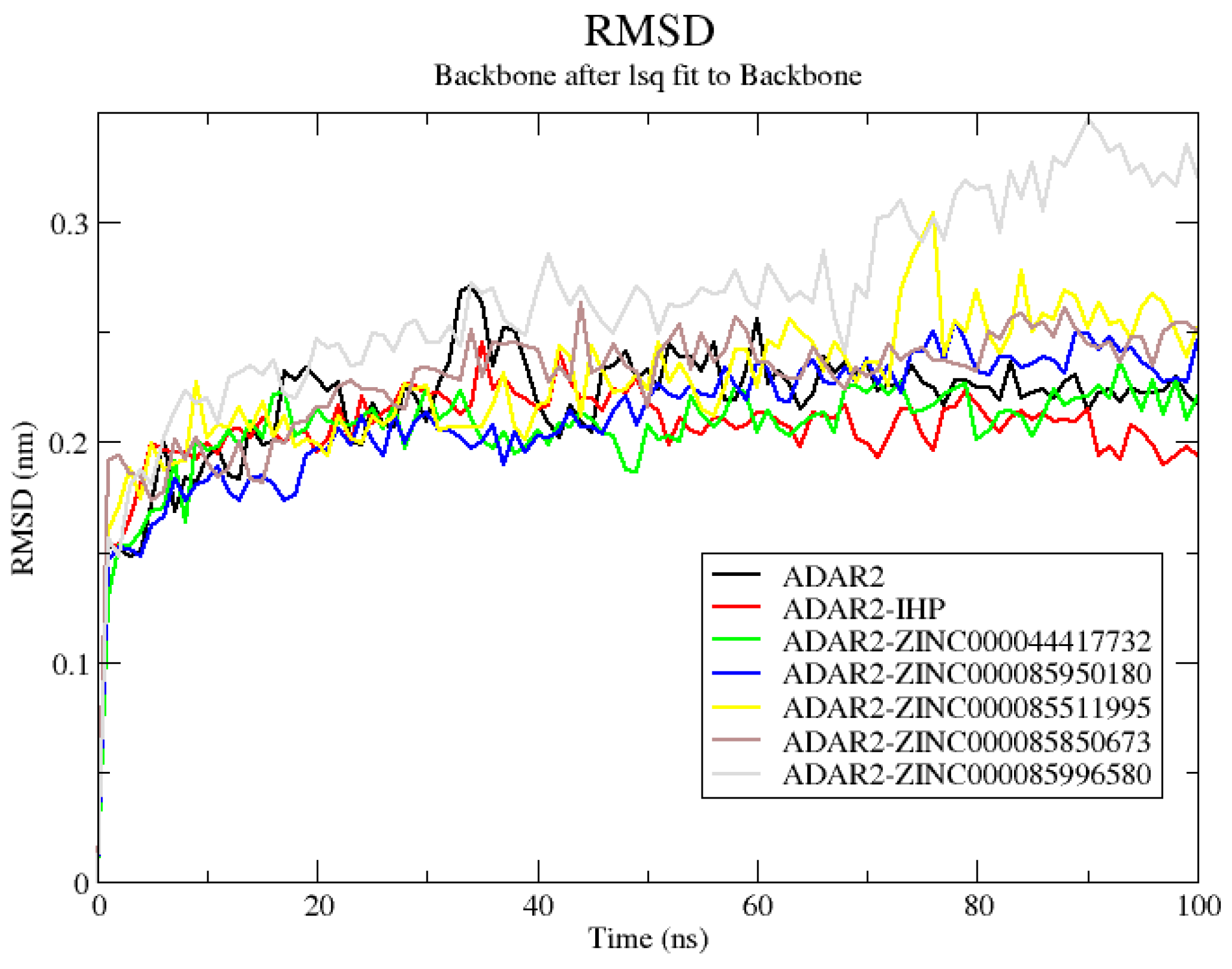
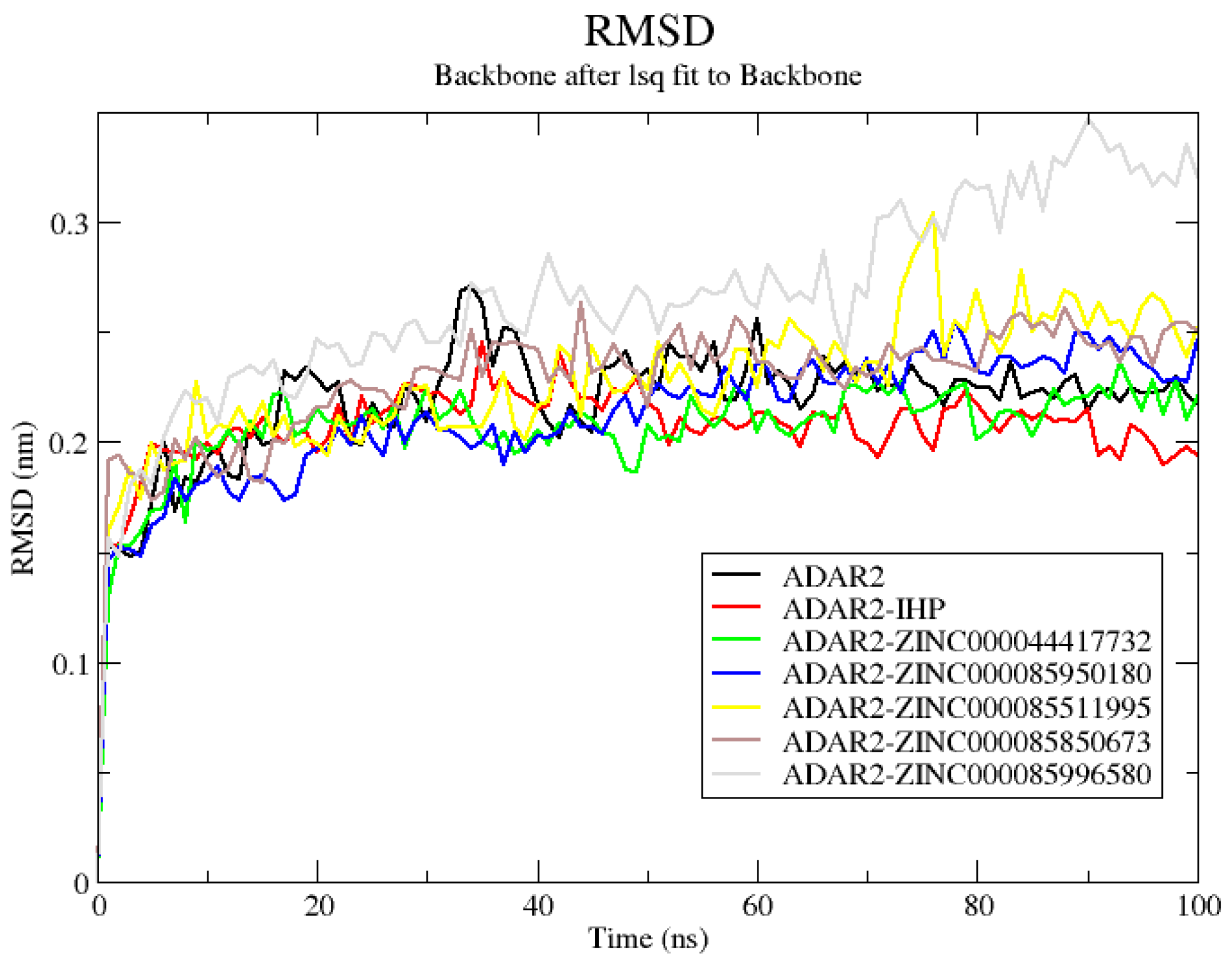
2.6.1. RMSD of ADAR2 and ADAR2-Ligand Complexes
2.6.2. Radius of Gyration of ADAR2 and ADAR2-Ligand Complexes
2.6.3. RMSF of ADAR2-Ligand Complexes
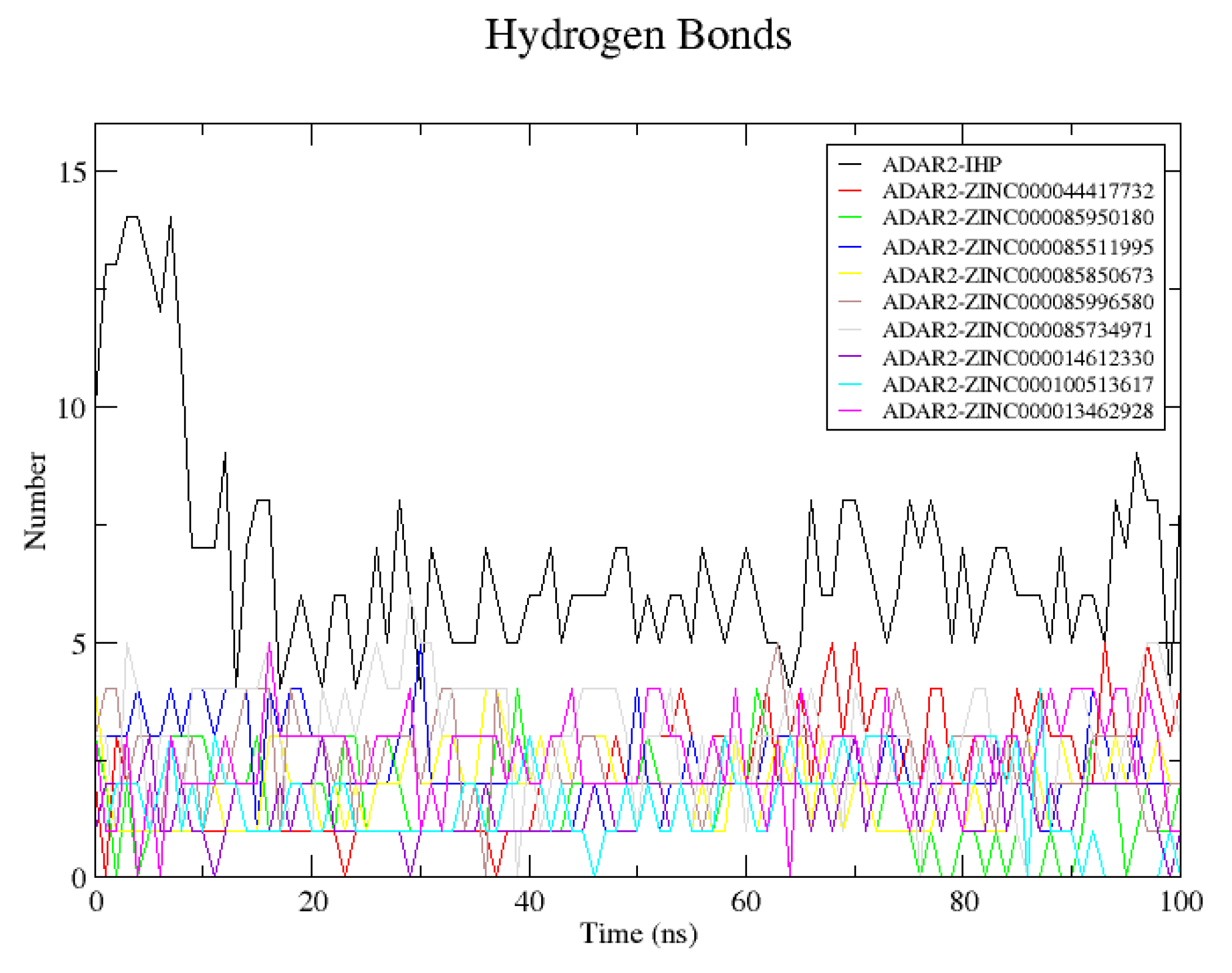
2.6.4. Snapshots and Hydrogen Bond Analysis of Complexes
2.7. Evaluating Potential Leads via MM/PBSA Calculation
Per-Residue Energy Decomposition
2.8. Re-Docking Predicted Hits against 5-HT2C Receptor
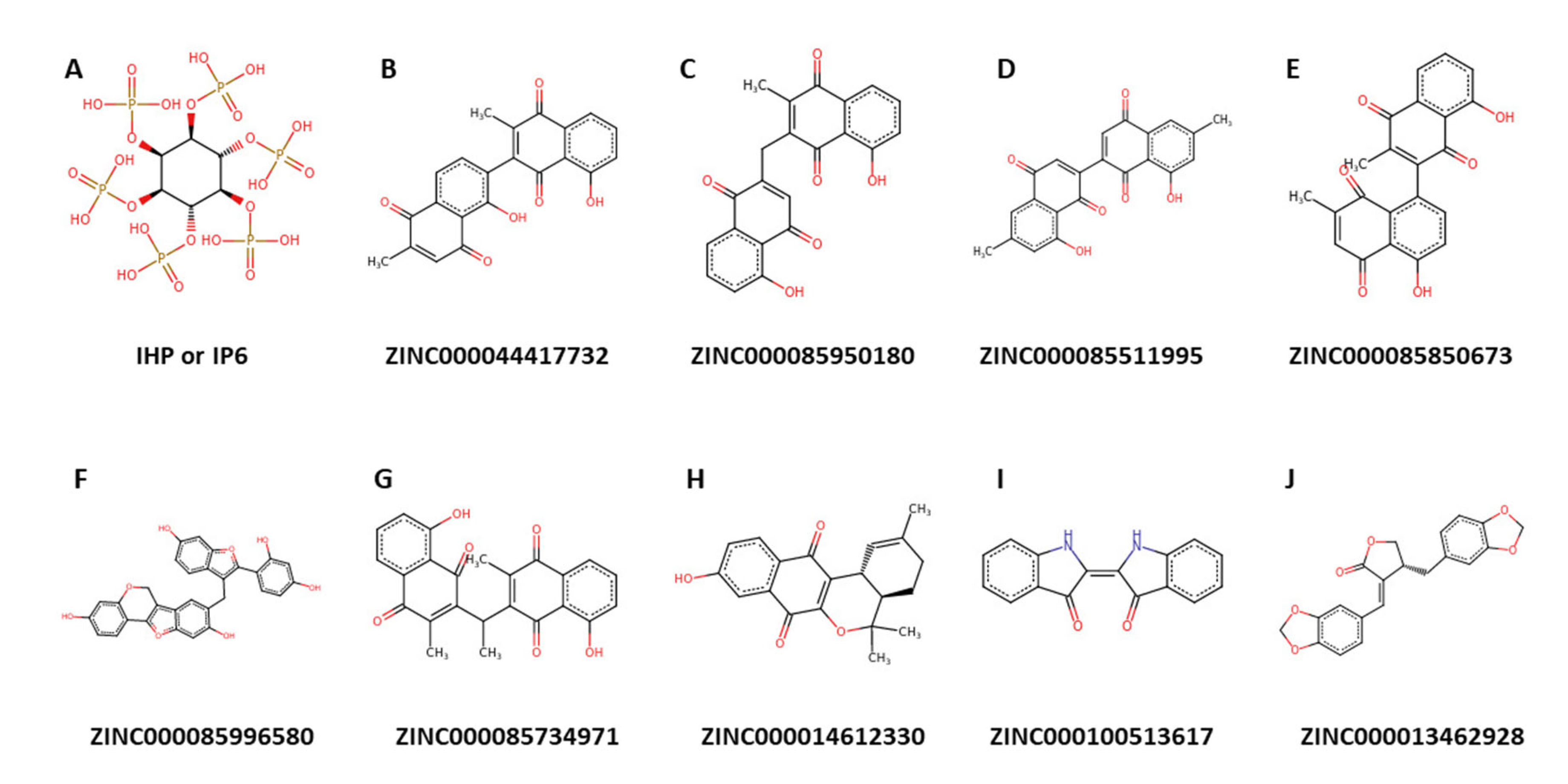
2.9. Origin and Source of the Potential Lead Compounds
3. Materials and Methods
3.1. Obtaining and Preparing Protein and Ligand Structures
3.2. Determining Binding Sites
3.3. Molecular Docking
3.3.1. Validation of Molecular Docking Protocols
3.3.2. Molecular Docking via AutoDock Vina
3.3.3. Molecular Docking via XGlide (Maestro)
3.3.4. Shortlisting Compounds using Consensus Scoring
3.4. Determining the Interactions between the ADAR2-Ligand Complexes
3.5. Determining ADMET Properties
3.6. Structural Similarity Search and Prediction of Biological Activity of Compounds
3.7. Molecular Dynamics Simulations
3.8. Molecular Mechanics Poisson–Boltzmann Surface Area (MM/PBSA) Computation of Potential Leads
3.9. Re-Docking Potential Leads against the 5-HT2CR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Okada, S.; Sakurai, M. Adenosine-to-inosine RNA editing in neurological development and disease. RNA Biol. 2021, 18, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, G.; Morris, K.V. All I’s on the RADAR: Role of ADAR in gene regulation. FEBS Lett. 2018, 592, 2860–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werry, T.D.; Loiacono, R.; Sexton, P.M.; Christopoulos, A. RNA editing of the serotonin 5HT2C receptor and its effects on cell signalling, pharmacology and brain function. Pharmacol. Ther. 2008, 119, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, W.; Nishikura, K. Adenosine-to-inosine RNA editing and human disease. Genome Med. 2013, 5, 105. [Google Scholar] [CrossRef]
- Morabito, M.V.; Abbas, A.I.; Hood, J.L.; Kesterson, R.A.; Jacobs, M.M.; Kump, D.S.; Hachey, D.L.; Roth, B.L.; Emeson, R.B. Mice with altered serotonin 2C receptor RNA editing display characteristics of Prader–Willi syndrome. Neurobiol. Dis. 2010, 39, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Paz-Yaacov, N.; Bazak, L.; Buchumenski, I.; Porath, H.T.; Danan-Gotthold, M.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015, 13, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Karagianni, K.; Pettas, S.; Christoforidou, G.; Kanata, E.; Bekas, N.; Xanthopoulos, K.; Dafou, D.; Sklaviadis, T. A Systematic Review of Common and Brain-Disease-Specific RNA Editing Alterations Providing Novel Insights into Neurological and Neurodegenerative Disease Manifestations. Biomolecules 2022, 12, 465. [Google Scholar] [CrossRef]
- Di Narzo, A.F.; Kozlenkov, A.; Roussos, P.; Hao, K.; Hurd, Y.; Lewis, D.A.; Sibille, E.; Siever, L.J.; Koonin, E.; Dracheva, S. A unique gene expression signature associated with serotonin 2C receptor RNA editing in the prefrontal cortex and altered in suicide. Hum. Mol. Genet. 2014, 23, 4801–4813. [Google Scholar] [CrossRef] [Green Version]
- Dracheva, S.; Patel, N.; Woo, D.A.; Marcus, S.M.; Siever, L.J.; Haroutunian, V. Increased serotonin 2C receptor mRNA editing: A possible risk factor for suicide. Mol. Psychiatry 2008, 13, 1001–1010. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, I.; Tamir, H.; Arango, V.; Dwork, A.J.; Mann, J.J.; Schmauss, C. Altered Editing of Serotonin 2C Receptor Pre-mRNA in the Prefrontal Cortex of Depressed Suicide Victims. Neuron 2002, 34, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Karanović, J.; Šviković, S.; Pantović, M.; Durica, S.; Brajušković, G.; Damjanović, A.; Jovanović, V.; Ivković, M.; Romac, S.; Savić Pavićević, D. Joint effect of ADARB1 gene, HTR2C gene and stressful life events on suicide attempt risk in patients with major psychiatric disorders. World J. Biol. Psychiatry 2015, 16, 261–271. [Google Scholar] [CrossRef]
- Lyddon, R.; Dwork, A.J.; Keddache, M.; Siever, L.J.; Dracheva, S. Serotonin 2c receptor RNA editing in major depression and suicide. World J. Biol. Psychiatry 2013, 14, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Niswender, C. RNA Editing of the Human Serotonin 5-HT2C Receptor Alterations in Suicide and Implications for Serotonergic Pharmacotherapy. Neuropsychopharmacology 2001, 24, 478–491. [Google Scholar] [CrossRef]
- Pandey, G.N.; Dwivedi, Y.; Ren, X.; Rizavi, H.S.; Faludi, G.; Sarosi, A.; Palkovits, M. Regional Distribution and Relative Abundance of Serotonin2c Receptors in Human Brain: Effect of Suicide. Neurochem. Res. 2006, 31, 167–176. [Google Scholar] [CrossRef]
- Weissmann, D.; van der Laan, S.; Underwood, M.D.; Salvetat, N.; Cavarec, L.; Vincent, L.; Molina, F.; Mann, J.J.; Arango, V.; Pujol, J.F. Region-specific alterations of A-to-I RNA editing of serotonin 2c receptor in the cortex of suicides with major depression. Transl. Psychiatry 2016, 6, e878. [Google Scholar] [CrossRef] [Green Version]
- Bombail, V.; Qing, W.; Chapman, K.E.; Holmes, M.C. Prevention of 5-hydroxytryptamine 2C receptor RNA editing and alternate splicing in C57BL/6 mice activates the hypothalamic-pituitary-adrenal axis and alters mood. Eur. J. Neurosci. 2014, 40, 3663–3673. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.B.P.; Ramond, F.; Farrington, D.T.; Aguiar, A.S.; Chevarin, C.; Berthiau, A.-S.; Caussanel, S.; Lanfumey, L.; Herrick-Davis, K.; Hamon, M.; et al. RNA splicing and editing modulation of 5-HT2C receptor function: Relevance to anxiety and aggression in VGV mice. Mol. Psychiatry 2013, 18, 656–665. [Google Scholar] [CrossRef]
- Règue, M.; Poilbout, C.; Martin, V.; Franc, B.; Lanfumey, L.; Mongeau, R. Increased 5-HT2C receptor editing predisposes to PTSD-like behaviors and alters BDNF and cytokines signaling. Transl. Psychiatry 2019, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Warhaftig, G.; Sokolik, C.M.; Khermesh, K.; Lichtenstein, Y.; Barak, M.; Bareli, T.; Levanon, E.Y.; Yadid, G. RNA editing of the 5-HT2C receptor in the central nucleus of the amygdala is involved in resilience behavior. Transl. Psychiatry 2021, 11, 137. [Google Scholar] [CrossRef]
- Sodhi, M.S.; Burnet, P.W.J.; Makoff, A.J.; Kerwin, R.W.; Harrison, P.J. RNA editing of the 5-HT2C receptor is reduced in schizophrenia. Mol. Psychiatry 2001, 6, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirle, N.T.; Goodman, R.A.; Krishnamurthy, M.; Beal, P.A. Selective inhibition of ADAR2-catalyzed editing of the serotonin 2c receptor pre-mRNA by a helix-threading peptide. Org. Biomol. Chem. 2010, 8, 4898. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shen, M.; Gresch, P.J.; Ghamari-Langroudi, M.; Rabchevsky, A.G.; Emeson, R.B.; Stamm, S. Oligonucleotide-induced alternative splicing of serotonin 2C receptor reduces food intake. EMBO Mol. Med. 2016, 8, 878–894. [Google Scholar] [CrossRef] [PubMed]
- Heisler, L.K.; Chu, H.-M.; Tecott, L.H. Epilepsy and Obesity in Serotonin 5-HT2C Receptor Mutant Mice. Ann. N. Y. Acad. Sci. 1998, 861, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Tecott, L.H.; Sun, L.M.; Akana, S.F.; Strack, A.M.; Lowenstein, D.H.; Dallman, M.F.; Julius, D. Eating disorder and epilepsy in mice lacking 5-HT2C serotonin receptors. Nature 1995, 374, 542–546. [Google Scholar] [CrossRef]
- Piontkivska, H.; Wales-McGrath, B.; Miyamoto, M.; Wayne, M.L. ADAR Editing in Viruses: An Evolutionary Force to Reckon with. Genome Biol. Evol. 2021, 13, evab240. [Google Scholar] [CrossRef]
- Matthews, M.M.; Thomas, J.M.; Zheng, Y.; Tran, K.; Phelps, K.J.; Scott, A.I.; Havel, J.; Fisher, A.J.; Beal, P.A. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 2016, 23, 426–433. [Google Scholar] [CrossRef] [Green Version]
- Stefl, R.; Oberstrass, F.C.; Hood, J.L.; Jourdan, M.; Zimmermann, M.; Skrisovska, L.; Maris, C.; Peng, L.; Hofr, C.; Emeson, R.B.; et al. The Solution Structure of the ADAR2 dsRBM-RNA Complex Reveals a Sequence-Specific Readout of the Minor Groove. Cell 2010, 143, 225–237. [Google Scholar] [CrossRef]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Mushtaq, S.; Abbasi, B.H.; Uzair, B.; Abbasi, R. Natural products as reservoirs of novel therapeutic agents. EXCLI J. 2018, 17, 420–451. [Google Scholar] [CrossRef]
- Li, G.; Lou, H.-X. Strategies to diversify natural products for drug discovery. Med. Res. Rev. 2018, 38, 1255–1294. [Google Scholar] [CrossRef]
- Kumar, G.B.; Nair, B.G.; Perry, J.J.P.; Martin, D.B.C. Recent insights into natural product inhibitors of matrix metalloproteinases. Medchemcomm 2019, 10, 2024–2037. [Google Scholar] [CrossRef]
- Ren, Z.-L.; Zuo, P.-P. Neural Regeneration: Role of Traditional Chinese Medicine in Neurological Diseases Treatment. J. Pharmacol. Sci. 2012, 120, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, L.; Chen, T.; Sun, Z.; Tang, W.; Wang, S.; Wang, T.; Wang, Y.; Zhang, H. Research Progress in the Effect of Traditional Chinese Medicine for Invigoration on Neurotransmitter Related Diseases. Evid.-Based Complement. Altern. Med. 2018, 2018, 4642018. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, S.; Kan, J.; Zhang, J.; Zhou, L.; Huang, Y.; Zhang, Y. Chinese Herbal Medicine Interventions in Neurological Disorder Therapeutics by Regulating Glutamate Signaling. Curr. Neuropharmacol. 2020, 18, 260–276. [Google Scholar] [CrossRef]
- Savva, Y.A.; Rieder, L.E.; Reenan, R.A. The ADAR protein family. Genome Biol. 2012, 13, 252. [Google Scholar] [CrossRef] [Green Version]
- Macbeth, M.R.; Lingam, A.T.; Bass, B.L. Evidence for auto-inhibition by the N terminus of hADAR2 and activation by dsRNA binding. RNA 2004, 10, 1563–1571. [Google Scholar] [CrossRef] [Green Version]
- Macbeth, M.R.; Schubert, H.L.; VanDemark, A.P.; Lingam, A.T.; Hill, C.P.; Bass, B.L. Inositol Hexakisphosphate Is Bound in the ADAR2 Core and Required for RNA Editing. Science 2005, 309, 1534–1539. [Google Scholar] [CrossRef] [Green Version]
- Broni, E.; Kwofie, S.K.; Asiedu, S.O.; Miller, W.A.; Wilson, M.D. A Molecular Modeling Approach to Identify Potential Antileishmanial Compounds Against the Cell Division Cycle (cdc)-2-Related Kinase 12 (CRK12) Receptor of Leishmania donovani. Biomolecules 2021, 11, 458. [Google Scholar] [CrossRef]
- Kwofie, S.K.; Broni, E.; Asiedu, S.O.; Kwarko, G.B.; Dankwa, B.; Enninful, K.S.; Tiburu, E.K.; Wilson, M.D. Cheminformatics-Based Identification of Potential Novel Anti-SARS-CoV-2 Natural Compounds of African Origin. Molecules 2021, 26, 406. [Google Scholar] [CrossRef]
- Wang, Y.; Beal, P.A. Probing RNA recognition by human ADAR2 using a high-throughput mutagenesis method. Nucleic Acids Res. 2016, 44, 9872–9880. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houston, D.R.; Walkinshaw, M.D. Consensus docking: Improving the reliability of docking in a virtual screening context. J. Chem. Inf. Model. 2013, 53, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.; Froufe, H.; Costa, A.; Santos, A.; Oliveira, L.; Osório, S.; Abreu, R.; Pintado, M.; Ferreira, I. Docking Studies in Target Proteins Involved in Antibacterial Action Mechanisms: Extending the Knowledge on Standard Antibiotics to Antimicrobial Mushroom Compounds. Molecules 2014, 19, 1672–1684. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.W.; Lindstrom, W.; Olson, A.J.; Belew, R.K. Analysis of HIV wild-type and mutant structures via in silico docking against diverse ligand libraries. J. Chem. Inf. Model. 2007, 47, 1258–1262. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Richard, D.M.; Dawes, M.A.; Mathias, C.W.; Acheson, A.; Hill-Kapturczak, N.; Dougherty, D.M. L -Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int. J. Tryptophan Res. 2009, 2, IJTR.S2129. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, T.; Nguyen, J.; Polglaze, K.; Bertrand, P. Influence of Tryptophan and Serotonin on Mood and Cognition with a Possible Role of the Gut-Brain Axis. Nutrients 2016, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Baixauli, E. Happiness: Role of Dopamine and Serotonin on Mood and Negative Emotions. Emerg. Med. Open Access 2017, 7, 33–51. [Google Scholar] [CrossRef] [Green Version]
- Yousef, I. Serotonin-Happiness and Satisfaction. Biomed. J. Sci. Tech. Res. 2021, 33, 25870–25871. [Google Scholar] [CrossRef]
- Alenina, N.; Klempin, F. The role of serotonin in adult hippocampal neurogenesis. Behav. Brain Res. 2015, 277, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Voracek, M.; Tran, U.S. Dietary tryptophan intake and suicide rate in industrialized nations. J. Affect. Disord. 2007, 98, 259–262. [Google Scholar] [CrossRef]
- Munir, S.; Shahid, A.; Aslam, B.; Ashfaq, U.A.; Akash, M.S.H.; Ali, M.A.; Almatroudi, A.; Allemailem, K.S.; Rajoka, M.S.R.; Khurshid, M. The Therapeutic Prospects of Naturally Occurring and Synthetic Indole Alkaloids for Depression and Anxiety Disorders. Evid.-Based Complement. Altern. Med. 2020, 2020, 8836983. [Google Scholar] [CrossRef]
- Melancon, M.O.; Lorrain, D.; Dionne, I.J. Exercise and sleep in aging: Emphasis on serotonin. Pathol. Biol. 2014, 62, 276–283. [Google Scholar] [CrossRef]
- Kohyama, J. Sleep, Serotonin, and Suicide in Japan. J. Physiol. Anthropol. 2011, 30, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Tricklebank, M.D. Serotonin and sleep. In The Serotonin System; Elsevier: Amsterdam, The Netherlands, 2019; pp. 181–192. ISBN 9780128133231. [Google Scholar]
- Ursin, R. Serotonin and sleep. Sleep Med. Rev. 2002, 6, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Fava, M.; Rosenbaum, J.F.; Hoog, S.L.; Tepner, R.G.; Kopp, J.B.; Nilsson, M.E. Fluoxetine versus sertraline and paroxetine in major depression: Tolerability and efficacy in anxious depression. J. Affect. Disord. 2000, 59, 119–126. [Google Scholar] [CrossRef]
- Cipriani, A.; La Ferla, T.; Furukawa, T.A.; Signoretti, A.; Nakagawa, A.; Churchill, R.; McGuire, H.; Barbui, C. Sertraline versus other antidepressive agents for depression. Cochrane Database Syst. Rev. 2010. [Google Scholar] [CrossRef]
- Bergeron, R.; Ravindran, A.V.; Chaput, Y.; Goldner, E.; Swinson, R.; van Ameringen, M.A.; Austin, C.; Hadrava, V. Sertraline and Fluoxetine Treatment of Obsessive-Compulsive Disorder: Results of a Double-Blind, 6-Month Treatment Study. J. Clin. Psychopharmacol. 2002, 22, 148–154. [Google Scholar] [CrossRef]
- Soorya, L.; Kiarashi, J.; Hollander, E. Psychopharmacologic Interventions for Repetitive Behaviors in Autism Spectrum Disorders. Child Adolesc. Psychiatr. Clin. N. Am. 2008, 17, 753–771. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Li, M.; Li, X. The new coumarin compound Bis 3 ameliorates cognitive disorder and suppresses brain-intestine-liver systematic oxidative stress in high-fat diet mice. Biomed. Pharmacother. 2021, 137, 111293. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Kong, L.; Xia, Y.; Liang, W.; Wang, L.; Song, J.; Yao, Y.; Lin, Y.; Yang, J. Osthole promotes endogenous neural stem cell proliferation and improved neurological function through Notch signaling pathway in mice acute mechanical brain injury. Brain. Behav. Immun. 2018, 67, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Yao, Y.; Xia, Y.; Liang, X.; Ni, Y.; Yang, J. Osthole alleviates inflammation by down-regulating NF-κB signaling pathway in traumatic brain injury. Immunopharmacol. Immunotoxicol. 2019, 41, 349–360. [Google Scholar] [CrossRef]
- Du, G.; Tu, H.; Li, X.; Pei, A.; Chen, J.; Miao, Z.; Li, J.; Wang, C.; Xie, H.; Xu, X.; et al. Daphnetin, a Natural Coumarin Derivative, Provides the Neuroprotection Against Glutamate-Induced Toxicity in HT22 Cells and Ischemic Brain Injury. Neurochem. Res. 2014, 39, 269–275. [Google Scholar] [CrossRef]
- Jin, X.; Wang, Y.; Li, X.; Tan, X.; Miao, Z.; Chen, Y.; Hamdy, R.C.; Chua, B.H.L.; Kong, J.; Zhao, H.; et al. 7,8-Dihydroxy-4-methylcoumarin Provides Neuroprotection by Increasing Hippocalcin Expression. Neurotox. Res. 2015, 27, 268–274. [Google Scholar] [CrossRef]
- Langdon, S.R.; Ertl, P.; Brown, N. Bioisosteric Replacement and Scaffold Hopping in Lead Generation and Optimization. Mol. Inform. 2010, 29, 366–385. [Google Scholar] [CrossRef]
- Wassermann, A.M.; Bajorath, J. Identification of target family directed bioisosteric replacements. Medchemcomm 2011, 2, 601–606. [Google Scholar] [CrossRef]
- Lazzara, P.R.; Moore, T.W. Scaffold-hopping as a strategy to address metabolic liabilities of aromatic compounds. RSC Med. Chem. 2020, 11, 18–29. [Google Scholar] [CrossRef]
- Sakyi, P.O.; Broni, E.; Amewu, R.K.; Miller, W.A.I.; Wilson, M.D.; Kwofie, S.K. Homology Modeling, de Novo Design of Ligands, and Molecular Docking Identify Potential Inhibitors of Leishmania donovani 24-Sterol Methyltransferase. Front. Cell. Infect. Microbiol. 2022, 12, 657. [Google Scholar] [CrossRef]
- Wen, C.C.; Kuo, Y.H.; Jan, J.T.; Liang, P.H.; Wang, S.Y.; Liu, H.G.; Lee, C.K.; Chang, S.T.; Kuo, C.J.; Lee, S.S.; et al. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem. 2007, 50, 4087–4095. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Barret, R. Lipinski’s Rule of Five. In Therapeutical Chemistry; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Lipinski, C.A. Rule of five in 2015 and beyond: Target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv. Drug Deliv. Rev. 2016, 101, 34–41. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Majerova, P.; Olesova, D.; Golisova, G.; Buralova, M.; Michalicova, A.; Vegh, J.; Piestansky, J.; Bhide, M.; Hanes, J.; Kovac, A. Analog of kynurenic acid decreases tau pathology by modulating astrogliosis in rat model for tauopathy. Biomed. Pharmacother. 2022, 152, 113257. [Google Scholar] [CrossRef]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef]
- Bors, L.; Erdő, F. Overcoming the Blood–Brain Barrier. Challenges and Tricks for CNS Drug Delivery. Sci. Pharm. 2019, 87, 6. [Google Scholar] [CrossRef] [Green Version]
- Hanson, L.R.; Frey, W.H. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008, 9, S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Jiang, C. Evolution of blood–brain barrier in brain diseases and related systemic nanoscale brain-targeting drug delivery strategies. Acta Pharm. Sin. B 2021, 11, 2306–2325. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; An, O.; Hong, H.Q.; Chan, T.H.M.; Song, Y.; Shen, H.; Tang, S.J.; Lin, J.S.; Ng, V.H.E.; Tay, D.J.T.; et al. Suppression of adenosine-to-inosine (A-to-I) RNA editome by death associated protein 3 (DAP3) promotes cancer progression. Sci. Adv. 2020, 6, eaba5136. [Google Scholar] [CrossRef] [PubMed]
- Valles, I.; Pajares, M.J.; Segura, V.; Guruceaga, E.; Gomez-Roman, J.; Blanco, D.; Tamura, A.; Montuenga, L.M.; Pio, R. Identification of Novel Deregulated RNA Metabolism-Related Genes in Non-Small Cell Lung Cancer. PLoS ONE 2012, 7, e42086. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-B.; Liao, X.-Y.; Zhang, J.-B.; Wang, F.; Qin, H.-D.; Zhang, L.; Shugart, Y.Y.; Zeng, Y.-X.; Jia, W.-H. ADAR2 functions as a tumor suppressor via editing IGFBP7 in esophageal squamous cell carcinoma. Int. J. Oncol. 2017, 50, 622–630. [Google Scholar] [CrossRef]
- Tomaselli, S.; Galeano, F.; Alon, S.; Raho, S.; Galardi, S.; Polito, V.A.; Presutti, C.; Vincenti, S.; Eisenberg, E.; Locatelli, F.; et al. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 2015, 16, 5. [Google Scholar] [CrossRef]
- Sakata, K.-I.; Maeda, K.; Sakurai, N.; Liang, S.; Nakazawa, S.; Yanagihara, K.; Kubo, T.; Yoshiyama, H.; Kitagawa, Y.; Hamada, J.-I.; et al. ADAR2 Regulates Malignant Behaviour of Mesothelioma Cells Independent of RNA-editing Activity. Anticancer Res. 2020, 40, 1307–1314. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the Biological Activity Spectra of Organic Compounds Using the Pass Online Web Resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Parasuraman, S. Prediction of activity spectra for substances. J. Pharmacol. Pharmacother. 2011, 2, 52–53. [Google Scholar] [CrossRef] [Green Version]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [Green Version]
- El-Said, K.S.; Atta, A.; Mobasher, M.A.; Germoush, M.O.; Mohamed, T.M.; Salem, M.M. Quercetin mitigates rheumatoid arthritis by inhibiting adenosine deaminase in rats. Mol. Med. 2022, 28, 24. [Google Scholar] [CrossRef]
- Li, G.; Nakagome, I.; Hirono, S.; Itoh, T.; Fujiwara, R. Inhibition of adenosine deaminase (ADA)-mediated metabolism of cordycepin by natural substances. Pharmacol. Res. Perspect. 2015, 3, e00121. [Google Scholar] [CrossRef]
- Lindell, S.D.; Maechling, S.; Klein, R.; Freigang, J.; Laber, B.; Blanazs, L.; Leonhardt, M.; Haupt, S.; Petry, T.; Sabina, R.L. Mechanism and structure based design of inhibitors of AMP and adenosine deaminase. Bioorganic Med. Chem. 2021, 43, 116272. [Google Scholar] [CrossRef]
- Lougiakis, N.; Marakos, P.; Pouli, N.; Fragopoulou, E.; Tenta, R. Synthesis of New Nebularine Analogues and Their Inhibitory Activity against Adenosine Deaminase. Chem. Pharm. Bull. 2015, 63, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Véliz, E.A.; Easterwood, L.M.; Beal, P.A. Substrate Analogues for an RNA-Editing Adenosine Deaminase: Mechanistic Investigation and Inhibitor Design. J. Am. Chem. Soc. 2003, 125, 10867–10876. [Google Scholar] [CrossRef]
- Samari, H.R.; Seglen, P.O. Inhibition of Hepatocytic Autophagy by Adenosine, Aminoimidazole-4-carboxamide Riboside, and N 6-Mercaptopurine Riboside. J. Biol. Chem. 1998, 273, 23758–23763. [Google Scholar] [CrossRef] [Green Version]
- Bojack, G.; Earnshaw, C.G.; Klein, R.; Lindell, S.D.; Lowinski, C.; Preuss, R. Design and Synthesis of Inhibitors of Adenosine and AMP Deaminases. Org. Lett. 2001, 3, 839–842. [Google Scholar] [CrossRef]
- Górska, N.; Słupski, J.; Cubała, W.J.; Wiglusz, M.S.; Gałuszko-Węgielnik, M. Antidepressants in epilepsy. Neurol. Neurochir. Pol. 2018, 52, 657–661. [Google Scholar] [CrossRef]
- Abboud, R.; Noronha, C.; Diwadkar, V.A. Motor system dysfunction in the schizophrenia diathesis: Neural systems to neurotransmitters. Eur. Psychiatry 2017, 44, 125–133. [Google Scholar] [CrossRef]
- Walther, S.; Strik, W. Motor Symptoms and Schizophrenia. Neuropsychobiology 2012, 66, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, S.; Pavlidou, A.; Walther, S. What is the potential of neurostimulation in the treatment of motor symptoms in schizophrenia? Expert Rev. Neurother. 2020, 20, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Varlet, M.; Marin, L.; Raffard, S.; Schmidt, R.C.; Capdevielle, D.; Boulenger, J.P.; Del-Monte, J.; Bardy, B.G. Impairments of social motor coordination in schizophrenia. PLoS ONE 2012, 7, e29772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posar, A.; Visconti, P. Early Motor Signs in Autism Spectrum Disorder. Children 2022, 9, 294. [Google Scholar] [CrossRef] [PubMed]
- Mohd Nordin, A.; Ismail, J.; Kamal Nor, N. Motor Development in Children With Autism Spectrum Disorder. Front. Pediatr. 2021, 9, 598276. [Google Scholar] [CrossRef]
- Tzeng, N.-S.; Hsu, Y.-H.; Ho, S.-Y.; Kuo, Y.-C.; Lee, H.-C.; Yin, Y.-J.; Chen, H.-A.; Chen, W.-L.; Chu, W.C.-C.; Huang, H.-L. Is schizophrenia associated with an increased risk of chronic kidney disease? A nationwide matched-cohort study. BMJ Open 2015, 5, e006777. [Google Scholar] [CrossRef] [Green Version]
- Tzur Bitan, D.; Krieger, I.; Berkovitch, A.; Comaneshter, D.; Cohen, A. Chronic kidney disease in adults with schizophrenia: A nationwide population-based study. Gen. Hosp. Psychiatry 2019, 58, 1–6. [Google Scholar] [CrossRef]
- Kissil, J.L.; Cohen, O.; Raveh, T.; Kimchi, A. Structure-function analysis of an evolutionary conserved protein, DAP3, which mediates TNF-α- and Fas-induced cell death. EMBO J. 1999, 18, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Kissil, J.L.; Deiss, L.P.; Bayewitch, M.; Raveh, T.; Khaspekov, G.; Kimchi, A. Isolation of DAP3, a Novel Mediator of Interferon-γ-induced Cell Death. J. Biol. Chem. 1995, 270, 27932–27936. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Ballheimer, Y.E.; Brackmann, F.; Zoglauer, D.; Geppert, C.-I.; Hartmann, A.; Trollmann, R. Seizure-induced neuronal apoptosis is related to dysregulation of the RNA-edited GluR2 subunit in the developing mouse brain. Brain Res. 2020, 1735, 146760. [Google Scholar] [CrossRef]
- Marcucci, R.; Brindle, J.; Paro, S.; Casadio, A.; Hempel, S.; Morrice, N.; Bisso, A.; Keegan, L.P.; Del Sal, G.; O’Connell, M.A. Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 2011, 30, 4211–4222. [Google Scholar] [CrossRef] [Green Version]
- Behm, M.; Wahlstedt, H.; Widmark, A.; Eriksson, M.; Öhman, M. Accumulation of nuclear ADAR2 regulates A-to-I RNA editing during neuronal development. J. Cell Sci. 2017, 130, 745–753. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wu, Y.; Yang, H.; Li, X.; Jie, M.; Hu, C.; Wu, Y.; Yang, S.; Yang, Y. Prolyl isomerase Pin1: A promoter of cancer and a target for therapy. Cell Death Dis. 2018, 9, 883. [Google Scholar] [CrossRef] [Green Version]
- Tay, D.J.T.; Song, Y.; Peng, B.; Toh, T.B.; Hooi, L.; Toh, D.-F.K.; Hong, H.; Tang, S.J.; Han, J.; Gan, W.L.; et al. Targeting RNA editing of antizyme inhibitor 1: A potential oligonucleotide-based antisense therapy for cancer. Mol. Ther. 2021, 29, 3258–3273. [Google Scholar] [CrossRef]
- Shen, X.; Wang, Y.; Han, X.; Sheng, L.; Wu, F.; Liu, X. Design, synthesis and anticancer activity of naphthoquinone derivatives. J. Enzyme Inhib. Med. Chem. 2020, 35, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Luo, Y.; Shen, G.; Piao, X.; Xu, W.; Zhang, Y.; Wang, J.; Feng, Y.; Li, J.; Zhang, Y.; et al. Two novel 1,4-naphthoquinone derivatives induce human gastric cancer cell apoptosis and cell cycle arrest by regulating reactive oxygen species-mediated MAPK/Akt/STAT3 signaling pathways. Mol. Med. Rep. 2019, 20, 2571–2582. [Google Scholar] [CrossRef] [Green Version]
- Kayashima, T.; Mori, M.; Yoshida, H.; Mizushina, Y.; Matsubara, K. 1,4-Naphthoquinone is a potent inhibitor of human cancer cell growth and angiogenesis. Cancer Lett. 2009, 278, 34–40. [Google Scholar] [CrossRef]
- Byrne, F.L.; Olzomer, E.M.; Marriott, G.R.; Quek, L.E.; Katen, A.; Su, J.; Nelson, M.E.; Hart-Smith, G.; Larance, M.; Sebesfi, V.F.; et al. Phenotypic screen for oxygen consumption rate identifies an anti-cancer naphthoquinone that induces mitochondrial oxidative stress. Redox Biol. 2020, 28, 101374. [Google Scholar] [CrossRef]
- Kumar, S.; Malachowski, W.P.; DuHadaway, J.B.; LaLonde, J.M.; Carroll, P.J.; Jaller, D.; Metz, R.; Prendergast, G.C.; Muller, A.J. Indoleamine 2,3-Dioxygenase Is the Anticancer Target for a Novel Series of Potent Naphthoquinone-Based Inhibitors. J. Med. Chem. 2008, 51, 1706–1718. [Google Scholar] [CrossRef] [Green Version]
- Presse, N.; Belleville, S.; Gaudreau, P.; Greenwood, C.E.; Kergoat, M.-J.; Morais, J.A.; Payette, H.; Shatenstein, B.; Ferland, G. Vitamin K status and cognitive function in healthy older adults. Neurobiol. Aging 2013, 34, 2777–2783. [Google Scholar] [CrossRef]
- Kiely, A.; Ferland, G.; Ouliass, B.; O’Toole, P.W.; Purtill, H.; O’Connor, E.M. Vitamin K status and inflammation are associated with cognition in older Irish adults. Nutr. Neurosci. 2020, 23, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Mokarizadeh, N.; Karimi, P.; Erfani, M.; Sadigh-Eteghad, S.; Fathi Maroufi, N.; Rashtchizadeh, N. β-Lapachone attenuates cognitive impairment and neuroinflammation in beta-amyloid induced mouse model of Alzheimer’s disease. Int. Immunopharmacol. 2020, 81, 106300. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Y.-Q.; Fang, J.-Y.; Li, P.; Li, F. Establishment of the concurrent experimental model of osteoporosis combined with Alzheimer’s disease in rat and the dual-effects of echinacoside and acteoside from Cistanche tubulosa. J. Ethnopharmacol. 2020, 257, 112834. [Google Scholar] [CrossRef] [PubMed]
- Feng, X. Selective protection of nigral dopaminergic neurons by echinacoside in a rat model of Parkinson disease induced by rotenone. J. Chin. Integr. Med. 2012, 10, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Han, G.; Xu, S.; Yuan, Y.; Zhao, C.; Ma, T. Echinacoside Suppresses Amyloidogenesis and Modulates F-actin Remodeling by Targeting the ER Stress Sensor PERK in a Mouse Model of Alzheimer’s Disease. Front. Cell Dev. Biol. 2020, 8, 593659. [Google Scholar] [CrossRef]
- Jia, C.; Shi, H.; Wu, X.; Li, Y.; Chen, J.; Tu, P. Determination of echinacoside in rat serum by reversed-phase high-performance liquid chromatography with ultraviolet detection and its application to pharmacokinetics and bioavailability. J. Chromatogr. B 2006, 844, 308–313. [Google Scholar] [CrossRef]
- Matthias, A.; Addison, R.S.; Penman, K.G.; Dickinson, R.G.; Bone, K.M.; Lehmann, R.P. Echinacea alkamide disposition and pharmacokinetics in humans after tablet ingestion. Life Sci. 2005, 77, 2018–2029. [Google Scholar] [CrossRef]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Chen, L.; Huang, C.; Shentu, J.; Wang, M.; Yan, S.; Zhou, F.; Zhang, Z.; Wang, C.; Han, Y.; Wang, Q.; et al. Indirubin Derivative 7-Bromoindirubin-3-Oxime (7Bio) Attenuates Aβ Oligomer-Induced Cognitive Impairments in Mice. Front. Mol. Neurosci. 2017, 10, 393. [Google Scholar] [CrossRef] [Green Version]
- Ribas, J.; Bettayeb, K.; Ferandin, Y.; Knockaert, M.; Garrofé-Ochoa, X.; Totzke, F.; Schächtele, C.; Mester, J.; Polychronopoulos, P.; Magiatis, P.; et al. 7-Bromoindirubin-3′-oxime induces caspase-independent cell death. Oncogene 2006, 25, 6304–6318. [Google Scholar] [CrossRef] [Green Version]
- Ribas, J.; Yuste, V.J.; Garrofé-Ochoa, X.; Meijer, L.; Esquerda, J.E.; Boix, J. 7-Bromoindirubin-3′-oxime uncovers a serine protease-mediated paradigm of necrotic cell death. Biochem. Pharmacol. 2008, 76, 39–52. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Piao, H.; Aosai, F.; Zeng, X.; Cheng, J.; Cui, Y.; Li, J.; Ma, J.; Piao, H.; Jin, X.; et al. Arctigenin protects against depression by inhibiting microglial activation and neuroinflammation via HMGB1/TLR4/NF-κB and TNF-α/TNFR1/NF-κB pathways. Br. J. Pharmacol. 2020, 177, 5224–5245. [Google Scholar] [CrossRef]
- Wei, L.; Xue, Z.; Lan, B.; Yuan, S.; Li, Y.; Guo, C.; Zhang, R.; Ding, R.; Shen, H. Arctigenin Exerts Neuroprotective Effect by Ameliorating Cortical Activities in Experimental Autoimmune Encephalomyelitis In Vivo. Front. Immunol. 2021, 12, 733101. [Google Scholar] [CrossRef]
- Xu, P.; Huang, M.-W.; Xiao, C.-X.; Long, F.; Wang, Y.; Liu, S.-Y.; Jia, W.-W.; Wu, W.-J.; Yang, D.; Hu, J.-F.; et al. Matairesinol Suppresses Neuroinflammation and Migration Associated with Src and ERK1/2-NF-κB Pathway in Activating BV2 Microglia. Neurochem. Res. 2017, 42, 2850–2860. [Google Scholar] [CrossRef]
- Mangat, H.K.; Rani, M.; Pathak, R.K.; Yadav, I.S.; Utreja, D.; Chhuneja, P.K.; Chhuneja, P. Virtual screening, molecular dynamics and binding energy-MM-PBSA studies of natural compounds to identify potential EcR inhibitors against Bemisia tabaci Gennadius. PLoS ONE 2022, 17, e0261545. [Google Scholar] [CrossRef]
- Sharma, J.; Kumar Bhardwaj, V.; Singh, R.; Rajendran, V.; Purohit, R.; Kumar, S. An in-silico evaluation of different bioactive molecules of tea for their inhibition potency against non structural protein-15 of SARS-CoV-2. Food Chem. 2021, 346, 128933. [Google Scholar] [CrossRef]
- De Vita, S.; Chini, M.G.; Bifulco, G.; Lauro, G. Insights into the Ligand Binding to Bromodomain-Containing Protein 9 (BRD9): A Guide to the Selection of Potential Binders by Computational Methods. Molecules 2021, 26, 7192. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Asiedu, S.O.; Kwofie, S.K.; Broni, E.; Wilson, M.D. Computational Identification of Potential Anti-Inflammatory Natural Compounds Targeting the p38 Mitogen-Activated Protein Kinase (MAPK): Implications for COVID-19-Induced Cytokine Storm. Biomolecules 2021, 11, 653. [Google Scholar] [CrossRef]
- Sgobba, M.; Caporuscio, F.; Anighoro, A.; Portioli, C.; Rastelli, G. Application of a post-docking procedure based on MM-PBSA and MM-GBSA on single and multiple protein conformations. Eur. J. Med. Chem. 2012, 58, 431–440. [Google Scholar] [CrossRef]
- Rifai, E.A.; Van Dijk, M.; Vermeulen, N.P.E.; Yanuar, A.; Geerke, D.P. A Comparative Linear Interaction Energy and MM/PBSA Study on SIRT1−Ligand Binding Free Energy Calculation. J. Chem. Inf. Model. 2019, 59, 4018–4033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Borkotoky, S.; Meena, C.K.; Murali, A. Interaction Analysis of T7 RNA Polymerase with Heparin and Its Low Molecular Weight Derivatives—An in Silico Approach. Bioinform. Biol. Insights 2016, 10, BBI.S40427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Pergolizzi, J.V.; Dahan, A.; Ann LeQuang, J.; Raffa, R.B. Overdoses due to fentanyl and its analogues (F/FAs) push naloxone to the limit. J. Clin. Pharm. Ther. 2021, 46, 1501–1504. [Google Scholar] [CrossRef]
- Hesselbarth, S.; Löwenstein, O.; Cegla, T. Effects of prolonged-release oxycodone/naloxone on pain control, bowel function and quality of life: A prospective observational study. Scand. J. Pain 2014, 5, 75–81. [Google Scholar] [CrossRef]
- DeLano, W.L. Unraveling hot spots in binding interfaces: Progress and challenges. Curr. Opin. Struct. Biol. 2002, 12, 14–20. [Google Scholar] [CrossRef]
- Zhong, S.; Macias, A.; MacKerell, A. Computational Identification of Inhibitors of Protein-Protein Interactions. Curr. Top. Med. Chem. 2007, 7, 63–82. [Google Scholar] [CrossRef] [Green Version]
- Grosdidier, S.; Fernández-Recio, J. Identification of hot-spot residues in protein-protein interactions by computational docking. BMC Bioinform. 2008, 9, 447. [Google Scholar] [CrossRef] [Green Version]
- Kwofie, S.; Dankwa, B.; Enninful, K.; Adobor, C.; Broni, E.; Ntiamoah, A.; Wilson, M. Molecular Docking and Dynamics Simulation Studies Predict Munc18b as a Target of Mycolactone: A Plausible Mechanism for Granule Exocytosis Impairment in Buruli Ulcer Pathogenesis. Toxins 2019, 11, 181. [Google Scholar] [CrossRef] [Green Version]
- Silhol, S.; Glin, L.; Gottesmann, C. Study of the 5-HT2 antagonist ritanserin on sleep-waking cycle in the rat. Pharmacol. Biochem. Behav. 1992, 41, 241–243. [Google Scholar] [CrossRef]
- Mayer, G. Ritanserin Improves Sleep Quality in Narcolepsy. Pharmacopsychiatry 2003, 36, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.U.; Brandenberger, G.; Toussaint, M.; Bouhours, P.; Paul Macher, J.; Luthringer, R. Ritanserin, a serotonin-2 receptor antagonist, improves ultradian sleep rhythmicity in young poor sleepers. Clin. Neurophysiol. 2002, 113, 429–434. [Google Scholar] [CrossRef]
- Henderson, J.; Yiannikas, C.; Graham, J.S. Effect of ritanserin, a highly selective 5-HT2 receptor antagonist, on Parkinson’s disease. Clin. Exp. Neurol. 1992, 29, 277–282. [Google Scholar]
- Poyurovsky, M.; Shardorodsky, M.; Fuchs, C.; Schneidman, M.; Weizman, A. Treatment of neuroleptic-induced akathisia with the 5-HT 2 antagonist mianserin. Br. J. Psychiatry 1999, 174, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Barone, J.A.; Bierman, R.H.; Cornish, J.W.; Hsuan, A.; Drake, N.D.; Colaizzi, J.L. Safety Evaluation of Ritanserin—An Investigational Serotonin Antagonist. Drug Intell. Clin. Pharm. 1986, 20, 770–775. [Google Scholar] [CrossRef]
- Campbell, S.T.; Franks, C.E.; Borne, A.L.; Shin, M.; Zhang, L.; Hsu, K.-L. Chemoproteomic Discovery of a Ritanserin-Targeted Kinase Network Mediating Apoptotic Cell Death of Lung Tumor Cells. Mol. Pharmacol. 2018, 94, 1246–1255. [Google Scholar] [CrossRef]
- Wesensten, N.J. Role of Pharmacological Interventions for Sleep Deprivation. Encycl. Sleep 2013, 366–370. [Google Scholar] [CrossRef]
- Daoui, S.; Kansiz, S.; Aktas, F.A.; Dege, N.; Saif, E.; Benchat, N.; Karrouchi, K. Crystal structure and molecular docking study of diethyl 2,2′-({[(1 E, 1′ E)-(hydrazine-1,2-diylidene)bis(methanylylidene)]bis(4,1-phenylene)}bis(oxy))diacetate. Acta Crystallogr. Sect. E Crystallogr. Commun. 2022, 78, 88–91. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [Green Version]
- Rutz, A.; Sorokina, M.; Galgonek, J.; Mietchen, D.; Willighagen, E.; Gaudry, A.; Graham, J.G.; Stephan, R.; Page, R.; Vondrášek, J.; et al. The LOTUS initiative for knowledge sharing in Natural Products research. In Proceedings of the GA—69th Annual Meeting 2021, Virtual, 5–8 September 2021. [Google Scholar]
- Nakamura, Y.; Mochamad Afendi, F.; Kawsar Parvin, A.; Ono, N.; Tanaka, K.; Hirai Morita, A.; Sato, T.; Sugiura, T.; Altaf-Ul-Amin, M.; Kanaya, S. KNApSAcK Metabolite Activity Database for Retrieving the Relationships Between Metabolites and Biological Activities. Plant Cell Physiol. 2014, 55, e7. [Google Scholar] [CrossRef] [Green Version]
- Afendi, F.M.; Okada, T.; Yamazaki, M.; Hirai-Morita, A.; Nakamura, Y.; Nakamura, K.; Ikeda, S.; Takahashi, H.; Altaf-Ul-Amin, M.; Darusman, L.K.; et al. KNApSAcK family databases: Integrated metabolite-plant species databases for multifaceted plant research. Plant Cell Physiol. 2012, 53, e1. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Zhang, P.; He, W.; Qin, C.; Chen, S.; Tao, L.; Wang, Y.; Tan, Y.; Gao, D.; Wang, B.; et al. NPASS: Natural product activity and species source database for natural product research, discovery and tool development. Nucleic Acids Res. 2018, 46, D1217–D1222. [Google Scholar] [CrossRef] [Green Version]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.P.B.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry And Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef] [Green Version]
- Broni, E.; Ashley, C.; Adams, J.; Manu, H.; Aikins, E.; Okom, M.; Miller, W.A.; Wilson, M.D.; Kwofie, S.K. Cheminformatics-Based Study Identifies Potential Ebola VP40 Inhibitors. Int. J. Mol. Sci. 2023, 24, 6298. [Google Scholar] [CrossRef]
- Baskaran, M.; Shanmugam, L.; Raman, V. Effect of Plumbago zeylanica administration on brain neurotransmitter level in Wistar albino rats. J. Appl. Pharm. Sci. 2015, 5, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.-C.; Yang, L.-L.; Chou, C.-J. Cytotoxic naphthoquinones and plumbagic acid glucosides from Plumbago zeylanica. Phytochemistry 2003, 62, 619–622. [Google Scholar] [CrossRef]
- Sreelatha, T.; Hymavathi, A.; Murthy, J.M.; Rani, P.U.; Rao, J.M.; Babu, K.S. Bioactivity-guided isolation of mosquitocidal constituents from the rhizomes of Plumbago capensis Thunb. Bioorg. Med. Chem. Lett. 2010, 20, 2974–2977. [Google Scholar] [CrossRef]
- Gu, J.-Q.; Graf, T.N.; Lee, D.; Chai, H.-B.; Mi, Q.; Kardono, L.B.S.; Setyowati, F.M.; Ismail, R.; Riswan, S.; Farnsworth, N.R.; et al. Cytotoxic and Antimicrobial Constituents of the Bark of Diospyros ma ritima Collected in Two Geographical Locations in Indonesia. J. Nat. Prod. 2004, 67, 1156–1161. [Google Scholar] [CrossRef]
- Higa, M.; Takashima, Y.; Yokaryo, H.; Harie, Y.; Suzuka, T.; Ogihara, K. Naphthoquinone Derivatives from Diospyros maritima. Chem. Pharm. Bull. 2017, 65, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Higa, M.; Noha, N.; Yokaryo, H.; Ogihara, K.; Yogi, S. Three New Naphthoquinone Derivatives from Diospyros maritima Blume. Chem. Pharm. Bull. 2002, 50, 590–593. [Google Scholar] [CrossRef]
- Sobhani, M.; Abbas-Mohammadi, M.; Ebrahimi, S.N.; Aliahmadi, A. Tracking leading anti-candida compounds in plant samples; Plumbago europaea. Iran. J. Microbiol. 2018, 10, 187–193. [Google Scholar]
- Sankaram, A.V.B.; Rao, A.S.; Shoolery, J.N. Zeylanone and Isozeylanone, two novel quinones from Plumbago Zeylanica. Tetrahedron 1979, 35, 1777–1782. [Google Scholar] [CrossRef]
- Wang, Y.C.; Huang, T.L. Anti-Helicobacter pylori activity of Plumbago zeylanica L. FEMS Immunol. Med. Microbiol. 2005, 43, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Mallavadhani, U.V.; Panda, A.K.; Rao, Y.R. Review article number 134 pharmacology and chemotaxonomy of diospyros. Phytochemistry 1998, 49, 901–951. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.C.; Costa, M.A.C.; Paul, M.I. Naphthaquinones of Diospyros batocana. Planta Med. 1983, 47, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Musgrave, O.C.; Skoyles, D. Ebenaceae extractives. Part IV. Diosindigo A, a blue pigment from several Diospyros species. J. Chem. Soc. Perkin Trans. 1974, 1, 1128. [Google Scholar] [CrossRef]
- Tezuka, M.; Kuroyanagi, M.; Yoshihira, K.; Natori, S. Naphthoquinone Derivatives from the Ebenaceae. IV. Naphthoquinone Derivatives from Diospyros kaki THUNB. and D. kaki THUNB. var. sylvestris MAKINO. Chem. Pharm. Bull. 1972, 20, 2029–2035. [Google Scholar] [CrossRef] [Green Version]
- Yoshihira, K.; Tezuka, M.; Natori, S. Naphthoquinone derivatives from SPP.: Bisisodiospyrin, a tetrameric naphthoquinone. Tetrahedron Lett. 1970, 11, 7–10. [Google Scholar] [CrossRef]
- YOSHIHIRA, K.; TEZUKA, M.; NATORI, S. Naphthoquinone Derivatives from the Ebenaceae. II. Isodiospyrin, Bisisodiospyrin, and Mamegakinone from Diospyros lotus L. and D. morrisiana HANCE. Chem. Pharm. Bull. 1971, 19, 2308–2313. [Google Scholar] [CrossRef] [Green Version]
- Marston, A.; Msonthi, J.; Hostettmann, K. Naphthoquinones of Diospyros usambarensis; their Molluscicidal and Fungicidal Activities. Planta Med. 1984, 50, 279–280. [Google Scholar] [CrossRef]
- Creveld, M. Diospyros kaki—Der Weltbaum—Ein neues homöopathisches Mittel. Allg. Homöopathische Zeitung 2004, 249, 246–250. [Google Scholar] [CrossRef]
- Miyase, T. Antioxidants from Lespedeza homoloba. (I). Phytochemistry 1999, 52, 303–310. [Google Scholar] [CrossRef]
- Onegi, B.; Kraft, C.; Köhler, I.; Freund, M.; Jenett-Siems, K.; Siems, K.; Beyer, G.; Melzig, M.F.; Bienzle, U.; Eich, E. Antiplasmodial activity of naphthoquinones and one anthraquinone from Stereospermum kunthianum. Phytochemistry 2002, 60, 39–44. [Google Scholar] [CrossRef]
- Mohn, T.; Plitzko, I.; Hamburger, M. A comprehensive metabolite profiling of Isatis tinctoria leaf extracts. Phytochemistry 2009, 70, 924–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.Y.; Leung, C.Y.; Wong, C.K.C.; Shen, X.L.; Wong, R.N.S.; Cai, Z.W.; Mak, N.K. Bisindigotin, a TCDD Antagonist from the Chinese Medicinal Herb Isatis indigotica. J. Nat. Prod. 2005, 68, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Bergman, J.; Lindström, J.-O.; Tilstam, U. The structure and properties of some indolic constituents in Couroupita guianensis aubl. Tetrahedron 1985, 41, 2879–2881. [Google Scholar] [CrossRef]
- Hirschmann, G.S.; Ferro, E. Indigo from Eupatorium laeve. J. Ethnopharmacol. 1989, 26, 93–94. [Google Scholar] [CrossRef]
- Hamburger, M. Isatis tinctoria—From the rediscovery of an ancient medicinal plant towards a novel anti-inflammatory phytopharmaceutical. Phytochem. Rev. 2002, 1, 333–344. [Google Scholar] [CrossRef]
- Chung, Y.-C.; Tang, F.-Y.; Liao, J.-W.; Chung, C.-H.; Jong, T.-T.; Chen, S.-S.; Tsai, C.-H.; Chiang, E.-P. Isatis indigotica Induces Hepatocellular Cancer Cell Death via Caspase-Independent Apoptosis-Inducing Factor Translocation Apoptotic Pathway In Vitro and In Vivo. Integr. Cancer Ther. 2011, 10, 201–214. [Google Scholar] [CrossRef]
- Xi, Y.-F.; Lou, L.-L.; Han, F.-Y.; Liu, S.-F.; Yao, G.-D.; Lin, B.; Huang, X.-X.; Wang, X.-B.; Song, S.-J. Four pairs of alkaloid enantiomers from Isatis indigotica Fortune Ex Land with neuroprotective effects against H2O2-induced SH-SY5Y cell injury. Bioorg. Chem. 2020, 96, 103650. [Google Scholar] [CrossRef]
- Liu, S.F.; Zhang, Y.Y.; Zhou, L.; Lin, B.; Huang, X.X.; Wang, X.B.; Song, S.J. Alkaloids with neuroprotective effects from the leaves of Isatis indigotica collected in the Anhui Province, China. Phytochemistry 2018, 149, 132–139. [Google Scholar] [CrossRef]
- Xi, Y.-F.; Lou, L.-L.; Han, F.-Y.; Wang, X.-B.; Huang, X.-X.; Yao, G.-D.; Song, S.-J. Discovery of alkaloids from the leaves of Isatis indigotica Fortune with neuroprotective activity. Chin. J. Nat. Med. 2021, 19, 680–685. [Google Scholar] [CrossRef]
- Xi, Y.-F.; Liu, S.-F.; Hong, W.; Song, X.-Y.; Lou, L.-L.; Zhou, L.; Yao, G.-D.; Lin, B.; Wang, X.-B.; Huang, X.-X.; et al. Discovery of cycloneolignan enantiomers from Isatis indigotica Fortune with neuroprotective effects against MPP+-induced SH-SY5Y cell injury. Bioorg. Chem. 2019, 88, 102926. [Google Scholar] [CrossRef]
- Xi, Y.-F.; Lou, L.-L.; Xu, Z.-Y.; Hou, Z.-L.; Wang, X.-B.; Huang, X.-X.; Song, S.-J. Alkaloid Enantiomers from Isatis tinctoria with Neuroprotective Effects against H2O2-Induced SH-SY5Y Cell Injury. Planta Med. 2019, 85, 1374–1382. [Google Scholar] [CrossRef]
- Shen, C.-C.; Ni, C.-L.; Huang, Y.-L.; Huang, R.-L.; Chen, C.-C. Furanolabdane Diterpenes from Hypoestes p urpurea. J. Nat. Prod. 2004, 67, 1947–1949. [Google Scholar] [CrossRef]
- Mohagheghzadeh, A.; Schmidt, T.; Bayindir, Ü.; Fuss, E.; Mehregan, I.; Alfermann, A. Diarylbutyrolactone Lignans from Linum corymbulosum in vitro Cultures. Planta Med. 2006, 72, 1165–1167. [Google Scholar] [CrossRef]
- Jim-Min, F.; Ching-Kuo, L.; Yu-Shia, C. Lignans from leaves of Juniperus chinensis. Phytochemistry 1992, 31, 3659–3661. [Google Scholar] [CrossRef]
- Niwa, A.M.; Marcarini, J.C.; Sartori, D.; Maistro, E.L.; Mantovani, M.S. Effects of (−)-cubebin (Piper cubeba) on cytotoxicity, mutagenicity and expression of p38 MAP kinase and GSTa2 in a hepatoma cell line. J. Food Compos. Anal. 2013, 30, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Rajalekshmi, D.S.; Kabeer, F.A.; Madhusoodhanan, A.R.; Bahulayan, A.K.; Prathapan, R.; Prakasan, N.; Varughese, S.; Nair, M.S. Anticancer activity studies of cubebin isolated from Piper cubeba and its synthetic derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 1767–1771. [Google Scholar] [CrossRef]
- Somani, G.S.; Nahire, M.S.; Parikh, A.D.; Mulik, M.B.; Ghumatkar, P.J.; Laddha, K.S.; Sathaye, S. Neuroprotective effect of Cubebin: A dibenzylbutyrolactone lignan on scopolamine-induced amnesia in mice. Indian J. Med. Res. 2017, 146, 255–259. [Google Scholar] [CrossRef]
- Bucciantini, M.; Leri, M.; Nardiello, P.; Casamenti, F.; Stefani, M. Olive Polyphenols: Antioxidant and Anti-Inflammatory Properties. Antioxidants 2021, 10, 1044. [Google Scholar] [CrossRef]
- Batista, Â.G.; Ferrari, A.S.; Da Cunha, D.C.; Da Silva, J.K.; Cazarin, C.B.B.; Correa, L.C.; Prado, M.A.; De Carvalho-Silva, L.B.; Esteves, E.A.; Maróstica Júnior, M.R. Polyphenols, antioxidants, and antimutagenic effects of Copaifera langsdorffii fruit. Food Chem. 2016, 197, 1153–1159. [Google Scholar] [CrossRef]
- Chen, G.-L.; Munyao Mutie, F.; Xu, Y.-B.; Saleri, F.D.; Hu, G.-W.; Guo, M.-Q. Antioxidant, Anti-inflammatory Activities and Polyphenol Profile of Rhamnus prinoides. Pharmaceuticals 2020, 13, 55. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Li, M.; Cao, Y.; Akan, O.D.; Guo, T.; Luo, F. Targeting AMPK Signaling by Dietary Polyphenols in Cancer Prevention. Mol. Nutr. Food Res. 2022, 66, 2100732. [Google Scholar] [CrossRef] [PubMed]
- Prakash, M.D.; Stojanovska, L.; Feehan, J.; Nurgali, K.; Donald, E.L.; Plebanski, M.; Flavel, M.; Kitchen, B.; Apostolopoulos, V. Anti-cancer effects of polyphenol-rich sugarcane extract. PLoS ONE 2021, 16, e0247492. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y.; Shida, Y.; Hakariya, T.; Sakai, H. Anti-Cancer Effects of Green Tea Polyphenols Against Prostate Cancer. Molecules 2019, 24, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakagami, H.; Shi, H.; Bandow, K.; Tomomura, M.; Tomomura, A.; Horiuchi, M.; Fujisawa, T.; Oizumi, T. Search of neuroprotective polyphenols using the “overlay” isolation method. Molecules 2018, 23, 1840. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, Y.; Li, J.; Fu, C.; Zhang, X. The Neuroprotective Effect of Tea Polyphenols on the Regulation of Intestinal Flora. Molecules 2021, 26, 3692. [Google Scholar] [CrossRef]
- Spagnuolo, C.; Napolitano, M.; Tedesco, I.; Moccia, S.; Milito, A.; Luigi Russo, G. Neuroprotective Role of Natural Polyphenols. Curr. Top. Med. Chem. 2016, 16, 1943–1950. [Google Scholar] [CrossRef]
- Galli, F. Interactions of Polyphenolic Compounds with Drug Disposition and Metabolism. Curr. Drug Metab. 2007, 8, 830–838. [Google Scholar] [CrossRef]
- Joshua, D.L.; Sang, S.; Anthony, Y.; Lu, H.; Chung, S. Yang Metabolism of Dietary Polyphenols and Possible Interactions with Drugs. Curr. Drug Metab. 2007, 8, 499–507. [Google Scholar] [CrossRef]
- Chalopin, M.; Tesse, A.; Martínez, M.C.; Rognan, D.; Arnal, J.F.; Andriantsitohaina, R. Estrogen receptor alpha as a key target of red wine polyphenols action on the endothelium. PLoS ONE 2010, 5, e8554. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, M.; Hazarika, J.; Sarma, S.; Bhuyan, P.; Mahanta, R. Estrogen receptor modulation of some polyphenols extracted from Daucus carota as a probable mechanism for antifertility effect: An in silico study. J. Theor. Comput. Chem. 2020, 19, 2041004. [Google Scholar] [CrossRef]
- Peng, N.; Clark, J.T.; Prasain, J.; Kim, H.; White, C.R.; Wyss, J.M. Antihypertensive and cognitive effects of grape polyphenols in estrogen-depleted, female, spontaneously hypertensive rats. Am. J. Physiol. Integr. Comp. Physiol. 2005, 289, R771–R775. [Google Scholar] [CrossRef]
- Cipolletti, M.; Solar Fernandez, V.; Montalesi, E.; Marino, M.; Fiocchetti, M. Beyond the Antioxidant Activity of Dietary Polyphenols in Cancer: The Modulation of Estrogen Receptors (ERs) Signaling. Int. J. Mol. Sci. 2018, 19, 2624. [Google Scholar] [CrossRef] [Green Version]
- Layrisse, M.; García-Casal, M.N.; Solano, L.; Barón, M.A.; Arguello, F.; Llovera, D.; Ramírez, J.; Leets, I.; Tropper, E. Iron Bioavailability in Humans from Breakfasts Enriched with Iron Bis-Glycine Chelate, Phytates and Polyphenols. J. Nutr. 2000, 130, 2195–2199. [Google Scholar] [CrossRef] [Green Version]
- Dabbagh-Bazarbachi, H.; Clergeaud, G.; Quesada, I.M.; Ortiz, M.; O’Sullivan, C.K.; Fernández-Larrea, J.B. Zinc ionophore activity of quercetin and epigallocatechin-gallate: From hepa 1-6 cells to a liposome model. J. Agric. Food Chem. 2014, 62, 8085–8093. [Google Scholar] [CrossRef]
- Ozdal, T.; Sela, D.A.; Xiao, J.; Boyacioglu, D.; Chen, F.; Capanoglu, E. The reciprocal interactions between polyphenols and gut microbiota and effects on bioaccessibility. Nutrients 2016, 8, 78. [Google Scholar] [CrossRef] [Green Version]
- Mithul Aravind, S.; Wichienchot, S.; Tsao, R.; Ramakrishnan, S.; Chakkaravarthi, S. Role of dietary polyphenols on gut microbiota, their metabolites and health benefits. Food Res. Int. 2021, 142, 110189. [Google Scholar] [CrossRef]
- Zhao, Y.; Jiang, Q. Roles of the Polyphenol–Gut Microbiota Interaction in Alleviating Colitis and Preventing Colitis-Associated Colorectal Cancer. Adv. Nutr. 2021, 12, 546–565. [Google Scholar] [CrossRef]
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Kanhere, H.S.; Rahangdale, Y.U.; Bodele, A.S.; Wadhwani, D.I.; Ghoshewar, A.R.; Karande, S.P. Karande Neurological disorders associated with impaired gut microbiota. GSC Biol. Pharm. Sci. 2021, 15, 29–39. [Google Scholar] [CrossRef]
- Tomás-Barberán, F. Interaction of polyphenols with gut microbiota: Role in human health. Planta Med. 2014, 80, PL7. [Google Scholar] [CrossRef]
- Zhang, Z.; Xie, L.; Ju, Y.; Dai, Y. Recent Advances in Metal-Phenolic Networks for Cancer Theranostics. Small 2021, 17, 2100314. [Google Scholar] [CrossRef] [PubMed]
- Repasky, M.P.; Shelley, M.; Friesner, R.A. Flexible Ligand Docking with Glide. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007. [Google Scholar]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Methods in Molecular Biology; Humana Press: Clifton, NJ, USA, 2015; Volume 1263, pp. 243–250. ISBN 9781118435762. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, EfficientOptimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Di Costanzo, L.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [CrossRef] [PubMed]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Chen, C.Y.-C. TCM Database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [Green Version]
- Kwofie, S.; Broni, E.; Yunus, F.; Nsoh, J.; Adoboe, D.; Miller, W.; Wilson, M. Molecular Docking Simulation Studies Identifies Potential Natural Product Derived-Antiwolbachial Compounds as Filaricides against Onchocerciasis. Biomedicines 2021, 9, 1682. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Chaput, L.; Mouawad, L. Efficient conformational sampling and weak scoring in docking programs? Strategy of the wisdom of crowds. J. Cheminform. 2017, 9, 1–18. [Google Scholar] [CrossRef]
- Gimeno, A.; Mestres-Truyol, J.; Ojeda-Montes, M.J.; Macip, G.; Saldivar-Espinoza, B.; Cereto-Massagué, A.; Pujadas, G.; Garcia-Vallvé, S. Prediction of novel inhibitors of the main protease (M-pro) of SARS-CoV-2 through consensus docking and drug reposition. Int. J. Mol. Sci. 2020, 21, 3793. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein–Ligand Interactions in Molecular Docking. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Dankwa, B.; Broni, E.; Enninful, K.S.; Kwofie, S.K.; Wilson, M.D. Consensus docking and MM-PBSA computations identify putative furin protease inhibitors for developing potential therapeutics against COVID-19. Struct. Chem. 2022, 33, 2221–2241. [Google Scholar] [CrossRef]
- Thomas, B.N.; Parrill, A.L.; Baker, D.L. Self-docking and cross-docking simulations of G protein-coupled receptor-ligand complexes: Impact of ligand type and receptor activation state. J. Mol. Graph. Model. 2022, 112, 108119. [Google Scholar] [CrossRef]
- Kwofie, S.K.; Dankwa, B.; Odame, E.A.; Agamah, F.E.; Doe, L.P.; Teye, J.; Agyapong, O.; Miller, W.; Mosi, L.; Wilson, M.D. In Silico Screening of Isocitrate Lyase for Novel Anti-Buruli Ulcer Natural Products Originating from Africa. Molecules 2018, 23, 1550. [Google Scholar] [CrossRef] [Green Version]
- Jaundoo, R.; Bohmann, J.; Gutierrez, G.; Klimas, N.; Broderick, G.; Craddock, T. Using a Consensus Docking Approach to Predict Adverse Drug Reactions in Combination Drug Therapies for Gulf War Illness. Int. J. Mol. Sci. 2018, 19, 3355. [Google Scholar] [CrossRef] [Green Version]
- Kapale, S.S.; Mali, S.N.; Chaudhari, H.K. Molecular modelling studies for 4-oxo-1,4-dihydroquinoline-3-carboxamide derivatives as anticancer agents. Med. Drug Discov. 2019, 2, 100008. [Google Scholar] [CrossRef]
- Hu, J.; Liu, Z.; Yu, D.J.; Zhang, Y. LS-align: An atom-level, flexible ligand structural alignment algorithm for high-throughput virtual screening. Bioinformatics 2018, 34, 2209–2218. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Seeliger, D.; De Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided. Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [Green Version]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. Consensus Scoring: A Method for Obtaining Improved Hit Rates from Docking Databases of Three-Dimensional Structures into Proteins. J. Med. Chem. 1999, 42, 5100–5109. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shortridge, M.D.; Bokemper, M.; Copeland, J.C.; Stark, J.L.; Powers, R. Correlation between Protein Function and Ligand Binding Profiles. J. Proteome Res. 2011, 10, 2538–2545. [Google Scholar] [CrossRef] [Green Version]
- Thorat, B.R.; Mali, S.N.; Rani, D.; Yamgar, R.S. Synthesis, In silico and In vitro Analysis of Hydrazones as Potential Antituberculosis Agents. Curr. Comput. Aided. Drug Des. 2021, 17, 294–306. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.; Viet, M.H.; Li, M.S. Effects of water models on binding affinity: Evidence from all-atom simulation of binding of tamiflu to A/H5N1 neuraminidase. Sci. World J. 2014, 2014, 536084. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Liu, L.; Zhao, L.; Wang, J. Effects of different force fields and temperatures on the structural character of abeta (12-28) peptide in aqueous solution. Int. J. Mol. Sci. 2011, 12, 8259–8274. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; McCorvy, J.D.; Harpsøe, K.; Lansu, K.; Yuan, S.; Popov, P.; Qu, L.; Pu, M.; Che, T.; Nikolajsen, L.F.; et al. 5-HT2C Receptor Structures Reveal the Structural Basis of GPCR Polypharmacology. Cell 2018, 172, 719–730.e14. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pocket No. | Area (Å2) | Volume (Å3) | Residues Lining the Pocket |
|---|---|---|---|
| 1 | 397.972 | 455.471 | Lys350, Val351, Gly374, Thr375, Lys376, Cys377, Ile378, Asn379, His394, Ala395, Glu396, Ile446, Thr448, Ser449, Pro450, Cys451, Gly452, Arg455, Ile456, Pro459, Lys483, Ile484, Glu485, Ser486, Gly487, Gln488, Gly489, Thr490, Leu511, Thr513, Cys516, Arg590, Lys594, and Ala595. |
| 2 | 541.281 | 342.976 | Ala389, Leu390, Asn391, Asp392, Ile397, Arg400, Arg401, Leu404, Tyr408, Gln500, Leu512, Thr513, Met514, Lys519, Arg522, Trp523, Val526, Gly527, Ile528, Gln529, Gly530, Ser531, Leu532, Leu533, Lys629, Leu632, Tyr658, His659, Lys662, Leu663, Tyr668, Gln669, Lys672, Phe676, Trp687, Val688, Glu689, Lys690, Pro691, Thr692, Gln694, and Asp695. |
| 3 | 120.812 | 129.477 | Ser458, His460, Glu461, Pro462, Ile463, Glu466, Pro467, Ala468, Asp469, Arg470, His471, His552, Asp554, and His555. |
| Compound | Binding Energy (kcal/mol) | Interacting Residues | ||||
|---|---|---|---|---|---|---|
| AutoDock Vina | Glide | Consensus Score | H-Bond | Pi-Cation | Salt Bridges | |
| ZINC000044417732 | −10.9 | −7.95 | −9.42 | Arg401, Leu532, Lys662 and Glu689 | Arg400 and Lys662 | Arg400, Arg401, Lys629 and Lys662 |
| ZINC000085950180 | −10.5 | −7.88 | −9.19 | Arg401, Ser531 (2), Lys629, Trp687 and Asp695 | Lys662 | - |
| ZINC000085511995 | −10.9 | −7.27 | −9.08 | Arg522, Lys629 and Trp687 | Arg400 and Lys662 | Arg401, Lys519, Lys629, Lys662 and Lys690 |
| ZINC000085850673 | −10.5 | −7.24 | −8.87 | Arg401, Arg522, Lys629, Tyr658 and Lys690 | Lys519 and Lys629. | Lys519 and Lys690 |
| ZINC000085996580 | −11 | −6.62 | −8.81 | Arg401 (2), Lys519 and Lys690 | Arg400, Arg522 and Lys662 (4) | - |
| ZINC000085734971 | −10.6 | −6.93 | −8.76 | Arg401, Ser531, Lys629, Lys662 (2) and Asp695 | Lys629 | - |
| ZINC000034517814 | −9.4 | −8.11 | −8.75 | Met514, Arg522, Ser531, Glu689 (2) and Lys690 | - | - |
| ZINC000014613520 | −9.9 | −7.39 | −8.64 | Arg401, Ser531, Lys662 and Trp687 | Arg522 and Lys662 | Arg522 |
| ZINC000095911588 | −9.4 | −7.86 | −8.63 | Arg401, Ser531, Arg522, Trp523 and Lys629 | Lys662 | Arg400 |
| ZINC000085569519 | −10.1 | −6.74 | −8.42 | Tyr408, Lys629, Tyr658, Lys662 and Lys690 | - | - |
| ZINC000008234342 | −9.7 | −7.08 | −8.39 | Leu532 | Arg400 and Lys662 | Arg400, Arg401 and Lys629 |
| ZINC000085569292 | −9.3 | −7.48 | −8.39 | Arg400 (2), Arg401 and Lys662 | - | Lys519 and Lys690 |
| ZINC000095911414 | −10.1 | −6.65 | −8.38 | Arg401 (2), Ser531, Lys629 and Trp687 | Arg400 and Lys662 | - |
| ZINC000086050572 | −9.8 | −6.94 | −8.37 | Arg401, Ser531, Lys629, Lys662 (2) and Asp695 | - | - |
| ZINC000014612330 | −10.2 | −6.51 | −8.36 | Arg401, Ser531 and Lys629 | - | - |
| ZINC000014814624 | −10.0 | −6.57 | −8.28 | Arg522, Ser531 and Lys690 | Arg400, Arg522 and Lys662 | Arg400, Arg401 and Lys629 |
| ZINC000004098700 | −9.0 | −7.50 | −8.25 | Arg401 (2), Arg522 and Ser531 | Arg400 and Lys662 | Lys519 |
| ZINC000095912516 | −9.8 | −6.57 | −8.18 | Arg401, Arg522, Leu532, Lys629, Tyr658 and Glu689 | - | - |
| ZINC000085488788 | −9.5 | −6.84 | −8.17 | Arg401, Ser531 and Lys629 | Lys519 | Arg401, Lys629 (2) and Lys662 |
| ZINC000070454227 | −9.0 | −7.34 | −8.17 | Arg401, Arg522, Ser531 and Lys690 | Lys662 | Lys519 and Lys690 |
| IHP | −8.6 | −7.97 | −8.26 | Asn391, Arg400, Arg401 (2), Lys519, Ser531, Lys672, Trp687, Val688, Glu689 and Lys690 | - | Arg401, Lys519, Arg522, Lys629, Lys662, Lys672 and Lys690 |
| Compound | MW (g/mol) | logP o/w | TPSA (Å2) | BBB Permeant | GI Absorption | ESOL Solubility Class | No of Lipinski’s Rule Violations | No. of Veber’s Rule Violations |
|---|---|---|---|---|---|---|---|---|
| IHP | 660.04 | –6.77 | 459.42 | No | Low | High | 3 | 2 |
| ZINC000044417732 | 374.34 | 2.88 | 108.74 | No | High | Moderate | 0 | 0 |
| ZINC000085950180 | 374.34 | 2.71 | 108.74 | No | High | Moderate | 0 | 0 |
| ZINC000085511995 | 374.34 | 2.82 | 108.74 | No | High | Moderate | 0 | 0 |
| ZINC000085850673 | 374.34 | 2.9 | 108.74 | No | High | Moderate | 0 | 0 |
| ZINC000085996580 | 508.48 | 4.47 | 136.66 | No | Low | Poor | 1 | 0 |
| ZINC000085734971 | 402.4 | 3.39 | 108.74 | Yes | High | Moderate | 0 | 0 |
| ZINC000014612330 | 324.37 | 3.05 | 63.6 | Yes | High | Moderate | 0 | 0 |
| ZINC000100513617 | 262.26 | 2.16 | 58.2 | Yes | High | Moderate | 0 | 0 |
| ZINC000013462928 | 352.34 | 3.22 | 63.22 | No | High | Moderate | 0 | 0 |
| Fluoxetine | 309.33 | 4.32 | 21.26 | Yes | High | Moderate | 0 | 0 |
| Nebularine | 252.23 | −1.16 | 113.52 | No | High | Very | 0 | 0 |
| Doxorubicin | 543.52 | 0.44 | 206.07 | No | Low | Soluble | 3 | 1 |
| Compound | vdW | Electrostatic Energy | Polar Solvation Energy | SASA Energy | Binding Energy |
|---|---|---|---|---|---|
| IHP | −138.816 ± 2.243 | −1597.111 ± 7.065 | 883.140 ± 5.474 | −20.866 ± 0.095 | −873.873 ± 6.225 |
| ZINC000044417732 | −164.45 ± 1.303 | −798.403 ± 4.671 | 340.231 ± 3.618 | −19.937 ± 0.089 | −642.856 ± 3.746 |
| ZINC000085950180 | −172.457 ± 1.278 | −66.957 ± 2.459 | 123.492 ± 2.987 | −19.151 ± 0.101 | −135.075 ± 3.285 |
| ZINC000085511995 | −157.3 ± 1.578 | −1550.773 ± 5.372 | 659.676 ± 5.323 | −19.816 ± 0.108 | −1068.26 ± 4.122 |
| ZINC000085850673 | −170.78 ± 1.79 | −818.946 ± 5.622 | 357.645 ± 5.284 | −18.914 ± 0.09 | −650.863 ± 4.925 |
| ZINC000085996580 | −174.655 ± 1.248 | −87.798 ± 1.888 | 203.977 ± 2.763 | −22.88 ± 0.144 | −81.304 ± 2.269 |
| ZINC000085734971 | −177.923 ± 1.177 | −37.521 ± 2.675 | 141.105 ± 3.714 | −20.808 ± 0.093 | −95.133 ± 4.263 |
| ZINC000014612330 | −167.284 ± 1.304 | −774.68 ± 2.258 | 310.493 ± 1.672 | −17.154 ± 0.074 | −648.56 ± 2.801 |
| ZINC000100513617 | −114.531 ± 1.602 | −764.552 ± 3.591 | 326.488 ± 4.91 | −14.949 ± 0.093 | −567.619 ± 4.003 |
| ZINC000013462928 | −177.25 ± 1.238 | −19.523 ± 1.104 | 56.374 ± 1.273 | −18.245 ± 0.108 | −158.661 ± 1.669 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broni, E.; Striegel, A.; Ashley, C.; Sakyi, P.O.; Peracha, S.; Velazquez, M.; Bebla, K.; Sodhi, M.; Kwofie, S.K.; Ademokunwa, A.; et al. Molecular Docking and Dynamics Simulation Studies Predict Potential Anti-ADAR2 Inhibitors: Implications for the Treatment of Cancer, Neurological, Immunological and Infectious Diseases. Int. J. Mol. Sci. 2023, 24, 6795. https://doi.org/10.3390/ijms24076795
Broni E, Striegel A, Ashley C, Sakyi PO, Peracha S, Velazquez M, Bebla K, Sodhi M, Kwofie SK, Ademokunwa A, et al. Molecular Docking and Dynamics Simulation Studies Predict Potential Anti-ADAR2 Inhibitors: Implications for the Treatment of Cancer, Neurological, Immunological and Infectious Diseases. International Journal of Molecular Sciences. 2023; 24(7):6795. https://doi.org/10.3390/ijms24076795
Chicago/Turabian StyleBroni, Emmanuel, Andrew Striegel, Carolyn Ashley, Patrick O. Sakyi, Saqib Peracha, Miriam Velazquez, Kristeen Bebla, Monsheel Sodhi, Samuel K. Kwofie, Adesanya Ademokunwa, and et al. 2023. "Molecular Docking and Dynamics Simulation Studies Predict Potential Anti-ADAR2 Inhibitors: Implications for the Treatment of Cancer, Neurological, Immunological and Infectious Diseases" International Journal of Molecular Sciences 24, no. 7: 6795. https://doi.org/10.3390/ijms24076795
APA StyleBroni, E., Striegel, A., Ashley, C., Sakyi, P. O., Peracha, S., Velazquez, M., Bebla, K., Sodhi, M., Kwofie, S. K., Ademokunwa, A., Khan, S., & Miller, W. A., III. (2023). Molecular Docking and Dynamics Simulation Studies Predict Potential Anti-ADAR2 Inhibitors: Implications for the Treatment of Cancer, Neurological, Immunological and Infectious Diseases. International Journal of Molecular Sciences, 24(7), 6795. https://doi.org/10.3390/ijms24076795