
The Ying and Yang of Sphingosine-1-Phosphate Signalling within the Bone

Abstract
:1. Introduction
2. Bone Remodelling
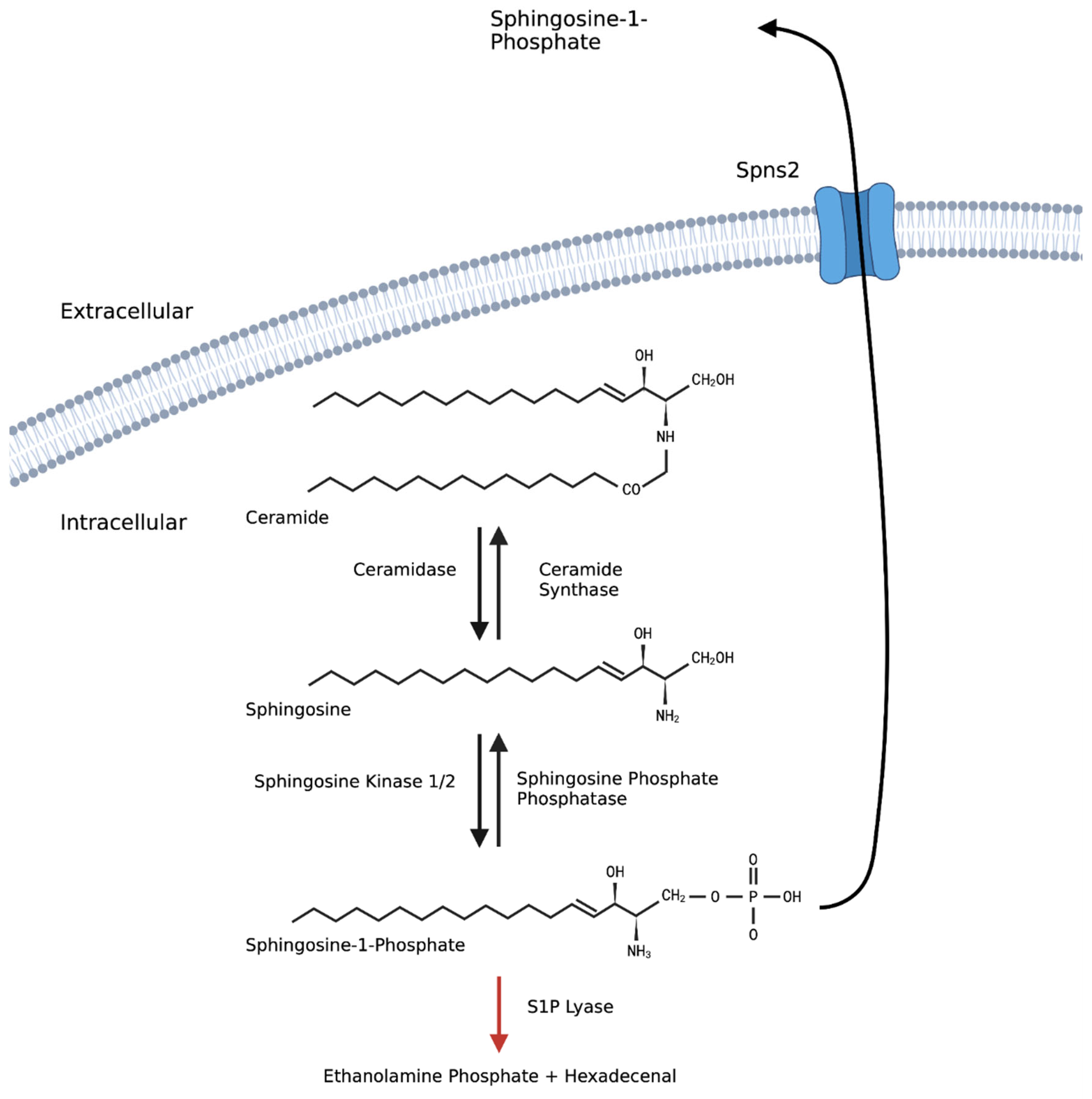
3. Sphingosine-1-Phosphate
4. S1P Signalling
4.1. Intracellular S1P Signalling
4.2. Extracellular S1P Signalling
5. S1P in Bone Precursors and Resident Cells
5.1. S1P as a Chemoattractant and Chemorepellent for Osteoclast Precursors
5.2. Osteoclast Progenitors
5.3. Osteoblast Progenitors
6. Role of S1P in Mature Bone-Resident Cells
6.1. Osteoclasts
6.2. Osteoblasts
7. Role in Osteogenic Communication
8. S1P in Catabolic Bone Diseases
8.1. Osteoporosis
8.2. Paget’s Disease
8.3. Inflammatory Diseases with Bone Damage
8.3.1. Rheumatoid Arthritis
8.3.2. Periodontitis
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jilka, R.L. Biology of the basic multicellular unit and the pathophysiology of osteoporosis. Med. Pediatr. Oncol. 2003, 41, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; McDonald, J.M. Disorders of bone remodeling. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 121–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutherford, C.; Childs, S.; Ohotski, J.; Mcglynn, L.; Riddick, M.; Macfarlane, S.; Tasker, D.; Pyne, S.; Pyne, N.J.; Edwards, J.; et al. Regulation of cell survival by sphingosine-1-phosphate receptor S1P1 via reciprocal ERK-dependent suppression of Bim and PI-3-kinase/protein kinase C-mediated upregulation of Mcl-1. Cell Death Dis. 2013, 4, e927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Huang, H.; Ding, S.F. Sphingosine-1-phosphate promotes the proliferation and attenuates apoptosis of Endothelial progenitor cells via S1PR1/S1PR3/PI3K/Akt pathway. Cell Biol. Int. 2018, 42, 1492–1502. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, Z.; Zhao, X.; Pan, F.; Cai, M.; Wang, T.; Zhang, H.; Lu, J.R.; Lei, M. Sphingosine-1-phosphate promotes the differentiation of human umbilical cord mesenchymal stem cells into cardiomyocytes under the designated culturing conditions. J. Biomed. Sci. 2011, 18, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Jeldres, T.; Alvarez-Lobos, M.; Rivera-Nieves, J. Targeting Sphingosine-1-Phosphate Signaling in Immune-Mediated Diseases: Beyond Multiple Sclerosis. Drugs 2021, 81, 985–1002. [Google Scholar] [CrossRef]
- Boyce, B.F. Advances in the regulation of osteoclasts and osteoclast functions. J. Dent. Res. 2013, 92, 860–867. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, Y.; Gou, W.; Lu, Q.; Peng, J.; Lu, S. Role of mesenchymal stem cells in bone regeneration and fracture repair: A review. Int. Orthop. 2013, 37, 2491. [Google Scholar] [CrossRef]
- Crockett, J.C.; Mellis, D.J.; Scott, D.I.; Helfrich, M.H. New knowledge on critical osteoclast formation and activation pathways from study of rare genetic diseases of osteoclasts: Focus on the RANK/RANKL axis. Osteoporos. Int. 2011, 22, 1–20. [Google Scholar] [CrossRef]
- Blumer, M.J.F.; Hausott, B.; Schwarzer, C.; Hayman, A.R.; Stempel, J.; Fritsch, H. Role of tartrate-resistant acid phosphatase (TRAP) in long bone development. Mech. Dev. 2012, 129, 162. [Google Scholar] [CrossRef]
- Saftig, P.; Hunziker, E.; Everts, V.; Jones, S.; Boyde, A.; Wehmeyer, O.; Suter, A.; von Figura, K. Functions of cathepsin K in bone resorption. Lessons from cathepsin K deficient mice. Adv. Exp. Med. Biol. 2000, 477, 293–303. [Google Scholar] [CrossRef]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef]
- Yoshiko, Y.; Candeliere, G.A.; Maeda, N.; Aubin, J.E. Osteoblast Autonomous Pi Regulation via Pit1 Plays a Role in Bone Mineralization. Mol. Cell. Biol. 2007, 27, 4465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, J.A.; Partridge, N.C. Physiological bone remodeling: Systemic regulation and growth factor involvement. Physiology 2016, 31, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Negishi-Koga, T.; Takayanagi, H. Bone cell communication factors and Semaphorins. Bonekey Rep. 2012, 1, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosla, S.; Oursler, M.J.; Monroe, D.G. Estrogen and the skeleton. Trends Endocrinol. Metab. 2012, 23, 576–581. [Google Scholar] [CrossRef] [Green Version]
- Qi, T.; Li, L.; Weidong, T. The Role of Sphingolipid Metabolism in Bone Remodeling. Front. Cell Dev. Biol. 2021, 9, 752540. [Google Scholar] [CrossRef]
- el Bawab, S.; Mao, C.; Obeid, L.M.; Hannun, Y.A. Ceramidases in the regulation of ceramide levels and function. Subcell. Biochem. 2002, 36, 187–205. [Google Scholar] [CrossRef]
- Ksia¸zek, M.; Chacińska, M.; Chabowski, A.; Baranowski, M. Sources, metabolism, and regulation of circulating sphingosine-1-phosphate. J. Lipid Res. 2015, 56, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Pralhada Rao, R.; Vaidyanathan, N.; Rengasamy, M.; Mammen Oommen, A.; Somaiya, N.; Jagannath, M.R. Sphingolipid Metabolic Pathway: An Overview of Major Roles Played in Human Diseases. J. Lipids 2013, 2013, 178910. [Google Scholar] [CrossRef] [Green Version]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and Intracellular Actions of Sphingosine-1-Phosphate. Adv. Exp. Med. Biol. 2010, 688, 141. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Harikumar, K.B.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012, 22, 50. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Zhou, Y.; Zhu, L.; Yang, S.; Huang, R.; Shi, W.; Peng, B.; Xiao, Y. SPHK1-S1PR1-RANKL Axis Regulates the Interactions Between Macrophages and BMSCs in Inflammatory Bone Loss. J. Bone Miner. Res. 2018, 33, 1090–1104. [Google Scholar] [CrossRef]
- Ryu, J.; Kim, H.J.; Chang, E.J.; Huang, H.; Banno, Y.; Kim, H.H. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J. 2006, 25, 5840–5851. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, S.E.; Milstien, S.; Spiegel, S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol. Metab. 2007, 18, 300–307. [Google Scholar] [CrossRef]
- Walker, J.M.; Yao, G.Q.; Siu, E.; Zhu, M.; Sun, B.H.; Simpson, C.; Insogna, K.L. An Unanticipated Role for Sphingosine Kinase-2 in Bone and in the Anabolic Effect of Parathyroid Hormone. Endocrinology 2021, 162, bqab042. [Google Scholar] [CrossRef]
- Bao, X.; Ogawa, T.; Se, S.; Akiyama, M.; Bahtiar, A.; Takeya, T.; Ishida-Kitagawa, N. Acid sphingomyelinase regulates osteoclastogenesis by modulating sphingosine kinases downstream of RANKL signaling. Biochem. Biophys. Res. Commun. 2011, 405, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Brizuela, L.; Martin, C.; Jeannot, P.; Ader, I.; Gstalder, C.; Andrieu, G.; Bocquet, M.; Laffosse, J.M.; Gomez-Brouchet, A.; Malavaud, B.; et al. Osteoblast-derived sphingosine 1-phosphate to induce proliferation and confer resistance to therapeutics to bone metastasis-derived prostate cancer cells. Mol. Oncol. 2014, 8, 1181–1195. [Google Scholar] [CrossRef]
- Keller, J.; Catala-Lehnen, P.; Huebner, A.K.; Jeschke, A.; Heckt, T.; Lueth, A.; Krause, M.; Koehne, T.; Albers, J.; Schulze, J.; et al. Calcitonin controls bone formation by inhibiting the release of sphingosine 1-phosphate from osteoclasts. Nat. Commun. 2014, 5, 5215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feehan, J.; Kassem, M.; Pignolo, R.J.; Duque, G. Bone From Blood: Characteristics and Clinical Implications of Circulating Osteogenic Progenitor (COP) Cells. J. Bone Miner. Res. 2021, 36, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Golan, K.; Kollet, O.; Lapidot, T. Dynamic cross talk between S1P and CXCL12 regulates hematopoietic stem cells migration, development and bone remodeling. Pharmaceuticals 2013, 6, 1145–1169. [Google Scholar] [CrossRef] [PubMed]
- Thuy, A.V.; Reimann, C.M.; Hemdan, N.Y.A.; Gräler, M.H. Sphingosine 1-phosphate in blood: Function, metabolism, and fate. Cell. Physiol. Biochem. 2014, 34, 158–171. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403. [Google Scholar] [CrossRef]
- Meshcheryakova, A.; Mechtcheriakova, D.; Pietschmann, P. Sphingosine 1-phosphate signaling in bone remodeling: Multifaceted roles and therapeutic potential. Expert Opin. Ther. Targets 2017, 21, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weske, S.; Vaidya, M.; Reese, A.; Von Wnuck Lipinski, K.; Keul, P.; Bayer, J.K.; Fischer, J.W.; Flögel, U.; Nelsen, J.; Epple, M.; et al. Targeting sphingosine-1-phosphate lyase as an anabolic therapy for bone loss. Nat. Med. 2018, 24, 667–678. [Google Scholar] [CrossRef]
- Ishii, T.; Shimazu, Y.; Nishiyama, I.; Kikuta, J.; Ishii, M. The role of sphingosine 1-phosphate in migration of osteoclast precursors; An application of intravital two-photon microscopy. Mol. Cells 2011, 31, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Egen, J.G.; Klauschen, F.; Meier-Schellersheim, M.; Saeki, Y.; Vacher, J.; Proia, R.L.; Germain, R.N. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature 2009, 458, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9 (Suppl. S1), 1–7. [Google Scholar] [CrossRef] [Green Version]
- Tomimori, Y.; Mori, K.; Koide, M.; Nakamichi, Y.; Ninomiya, T.; Udagawa, N.; Yasuda, H. Evaluation of pharmaceuticals with a novel 50-hour animal model of bone loss. J. Bone Miner. Res. 2009, 24, 1194–1205. [Google Scholar] [CrossRef]
- Hsu, L.C.; Reddy, S.V.; Yilmaz, Ö.; Yu, H. Sphingosine-1-phosphate receptor 2 controls podosome components induced by RANKL affecting osteoclastogenesis and bone resorption. Cells 2019, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Price, S.T. Sphingosine 1-Phosphate Receptor 2 Regulates the Migration, Proliferation, and Differentiation of Mesenchymal Stem Cells. Int. J. Stem Cell Res. Ther. 2015, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassoli, C.; Pierucci, F.; Zecchi-Orlandini, S.; Meacci, E. Sphingosine 1-phosphate (S1P)/S1P receptor signaling and mechanotransduction: Implications for intrinsic tissue repair/regeneration. Int. J. Mol. Sci. 2019, 20, 5545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Wang, H.; Lin, T.; Wang, S. Sphingosine-1-phosphate/s1p receptors signaling modulates cell migration in human bone marrow-derived mesenchymal stem cells. Mediat. Inflamm. 2014, 2014, 565369. [Google Scholar] [CrossRef] [Green Version]
- Quint, P.; Ruan, M.; Pederson, L.; Kassem, M.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Sphingosine 1-phosphate (S1P) receptors 1 and 2 coordinately induce mesenchymal cell migration through s1p activation of complementary kinase pathways. J. Biol. Chem. 2013, 288, 5398–5406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousselle, A.V.; Heymann, D. Osteoclastic acidification pathways during bone resorption. Bone 2002, 30, 533–540. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Xie, X.; Gu, F.; Sui, Z.; Zhang, K.; Yu, T. Recent Advances in Osteoclast Biological Behavior. Front. Cell Dev. Biol. 2021, 9, 1–19. [Google Scholar] [CrossRef]
- Choi, B.; Kim, J.E.; Park, S.O.; Kim, E.Y.; Oh, S.; Choi, H.; Yoon, D.; Min, H.J.; Kim, H.R.; Chang, E.J. Sphingosine-1-phosphate hinders the osteogenic differentiation of dental pulp stem cells in association with AKT signaling pathways. Int. J. Oral Sci. 2022, 14, 21. [Google Scholar] [CrossRef]
- Kikuta, J.; Kawamura, S.; Okiji, F.; Shirazaki, M.; Sakai, S.; Saito, H.; Ishii, M. Sphingosine-1-phosphate-mediated osteoclast precursor monocyte migration is a critical point of control in antibone-resorptive action of active vitamin D. Proc. Natl. Acad. Sci. USA 2013, 110, 7009–7013. [Google Scholar] [CrossRef] [Green Version]
- Grey, A.; Xu, X.; Hill, B.; Watson, M.; Callon, K.; Reid, I.R.; Cornish, J. Osteoblastic cells express phospholipid receptors and phosphatases and proliferate in response to sphingosine-1-phosphate. Calcif. Tissue Int. 2004, 74, 542–550. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Matsuzaki, E.; Higashi, K.; Takahashi-Yanaga, F.; Takano, A.; Hirata, M.; Nishimura, F. Sphingosine-1-phosphate inhibits differentiation of C3H10T1/2 cells into adipocyte. Mol. Cell. Biochem. 2015, 401, 39–47. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Kobayashi, M.; Matsuzaki, E.; Higashi, K.; Takahashi-Yanaga, F.; Takano, A.; Hirata, M.; Nishimura, F. Sphingosine-1-phosphate-enhanced Wnt5a promotes osteogenic differentiation in C3H10T1/2 cells. Cell Biol. Int. 2016, 40, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Carpio, L.C.; Stephan, E.; Kamer, A.; Dziak, R. Sphingolipids stimulate cell growth via MAP kinase activation in osteoblastic cells. Prostaglandins Leukot. Essent. Fat. Acids 1999, 61, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, E.; Hiratsuka, S.; Hamachi, T.; Takahashi-Yanaga, F.; Hashimoto, Y.; Higashi, K.; Kobayashi, M.; Hirofuji, T.; Hirata, M.; Maeda, K. Sphingosine-1-phosphate promotes the nuclear translocation of β-catenin and thereby induces osteoprotegerin gene expression in osteoblast-like cell lines. Bone 2013, 55, 315–324. [Google Scholar] [CrossRef]
- Higashi, K.; Matsuzaki, E.; Hashimoto, Y.; Takahashi-Yanaga, F.; Takano, A.; Anan, H.; Hirata, M.; Nishimura, F. Sphingosine-1-phosphate/S1PR2-mediated signaling triggers Smad1/5/8 phosphorylation and thereby induces Runx2 expression in osteoblasts. Bone 2016, 93, 1–11. [Google Scholar] [CrossRef]
- Nishimura, R.; Kato, Y.; Chen, D.; Harris, S.E.; Mundy, G.R.; Yoneda, T. Smad5 and DPC4 Are Key Molecules in Mediating BMP-2-induced Osteoblastic Differentiation of the Pluripotent Mesenchymal Precursor Cell Line C2C12. J. Biol. Chem. 1998, 273, 1872–1879. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells 2020, 9, 2073. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Linardi, D.; Decimo, I.; Mehboob, R.; Gebrie, M.A.; Innamorati, G.; Luciani, G.B.; Faggian, G.; Rungatscher, A. Characterization and expression of sphingosine 1-phosphate receptors in human and rat heart. Front. Pharmacol. 2017, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Spatz, J.M.; Wein, M.N.; Gooi, J.H.; Qu, Y.; Garr, J.L.; Liu, S.; Barry, K.J.; Uda, Y.; Lai, F.; Dedic, C.; et al. The Wnt Inhibitor Sclerostin Is Up-regulated by Mechanical Unloading in Osteocytes in Vitro. J. Biol. Chem. 2015, 290, 16744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.N.; Zhao, Y.; Liu, C.; Han, E.S.; Yu, X.; Lidington, D.; Bolz, S.S.; You, L. The role of the sphingosine-1-phosphate signaling pathway in osteocyte mechanotransduction. Bone 2015, 79, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Dobrosak, C.; Gooi, J.H. Increased sphingosine-1-phosphate production in response to osteocyte mechanotransduction. Bone Rep. 2017, 7, 114. [Google Scholar] [CrossRef]
- Xu, L.H.; Shao, H.; Ma, Y.H.V.; You, L. OCY454 Osteocytes as an in Vitro Cell Model for Bone Remodeling Under Mechanical Loading. J. Orthop. Res. 2019, 37, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Salari, N.; Ghasemi, H.; Mohammadi, L.; Behzadi, M.H.; Rabieenia, E.; Shohaimi, S.; Mohammadi, M. The global prevalence of osteoporosis in the world: A comprehensive systematic review and meta-analysis. J. Orthop. Surg. Res. 2021, 16, 1–20. [Google Scholar] [CrossRef]
- Riggs, B.L. The mechanisms of estrogen regulation of bone resorption. J. Clin. Investig. 2000, 106, 1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardawi, M.-S.M.; Rouzi, A.A.; Al-Senani, N.S.; Qari, M.H.; Elsamanoudy, A.Z.; Mousa, S.A. High Plasma Sphingosine 1-phosphate Levels Predict Osteoporotic Fractures in Postmenopausal Women: The Center of Excellence for Osteoporosis Research Study. J. Bone Metab. 2018, 25, 87. [Google Scholar] [CrossRef] [PubMed]
- Song, H.E.; Lee, S.H.; Kim, S.J.; Kim, B.J.; Yoo, H.J.; Koh, J.M. Association of Circulating Levels of Total and Protein-Bound Sphingosine 1-Phosphate with Osteoporotic Fracture. J. Investig. Med. 2020, 68, 1295–1299. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, J.Y.; Lim, K.H.; Lee, Y.S.; Kim, S.H.; Choi, S.; Cho, S.H.; Kim, J.S.; Koh, J.M. Associations of Circulating Levels of Sphingosine 1-Phosphate with the Trabecular Bone Score and Bone Mineral Density in Postmenopausal Women. J. Clin. Densitom. 2021, 24, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-J.; Shin, K.-O.; Kim, H.; Ahn, S.H.; Lee, S.H.; Seo, C.-H.; Byun, S.-E.; Chang, J.S.; Koh, J.-M.; Lee, Y.-M. The effect of sphingosine-1-phosphate on bone metabolism in humans depends on its plasma/bone marrow gradient. J. Endocrinol. Investig. 2016, 39, 297–303. [Google Scholar] [CrossRef]
- Bae, S.J.; Lee, S.H.; Ahn, S.H.; Kim, H.M.; Kim, B.J.; Koh, J.M. The circulating sphingosine-1-phosphate level predicts incident fracture in postmenopausal women: A 3.5-year follow-up observation study. Osteoporos. Int. 2016, 27, 2533–2541. [Google Scholar] [CrossRef]
- Ahn, S.H.; Koh, J.-M.; Gong, E.J.; Byun, S.; Lee, S.-Y.; Kim, B.-J.; Lee, S.H.; Chang, J.S.; Kim, G.S. Association of Bone Marrow Sphingosine 1-phosphate Levels with Osteoporotic Hip Fractures. J. Bone Metab. 2013, 20, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Tantikanlayaporn, D.; Tourkova, I.L.; Larrouture, Q.; Luo, J.; Piyachaturawat, P.; Witt, M.R.; Blair, H.C.; Robinson, L.J. Sphingosine-1-Phosphate Modulates the Effect of Estrogen in Human Osteoblasts. JBMR Plus 2018, 2, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Shahifar, N.; Samsudin, I.N.; Thambiah, S.C.; Yeap, S.S.; Sham, S.Y.Z.; Apannah, G.; Said, S.M.; Hew, F.L. Bone turnover markers and sphingosine-1-phosphate levels among the Chinese community in selangor, Malaysia and its correlation with bone density. Malays. J. Med. Health Sci. 2020, 16, 46–51. [Google Scholar]
- Lee, S.H.; Lee, S.Y.; Lee, Y.S.; Kim, B.J.; Lim, K.H.; Cho, E.H.; Kim, S.W.; Koh, J.M.; Kim, G.S. Higher circulating sphingosine 1-phosphate levels are associated with lower bone mineral density and higher bone resorption marker in humans. J. Clin. Endocrinol. Metab. 2012, 97, 1421–1428. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Kim, H.-E. Implications of Sphingolipids on Aging and Age-Related Diseases. Front. Aging 2021, 2, 797320. [Google Scholar] [CrossRef]
- Paget, J. On a Form of Chronic Inflammation of Bones (Osteitis Deformans). Med. Chir. Trans. 1877, 60, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Ralston, S.H. Pathogenesis of Paget’s disease of bone. Bone 2008, 43, 819–825. [Google Scholar] [CrossRef]
- Shaker, J.L. Paget’s Disease of Bone: A Review of Epidemiology, Pathophysiology and Management. Ther. Adv. Musculoskelet. Dis. 2009, 1, 107. [Google Scholar] [CrossRef] [PubMed]
- Roodman, G.D.; Windle, J.J. Paget disease of bone. J. Clin. Investig. 2005, 115, 200–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, L.; Gray, T.; Beneton, M.N.C.; Douglas, D.L.; Kanis, J.A.; Russell, R.G.G. Ultrastructural features of the osteoclasts from Paget’s disease of bone in relation to a viral aetiology. J. Clin. Pathol. 1982, 35, 771. [Google Scholar] [CrossRef] [Green Version]
- Nagata, Y.; Miyagawa, K.; Ohata, Y.; Petrusca, D.N.; Pagnotti, G.M.; Mohammad, K.S.; Guise, T.A.; Windle, J.J.; David Roodman, G.; Kurihara, N. Increased S1P expression in osteoclasts enhances bone formation in an animal model of Paget’s disease. J. Cell. Biochem. 2021, 122, 335. [Google Scholar] [CrossRef]
- Nagahashi, M.; Abe, M.; Sakimura, K.; Takabe, K.; Wakai, T. The role of sphingosine-1-phosphate in inflammation and cancer progression. Cancer Sci. 2018, 109, 3671. [Google Scholar] [CrossRef] [Green Version]
- Redlich, K.; Smolen, J.S. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 2012, 11, 234–250. [Google Scholar] [CrossRef]
- Lai, W.-Q.; Irwan, A.W.; Goh, H.H.; Howe, H.S.; Yu, D.T.; Valle-Oñate, R.; McInnes, I.B.; Melendez, A.J.; Leung, B.P. Anti-Inflammatory Effects of Sphingosine Kinase Modulation in Inflammatory Arthritis. J. Immunol. 2008, 181, 8010–8017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burg, N.; Salmon, J.E.; Hla, T. Sphingosine 1-phosphate receptor-targeted therapeutics in rheumatic diseases. Nat. Rev. Rheumatol. 2022, 18, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Hutami, I.R.; Tanaka, E.; Izawa, T. Crosstalk between Fas and S1P1 signaling via NF-kB in osteoclasts controls bone destruction in the TMJ due to rheumatoid arthritis. Jpn. Dent. Sci. Rev. 2019, 55, 12. [Google Scholar] [CrossRef]
- Baker, D.A.; Barth, J.; Chang, R.; Obeid, L.M.; Gilkeson, G.S. Genetic sphingosine kinase 1 deficiency significantly decreases synovial inflammation and joint erosions in murine TNFα-induced arthritis. J. Immunol. 2010, 185, 2570. [Google Scholar] [CrossRef] [Green Version]
- Tsunemi, S.; Iwasaki, T.; Kitano, S.; Imado, T.; Miyazawa, K.; Sano, H. Effects of the novel immunosuppressant FTY720 in a murine rheumatoid arthritis model. Clin. Immunol. 2010, 136, 197–204. [Google Scholar] [CrossRef]
- Hutami, I.R.; Izawa, T.; Mino-Oka, A.; Shinohara, T.; Mori, H.; Iwasa, A.; Tanaka, E. Fas/S1P1 crosstalk via NF-κB activation in osteoclasts controls subchondral bone remodeling in murine TMJ arthritis. Biochem. Biophys. Res. Commun. 2017, 490, 1274–1281. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Takahashi, T.; Hata, H.; Yoshitomi, H.; Tanaka, S.; Hirota, K.; Nomura, T.; Sakaguchi, N. SKG mice, a monogenic model of autoimmune arthritis due to altered signal transduction in T-cells. Hered. Basis Rheum. Dis. 2006, 147–159. [Google Scholar] [CrossRef]
- Moritz, E.; Jedlitschky, G.; Negnal, J.; Tzvetkov, M.V.; Daum, G.; Dörr, M.; Felix, S.B.; Völzke, H.; Nauck, M.; Schwedhelm, E.; et al. Increased Sphingosine-1-Phosphate Serum Concentrations in Subjects with Periodontitis: A Matter of Inflammation. J. Inflamm. Res. 2021, 14, 2883. [Google Scholar] [CrossRef]
- Al Kawas, S.; Al-Marzooq, F.; Rahman, B.; Shearston, J.A.; Saad, H.; Benzina, D.; Weitzman, M. The impact of smoking different tobacco types on the subgingival microbiome and periodontal health: A pilot study. Sci. Rep. 2021, 11, 1113. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Terao, C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells 2020, 9, 475. [Google Scholar] [CrossRef] [Green Version]
- Radwan-Oczko, M.; Duś-Ilnicka, I.; Richards, P.; Thomsen, A.M.; Rasmussen, C. Rheumatoid arthritis patients’ oral health and disease activity. Int. J. Rheum. Dis. 2019, 22, 1538–1543. [Google Scholar] [CrossRef]
- Al-Bashaireh, A.M.; Haddad, L.G.; Weaver, M.; Chengguo, X.; Lynch Kelly, D.; Yoon, S. The Effect of Tobacco Smoking on Bone Mass: An Overview of Pathophysiologic Mechanisms. J. Osteoporos. 2018, 2018, 1206235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cunto, G.; Brancaleone, V.; Riemma, M.A.; Cerqua, I.; Vellecco, V.; Spaziano, G.; Cavarra, E.; Bartalesi, B.; D’Agostino, B.; Lungarella, G.; et al. Functional contribution of sphingosine-1-phosphate to airway pathology in cigarette smoke-exposed mice. Br. J. Pharmacol. 2020, 177, 267–281. [Google Scholar] [CrossRef]
- Goel, K.; Schweitzer, K.S.; Serban, K.A.; Bittman, R.; Petrache, I. Pharmacological sphingosine-1 phosphate receptor 1 targeting in cigarette smoke-induced emphysema in mice. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2022, 322, L794–L803. [Google Scholar] [CrossRef] [PubMed]
- Kolkesen Şahin, Ö.; Çina Aksoy, M.; Cihat AVUNDUK, M. Effects of resveratrol and cigarette smoking on bone healing: Histomorphometric evaluation. Turk. J. Med. Sci. 2016, 46, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Acikan, I.; Yaman, F.; Dundar, S.; Ozercan, I.H.; Atilgan, S.S. Protective effects of caffeic acid phenethyl ester (CAPE) and thymoquinone against cigarette smoke in experimental bone fracture healing. J. Oral Biol. Craniofacial Res. 2022, 12, 610–616. [Google Scholar] [CrossRef]
- Barnawi, J.; Tran, H.B.; Roscioli, E.; Hodge, G.; Jersmann, H.; Haberberger, R.; Hodge, S. Pro-phagocytic Effects of Thymoquinone on Cigarette Smoke-exposed Macrophages Occur by Modulation of the Sphingosine-1-phosphate Signalling System. COPD J. Chronic Obstr. Pulm. Dis. 2016, 13, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Bingham, C.O., III; Moni, M. Periodontal disease and rheumatoid arthritis: The evidence accumulates for complex pathobiologic interactions. Curr. Opin. Rheumatol. 2013, 25, 345–353. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type, Function | Effect of S1P Signalling | Therapeutic Application in Catabolic Bone Disorders | Ref |
|---|---|---|---|
| Osteoblast migration | S1PR1 and S1PR3 mediate chemoattraction to S1P—supports migration of precursors to the bone surface. S1PR2 has dual actions: low concentrations promote migration towards S1P. High concentrations cause chemo-repulsion. | Direct targeting of receptors is contraindicated due to expected off-target effects on osteoclasts. Targeting of downstream signalling could prove effective. | [43,44] |
| Osteoblast proliferation | Exogenous application of S1P increases proliferation. Inhibition of S1PR2 enhances proliferation but not differentiation. | Inhibition of S1PR2 enhances total osteoblast number but not their maturation. Unlikely to be effective therapeutically. | [41,52] |
| Osteoblast differentiation | S1P increases osteoblast differentiation. S1PR2 is required for osteoblast differentiation and maturation. Inhibition reduces differentiation. S1P signalling via S1PR3 is required for bone formation and maintenance. | S1PR3 is an interesting potential therapeutic target as (unlike, e.g., S1PR2) it has no alternative effects on osteoclasts. S1PR3 agonists likely to increase osteoblast differentiation and increase bone formation. | [29,41,47,50,51,53] |
| Osteoclast migration | S1PR1 is required for chemoattraction to S1P, maintaining osteoclast precursors within the circulation. S1PR2 is required for chemorepulsion towards S1P, promoting osteoclast precursors migration to tissue. | Inhibition of S1PR2 via antagonists (e.g., JTE013), may lower the number of osteoclasts present within bone tissue, reducing resorption. | [36,37,39,40] |
| Monocyte fusion | S1PR2 is required for monocyte fusion into osteoclasts, via regulation of podosome-adhesive proteins. | Inhibition of S1PR2 via antagonists (e.g., JTE013) prevents osteoclast formation, reducing resorption. | [40] |
| Osteoclast differentiation | SPHK1 is required for negative regulation of RANKL-mediated differentiation, through suppression of p38 signalling. The impact of exogenous S1P on osteoclast differentiation is unknown (incomplete data available). | Therapeutics that upregulate SPHK1 function or suppress p38 signalling may reduce mature osteoclasts. | [24,48] |
| RANKL production | Activation of S1PR1/3 on osteoblasts induces RANKL release, via activation of ERK/p38. | Reduction of RANKL-mediated osteoclast differentiation could be achieved through inhibition of osteoblast S1PR1/S1PR3 receptors or targeting the downstream signalling pathways. | [23] |
| OPG Production | Activation of S1PR2 on osteoblasts induces OPG release via GSK3β and β-catenin. | S1PR2 agonists or targeting of β-catenin-mediated secretion of OPG may reduce osteoclast numbers to prevent bone resorption. | [23,35,54] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frost, K.; Naylor, A.J.; McGettrick, H.M. The Ying and Yang of Sphingosine-1-Phosphate Signalling within the Bone. Int. J. Mol. Sci. 2023, 24, 6935. https://doi.org/10.3390/ijms24086935
Frost K, Naylor AJ, McGettrick HM. The Ying and Yang of Sphingosine-1-Phosphate Signalling within the Bone. International Journal of Molecular Sciences. 2023; 24(8):6935. https://doi.org/10.3390/ijms24086935
Chicago/Turabian StyleFrost, Kathryn, Amy J. Naylor, and Helen M. McGettrick. 2023. "The Ying and Yang of Sphingosine-1-Phosphate Signalling within the Bone" International Journal of Molecular Sciences 24, no. 8: 6935. https://doi.org/10.3390/ijms24086935