

R-Loops and R-Loop-Binding Proteins in Cancer Progression and Drug Resistance

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. R-Loops—Physiological Appearance and Regulation
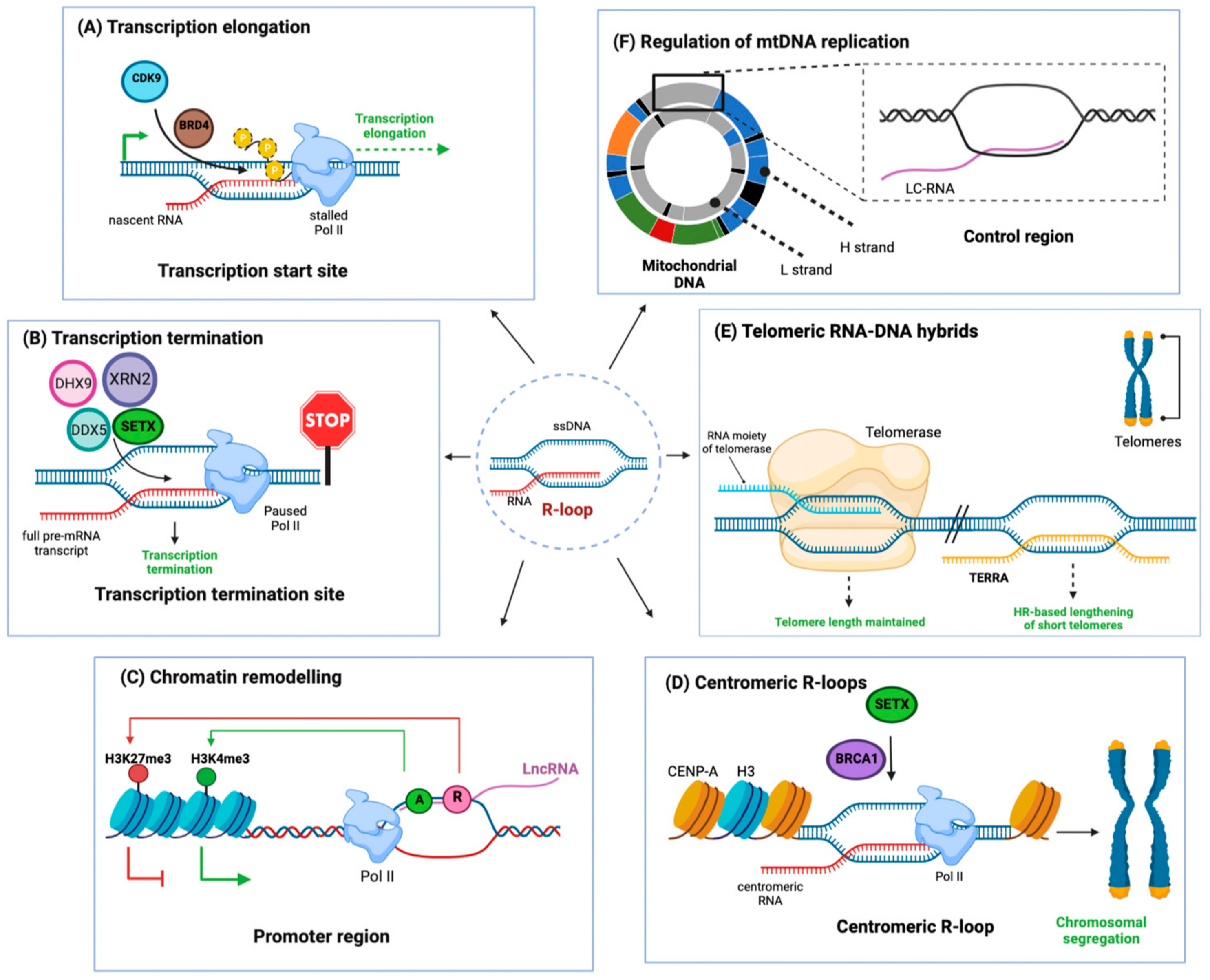
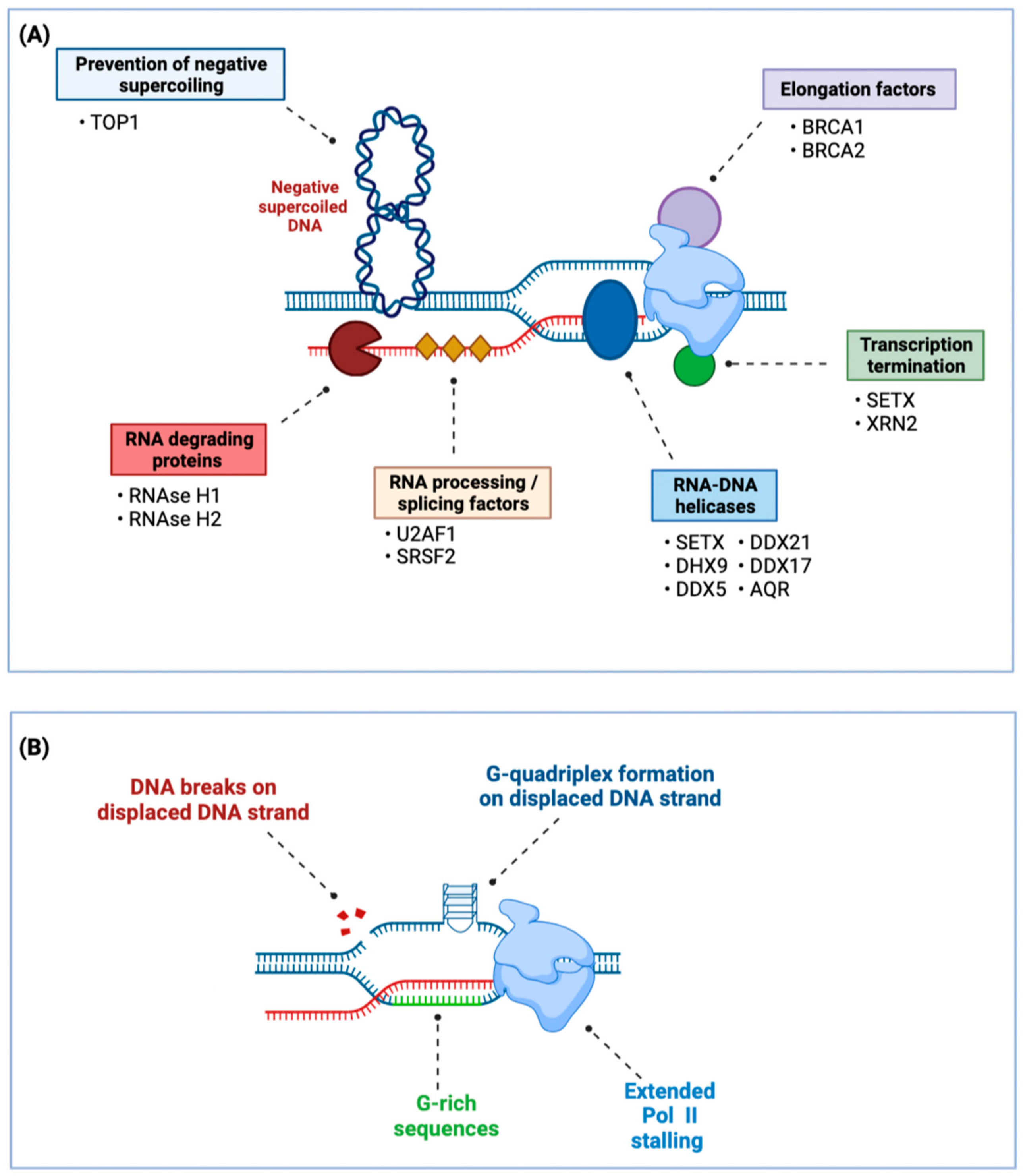
2. Pro-Tumorigenic Effects of R-Loops in BRCA1/2 Mutant Cancers
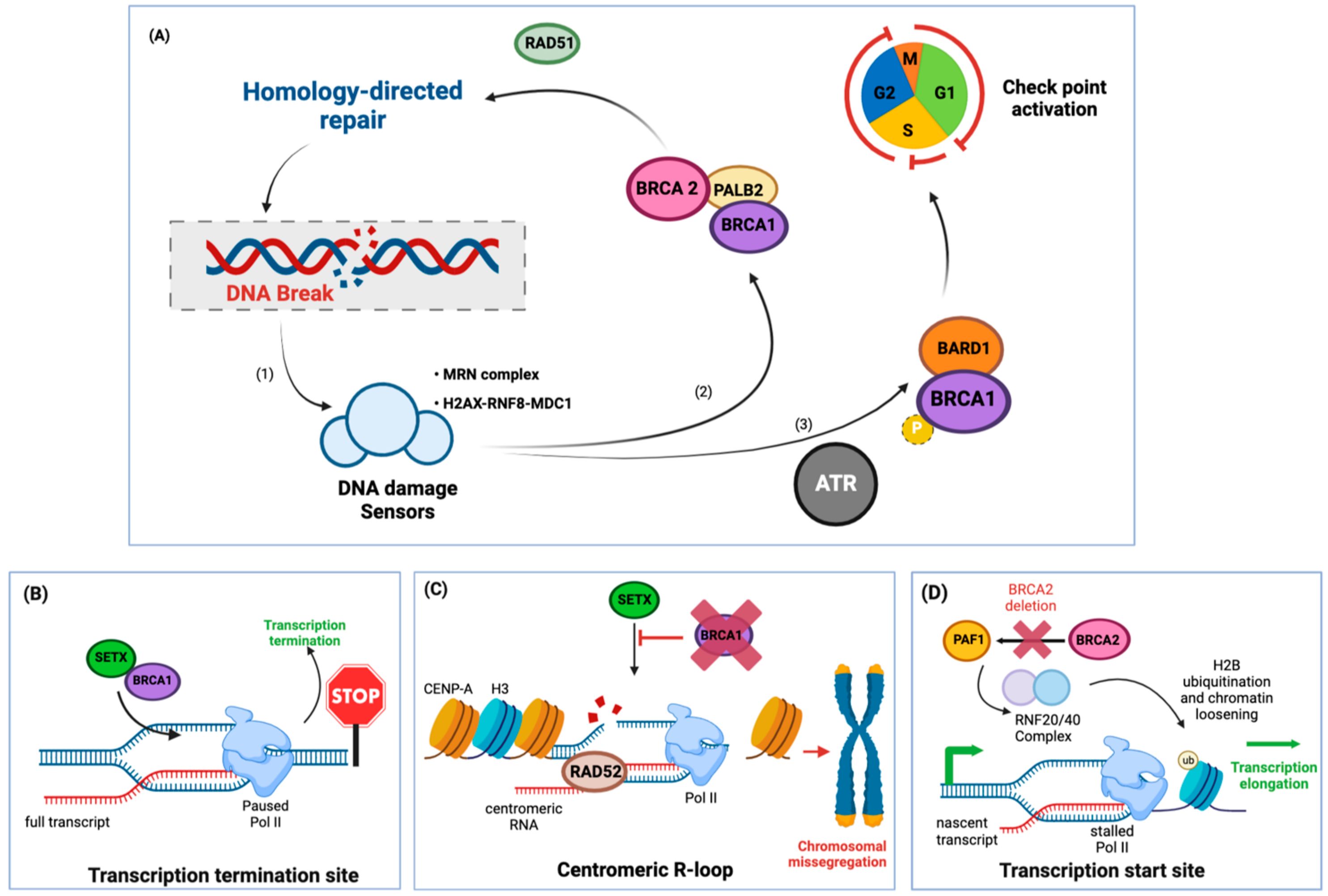
2.1. R-Loops at the Site of Active Transcription in BRCA1/2 Mutant Cancers
2.2. R-Loop-Driven Control of Luminal Differentiation
2.3. The Role of R-Loops in Telomere Maintenance
2.4. R-Loops in Other Breast Cancers
2.5. BRCA2 and R-Loops in the Mitochondrial Genome
3. R-Loop Sensing through ATR and the Activation of DNA Repair Pathways
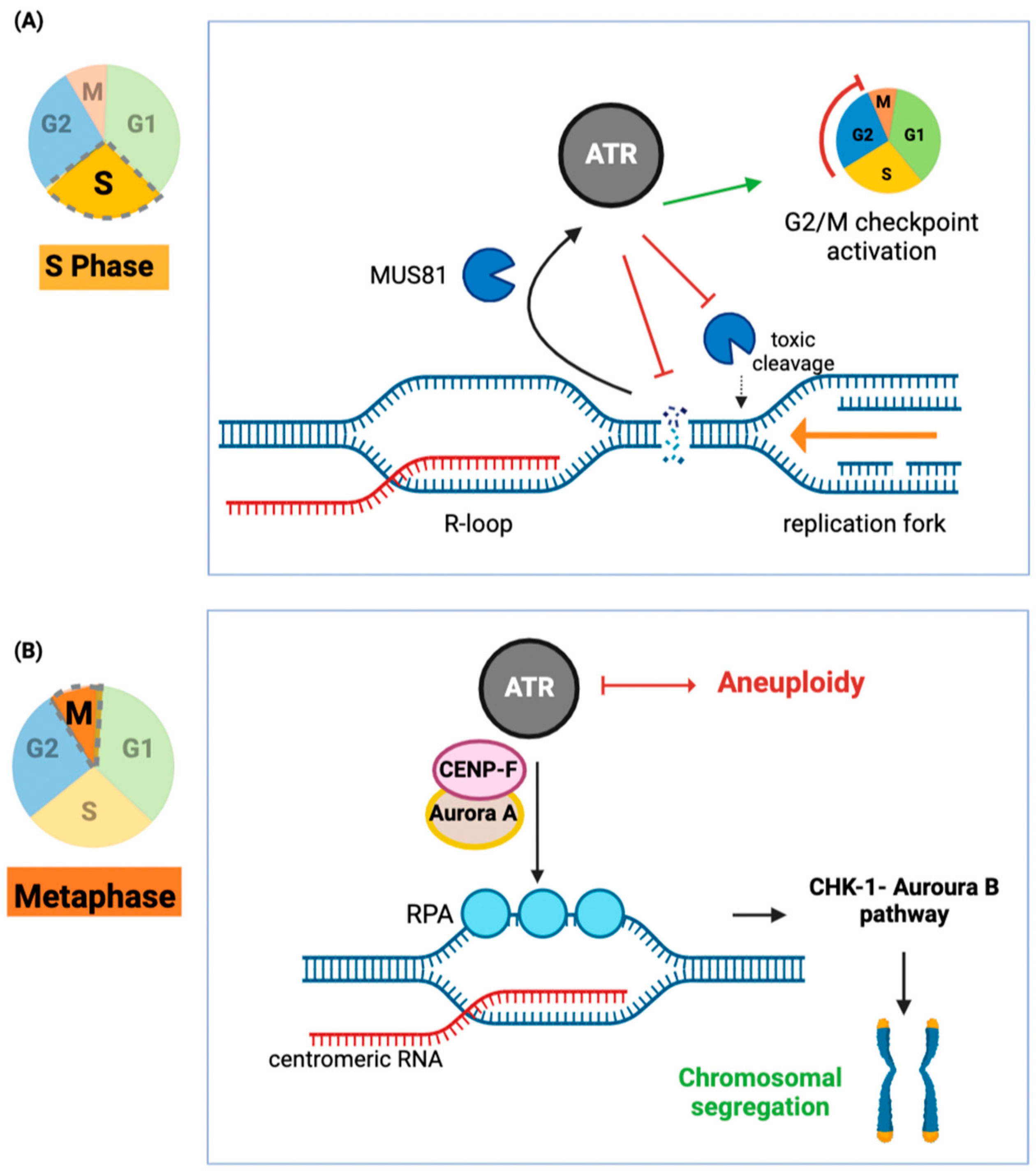
3.1. ATR-Activation through R-Loops
3.2. ATR Inhibition Sensitizes Cancer Cells to R-Loop-Induced DNA Damage
4. Anticancer Effects of R-Loops and Their Exploitation for Therapy
4.1. Inhibition of the DNA Damage Response through R-Loops
4.2. Cancers with Elevated R-Loop Formation Are Susceptible to DNA Damage
4.3. Induction of R-Loop Formation in Anticancer Therapy
5. R-Loops as Targets for Anticancer Drugs to Combat Chemoresistance
5.1. Inhibition of R-Loop Unwinding
5.2. Inhibition of R-Loop Cleavage
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petermann, E.; Lan, L.; Zou, L. Sources, resolution and physiological relevance of R-loops and RNA–DNA hybrids. Nat. Rev. Mol. Cell Biol. 2022, 23, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chédin, F. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Manley, J.L. Inactivation of the SR Protein Splicing Factor ASF/SF2 Results in Genomic Instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, R.; Lee, Y.; Wischnewski, H.; Brun, C.M.; Schwarz, T.; Azzalin, C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014, 5, 5220. [Google Scholar] [CrossRef] [Green Version]
- Crossley, M.P.; Song, C.; Bocek, M.J.; Choi, J.-H.; Kousorous, J.; Sathirachinda, A.; Lin, C.; Brickner, J.R.; Bai, G.; Lans, H.; et al. R-loop-derived cytoplasmic RNA–DNA hybrids activate an immune response. Nature 2022, 613, 187–194. [Google Scholar] [CrossRef]
- Brickner, J.R.; Garzon, J.L.; Cimprich, K.A. Walking a tightrope: The complex balancing act of R-loops in genome stability. Mol. Cell 2022, 82, 2267–2297. [Google Scholar] [CrossRef]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Rutkauskas, M.; Sinkunas, T.; Songailiene, I.; Tikhomirova, M.; Siksnys, V.; Seidel, R. Directional R-Loop Formation by the CRISPR-Cas Surveillance Complex Cascade Provides Efficient Off-Target Site Rejection. Cell Rep. 2015, 10, 1534–1543. [Google Scholar] [CrossRef] [Green Version]
- Tuminauskaite, D.; Norkunaite, D.; Fiodorovaite, M.; Tumas, S.; Songailiene, I.; Tamulaitiene, G.; Sinkunas, T. DNA interference is controlled by R-loop length in a type I-F1 CRISPR-Cas system. BMC Biol. 2020, 18, 65. [Google Scholar] [CrossRef]
- El Hage, A.; Webb, S.; Kerr, A.; Tollervey, D. Genome-Wide Distribution of RNA-DNA Hybrids Identifies RNase H Targets in tRNA Genes, Retrotransposons and Mitochondria. PLoS Genet. 2014, 10, e1004716. [Google Scholar] [CrossRef]
- Wahba, L.; Costantino, L.; Tan, F.J.; Zimmer, A.; Koshland, D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev. 2016, 30, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Sharifi, S.; Bierhoff, H. Regulation of RNA Polymerase I Transcription in Development, Disease, and Aging. Annu. Rev. Biochem. 2018, 87, 51–73. [Google Scholar] [CrossRef]
- White, R.J. Transcription by RNA polymerase III: More complex than we thought. Nat. Rev. Genet. 2011, 12, 459–463. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.-Y.; Zhang, X.; Gu, Y.; Xiao, R.; Shao, C.; Tang, P.; Qian, H.; Luo, D.; Li, H.; et al. R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol. Cell 2017, 68, 745–757.e5. [Google Scholar] [CrossRef] [Green Version]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Liu, X.; Liu, J. Detection of Single Nucleotide Polymorphism by RNase H-Cleavage Mediated Allele-Specific Extension Method. Biotechnol. Biotechnol. Equip. 2012, 26, 3148–3154. [Google Scholar] [CrossRef] [Green Version]
- Cerritelli, S.M.; Sakhuja, K.; Crouch, R.J. RNase H1, the Gold Standard for R-Loop Detection; Springer: New York, NY, USA, 2022; pp. 91–114. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human Senataxin Resolves RNA/DNA Hybrids Formed at Transcriptional Pause Sites to Promote Xrn2-Dependent Termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Bader, A.S.; Luessing, J.; Hawley, B.R.; Skalka, G.L.; Lu, W.-T.; Lowndes, N.F.; Bushell, M. DDX17 Is Required for Efficient DSB Repair at DNA:RNA Hybrid Deficient Loci. Nucleic Acids Res. 2022, 50, 10487–10502. [Google Scholar] [CrossRef]
- Khan, E.S.; Danckwardt, S. Pathophysiological Role and Diagnostic Potential of R-Loops in Cancer and Beyond. Genes 2022, 13, 2181. [Google Scholar] [CrossRef]
- Sollier, J.; Cimprich, K.A. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015, 25, 514–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, C.S.; Zhao, H.; Barlow, J.H. Transcription-Replication Collisions—A Series of Unfortunate Events. Biomolecules 2021, 11, 1249. [Google Scholar] [CrossRef] [PubMed]
- Chédin, F. Nascent Connections: R-Loops and Chromatin Patterning. Trends Genet. 2016, 32, 828–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abakir, A.; Giles, T.C.; Cristini, A.; Foster, J.M.; Dai, N.; Starczak, M.; Rubio-Roldan, A.; Li, M.; Eleftheriou, M.; Crutchley, J.; et al. N6-methyladenosine regulates the stability of RNA:DNA hybrids in human cells. Nat. Genet. 2020, 52, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Perego, M.G.L.; Taiana, M.; Bresolin, N.; Comi, G.P.; Corti, S. R-Loops in Motor Neuron Diseases. Mol. Neurobiol. 2019, 56, 2579–2589. [Google Scholar] [CrossRef]
- Richard, P.; Manley, J.L. R Loops and Links to Human Disease. J. Mol. Biol. 2017, 429, 3168–3180. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.P.; White, J.; Stirling, P.C. R Loops and Their Composite Cancer Connections. Trends Cancer 2019, 5, 619–631. [Google Scholar] [CrossRef]
- Promonet, A.; Padioleau, I.; Liu, Y.; Sanz, L.; Biernacka, A.; Schmitz, A.-L.; Skrzypczak, M.; Sarrazin, A.; Mettling, C.; Rowicka, M.; et al. Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites. Nat. Commun. 2020, 11, 3940. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Leong, W.Y.; Li, W.; Reddy, P.N.; Sullivan, J.D.; Walter, M.J.; Zou, L.; Graubert, T.A. Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res. 2018, 78, 5363–5374. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, J.-Y.; Huang, Y.-J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef] [Green Version]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 Recruitment to Transcriptional Pause Sites Is Required for R-Loop-Driven DNA Damage Repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chiang, H.-C.; Wang, Y.; Zhang, C.; Smith, S.; Zhao, X.; Nair, S.J.; Michalek, J.; Jatoi, I.; Lautner, M.; et al. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017, 8, 15908. [Google Scholar] [CrossRef] [Green Version]
- Shivji, M.K.; Renaudin, X.; Williams, H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Howlader, N.; Noone, A.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; MAriotto, A.; Lewis, D.; et al. SEER Cancer Statistics Review, 1975–2017; National Cancer Institute: Bethesda, MD, USA, 2020. [Google Scholar]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Racca, C.; Britton, S.; Hédouin, S.; Francastel, C.; Calsou, P.; Larminat, F. BRCA1 prevents R-loop-associated centromeric instability. Cell Death Dis. 2021, 12, 896. [Google Scholar] [CrossRef]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING Heterodimer BRCA1-BARD1 Is a Ubiquitin Ligase Inactivated by a Breast Cancer-derived Mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Chini, C.C.S.; He, M.; Mer, G.; Chen, J. The BRCT Domain Is a Phospho-Protein Binding Domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of BRCA1, BRCA2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.-S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Li, S.; Boyer, T.G.; Lee, W.-H. Lessons learned from BRCA1 and BRCA2. Oncogene 2000, 19, 6159–6175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, K.P.; Cortez, D. RPA and RAD51: Fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 2018, 25, 446–453. [Google Scholar] [CrossRef]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Szabo, C.I.; Worley, T.; Monteiro, A.N. Understanding Germ-Line Mutations in BRCA1. Cancer Biol. Ther. 2004, 3, 515–520. [Google Scholar] [CrossRef] [Green Version]
- Alonso, M.S.M.; Noordermeer, S.M. Untangling the crosstalk between BRCA1 and R-loops during DNA repair. Nucleic Acids Res. 2021, 49, 4848–4863. [Google Scholar] [CrossRef]
- Hopkins, J.L.; Lan, L.; Zou, L. DNA repair defects in cancer and therapeutic opportunities. Genes Dev. 2022, 36, 278–293. [Google Scholar] [CrossRef]
- McKinley, K.; Cheeseman, I.M. The molecular basis for centromere identity and function. Nat. Rev. Mol. Cell Biol. 2016, 17, 16–29. [Google Scholar] [CrossRef]
- Adelman, K.; Lis, J.T. Promoter-proximal pausing of RNA polymerase II: Emerging roles in metazoans. Nat. Rev. Genet. 2012, 13, 720–731. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Li, T.; Price, D.H. RNA Polymerase II Elongation Control. Annu. Rev. Biochem. 2012, 81, 119–143. [Google Scholar] [CrossRef] [Green Version]
- Buckley, N.; Tsaoir, C.B.N.A.; Blayney, J.K.; Oram, L.C.; Crawford, N.T.; D’Costa, Z.C.; Quinn, J.E.; Kennedy, R.D.; Harkin, D.P.; Mullan, P.B. BRCA1 is a key regulator of breast differentiation through activation of Notch signalling with implications for anti-endocrine treatment of breast cancers. Nucleic Acids Res. 2013, 41, 8601–8614. [Google Scholar] [CrossRef]
- Furuta, S.; Jiang, X.; Gu, B.; Cheng, E.; Chen, P.-L.; Lee, W.-H. Depletion of BRCA1 impairs differentiation but enhances proliferation of mammary epithelial cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9176–9181. [Google Scholar] [CrossRef] [Green Version]
- Chiang, H.-C.; Chiang, H.-C.; Zhang, X.; Zhang, X.; Li, J.; Li, J.; Zhao, X.; Zhao, X.; Chen, J.; Chen, J.; et al. BRCA1-associated R-loop affects transcription and differentiation in breast luminal epithelial cells. Nucleic Acids Res. 2019, 47, 5086–5099. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, T.B.; Sá, A.; Lopes, J.M.; Sobrinho-Simões, M.; Soares, P.; Vinagre, J. Telomere Maintenance Mechanisms in Cancer. Genes 2018, 9, 241. [Google Scholar] [CrossRef] [Green Version]
- Bejarano, L.; Bosso, G.; Louzame, J.; Serrano, R.; Gómez-Casero, E.; Martinez-Torrecuadrada, J.L.; Martínez, S.; Blanco-Aparicio, C.; Pastor, J.; A Blasco, M. Multiple cancer pathways regulate telomere protection. EMBO Mol. Med. 2019, 11, e10292. [Google Scholar] [CrossRef]
- Feretzaki, M.; Pospisilova, M.; Fernandes, R.V.; Lunardi, T.; Krejci, L.; Lingner, J. RAD51-dependent recruitment of TERRA lncRNA to telomeres through R-loops. Nature 2020, 587, 303–308. [Google Scholar] [CrossRef]
- Vohhodina, J.; Goehring, L.J.; Liu, B.; Kong, Q.; Botchkarev, V.V.; Huynh, M.; Liu, Z.; Abderazzaq, F.O.; Clark, A.P.; Ficarro, S.B.; et al. BRCA1 binds TERRA RNA and suppresses R-Loop-based telomeric DNA damage. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Pompili, L.; Maresca, C.; Stritto, A.D.; Biroccio, A.; Salvati, E. BRCA2 Deletion Induces Alternative Lengthening of Telomeres in Telomerase Positive Colon Cancer Cells. Genes 2019, 10, 697. [Google Scholar] [CrossRef] [Green Version]
- Shiromoto, Y.; Sakurai, M.; Minakuchi, M.; Ariyoshi, K.; Nishikura, K. ADAR1 RNA editing enzyme regulates R-loop formation and genome stability at telomeres in cancer cells. Nat. Commun. 2021, 12, 1–18. [Google Scholar] [CrossRef]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; A Sanz, L.; Chédin, F.; Swigut, T.; A Cimprich, K. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. eLife 2016, 5, e17548. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J. R-Loops and Mitochondrial DNA Metabolism. Methods Mol. Biol. 2022, 2528, 173–202. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Clayton, D.A. RNA-DNA hybrid formation at the human mitochondrial heavy-strand origin ceases at replication start sites: An implication for RNA-DNA hybrids serving as primers. EMBO J. 1996, 15, 3135–3143. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, R.J.; Borowski, L.S.; Malecki, M.; Wojcik, M.A.; Stepien, P.P.; Golik, P. RNA Degradation in Yeast and Human Mitochondria. Biochim. Biophys. Acta (BBA) Gene Regul. Mech. 2012, 1819, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Camino, L.P.; Aguilera, A. Human mitochondrial degradosome prevents harmful mitochondrial R loops and mitochondrial genome instability. Proc. Natl. Acad. Sci. USA 2018, 115, 11024–11029. [Google Scholar] [CrossRef] [Green Version]
- Holmes, J.B.; Akman, G.; Wood, S.R.; Sakhuja, K.; Cerritelli, S.M.; Moss, C.; Bowmaker, M.R.; Jacobs, H.T.; Crouch, R.J.; Holt, I.J. Primer retention owing to the absence of RNase H1 is catastrophic for mitochondrial DNA replication. Proc. Natl. Acad. Sci. USA 2015, 112, 9334–9339. [Google Scholar] [CrossRef] [Green Version]
- Boland, M.L.; Chourasia, A.H.; MacLeod, K.F. Mitochondrial Dysfunction in Cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.W.; Kang, H.J.; Bae, I. BRCA1 and Oxidative Stress. Cancers 2014, 6, 771–795. [Google Scholar] [CrossRef] [Green Version]
- Renaudin, X.; Lee, M.; Shehata, M.; Surmann, E.-M.; Venkitaraman, A.R. BRCA2 deficiency reveals that oxidative stress impairs RNaseH1 function to cripple mitochondrial DNA maintenance. Cell Rep. 2021, 36, 109478. [Google Scholar] [CrossRef]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef]
- Jo, U.; Senatorov, I.S.; Zimmermann, A.; Saha, L.K.; Murai, Y.; Kim, S.H.; Rajapakse, V.N.; Elloumi, F.; Takahashi, N.; Schultz, C.W.; et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells with Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol. Cancer Ther. 2021, 20, 1431–1441. [Google Scholar] [CrossRef]
- Plummer, R.; Dean, E.; Arkenau, H.-T.; Redfern, C.; Spira, A.I.; Melear, J.M.; Chung, K.Y.; Ferrer-Playan, J.; Goddemeier, T.; Locatelli, G.; et al. A phase 1b study evaluating the safety and preliminary efficacy of berzosertib in combination with gemcitabine in patients with advanced non-small cell lung cancer. Lung Cancer 2022, 163, 19–26. [Google Scholar] [CrossRef]
- Kwon, M.; Kim, G.; Kim, R.; Kim, K.-T.; Kim, S.T.; Smith, S.; Mortimer, P.G.S.; Hong, J.Y.; Loembé, A.-B.; Irurzun-Arana, I.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced gastric cancer. J. Immunother. Cancer 2022, 10, e005041. [Google Scholar] [CrossRef]
- Middleton, M.R.; Dean, E.; Evans, T.R.J.; Shapiro, G.I.; Pollard, J.; Hendriks, B.S.; Falk, M.; Diaz-Padilla, I.; Plummer, R. Phase 1 study of the ATR inhibitor berzosertib (formerly M6620, VX-970) combined with gemcitabine ± cisplatin in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 510–519. [Google Scholar] [CrossRef]
- Matos, D.A.; Zhang, J.-M.; Ouyang, J.; Nguyen, H.D.; Genois, M.-M.; Zou, L. ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol. Cell 2020, 77, 514–527.e4. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, P.; Li, C.; Liu, W.; Shen, Q.; Yang, L.; Xie, G.; Bai, J.; Li, R.; Tao, K.; et al. MUS81 Inhibition Enhances the Anticancer Efficacy of Talazoparib by Impairing ATR/CHK1 Signaling Pathway in Gastric Cancer. Front. Oncol. 2022, 12, 844135. [Google Scholar] [CrossRef]
- Kabeche, L.; Nguyen, H.D.; Buisson, R.; Zou, L. A mitosis-specific and R loop–driven ATR pathway promotes faithful chromosome segregation. Science 2018, 359, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS–FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Morel, D.; Eychenne, T.; Colmet-Daage, L.; Bajrami, I.; Dorvault, N.; Garrido, M.; Meisenberg, C.; Lamb, A.; Ngo, C.; et al. PBRM1 Deficiency Confers Synthetic Lethality to DNA Repair Inhibitors in Cancer. Cancer Res. 2021, 81, 2888–2902. [Google Scholar] [CrossRef]
- Paronetto, M.P. Ewing Sarcoma Protein: A Key Player in Human Cancer. Int. J. Cell Biol. 2013, 2013, 642853. [Google Scholar] [CrossRef] [Green Version]
- Abraham, K.J.; Khosraviani, N.; Chan, J.N.Y.; Gorthi, A.; Samman, A.; Zhao, D.Y.; Wang, M.; Bokros, M.; Vidya, E.; Ostrowski, L.A.; et al. Nucleolar RNA polymerase II drives ribosome biogenesis. Nature 2020, 585, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Kanellis, D.C.; Espinoza, J.A.; Zisi, A.; Sakkas, E.; Bartkova, J.; Katsori, A.-M.; Boström, J.; Dyrskjøt, L.; Broholm, H.; Altun, M.; et al. The exon-junction complex helicase eIF4A3 controls cell fate via coordinated regulation of ribosome biogenesis and translational output. Sci. Adv. 2021, 7, eabf7561. [Google Scholar] [CrossRef] [PubMed]
- Lindström, M.S.; Bartek, J.; Maya-Mendoza, A. P53 at the crossroad of DNA replication and ribosome biogenesis stress pathways. Cell Death Differ. 2022, 29, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs—Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Brownlee, P.M.; Chambers, A.L.; Cloney, R.; Bianchi, A.; Downs, J.A. BAF180 Promotes Cohesion and Prevents Genome Instability and Aneuploidy. Cell Rep. 2014, 6, 973–981. [Google Scholar] [CrossRef] [Green Version]
- Krais, J.J.; Wang, Y.; Patel, P.; Basu, J.; Bernhardy, A.J.; Johnson, N. RNF168-mediated localization of BARD1 recruits the BRCA1-PALB2 complex to DNA damage. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Patel, P.S.; Abraham, K.J.; Guturi, K.K.N.; Halaby, M.-J.; Khan, Z.; Palomero, L.; Ho, B.; Duan, S.; St-Germain, J.; Algouneh, A.; et al. RNF168 regulates R-loop resolution and genomic stability in BRCA1/2-deficient tumors. J. Clin. Investig. 2021, 131, 140105. [Google Scholar] [CrossRef]
- Lam, F.C.; Kong, Y.W.; Huang, Q.; Han, T.-L.V.; Maffa, A.D.; Kasper, E.M.; Yaffe, M.B. BRD4 prevents the accumulation of R-loops and protects against transcription–replication collision events and DNA damage. Nat. Commun. 2020, 11, 4083. [Google Scholar] [CrossRef]
- Edwards, D.S.; Maganti, R.; Tanksley, J.P.; Luo, J.; Park, J.J.; Balkanska-Sinclair, E.; Ling, J.; Floyd, S.R. BRD4 Prevents R-Loop Formation and Transcription-Replication Conflicts by Ensuring Efficient Transcription Elongation. Cell Rep. 2020, 32, 108166. [Google Scholar] [CrossRef]
- Fagan-Solis, K.D.; Simpson, D.A.; Kumar, R.J.; Martelotto, L.G.; Mose, L.E.; Rashid, N.U.; Ho, A.Y.; Powell, S.N.; Wen, Y.H.; Parker, J.S.; et al. A P53-Independent DNA Damage Response Suppresses Oncogenic Proliferation and Genome Instability. Cell Rep. 2020, 30, 1385–1399.e7. [Google Scholar] [CrossRef]
- Bai, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Triple-negative breast cancer therapeutic resistance: Where is the Achilles’ heel? Cancer Lett. 2021, 497, 100–111. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Hung, A.C.; Lo, S.; Hsieh, Y.-C.; Yuan, S.-S.F. MRE11 as a molecular signature and therapeutic target for cancer treatment with radiotherapy. Cancer Lett. 2021, 514, 1–11. [Google Scholar] [CrossRef]
- Lambo, S.; Gröbner, S.N.; Rausch, T.; Waszak, S.M.; Schmidt, C.; Gorthi, A.; Romero, J.C.; Mauermann, M.; Brabetz, S.; Krausert, S.; et al. The molecular landscape of ETMR at diagnosis and relapse. Nature 2019, 576, 274–280. [Google Scholar] [CrossRef]
- Lu, W.-T.; Hawley, B.R.; Skalka, G.L.; Baldock, R.A.; Smith, E.M.; Bader, A.S.; Malewicz, M.; Watts, F.Z.; Wilczynska, A.; Bushell, M. Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair. Nat. Commun. 2018, 9, 532. [Google Scholar] [CrossRef] [Green Version]
- Smolinski, D. R-loops at microRNA encoding loci promote co-transcriptional processing of pri-miRNAs in plants. Nat. Plants 2022, 8, 402–418. [Google Scholar] [CrossRef]
- Itzen, F.; Greifenberg, A.K.; Bösken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef] [Green Version]
- Muhar, M.; Ebert, A.; Neumann, T.; Umkehrer, C.; Jude, J.; Wieshofer, C.; Rescheneder, P.; Lipp, J.J.; Herzog, V.A.; Reichholf, B.; et al. SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 2018, 360, 800–805. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.-W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [Green Version]
- Tajima, N.; Fukui, K.; Uesato, N.; Maruhashi, J.; Yoshida, T.; Watanabe, Y.; Tojo, A. JTE-607, a multiple cytokine production inhibitor, induces apoptosis accompanied by an increase in p21waf1/cip1 in acute myelogenous leukemia cells. Cancer Sci. 2010, 101, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Uesato, N.; Fukui, K.; Maruhashi, J.; Tojo, A.; Tajima, N. JTE-607, a multiple cytokine production inhibitor, ameliorates disease in a SCID mouse xenograft acute myeloid leukemia model. Exp. Hematol. 2006, 34, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Ross, N.T.; Lohmann, F.; Carbonneau, S.; Fazal, A.; Weihofen, W.A.; Gleim, S.; Salcius, M.; Sigoillot, F.; Henault, M.; Carl, S.H.; et al. CPSF3-dependent pre-mRNA processing as a druggable node in AML and Ewing’s sarcoma. Nat. Chem. Biol. 2020, 16, 50–59. [Google Scholar] [CrossRef] [PubMed]
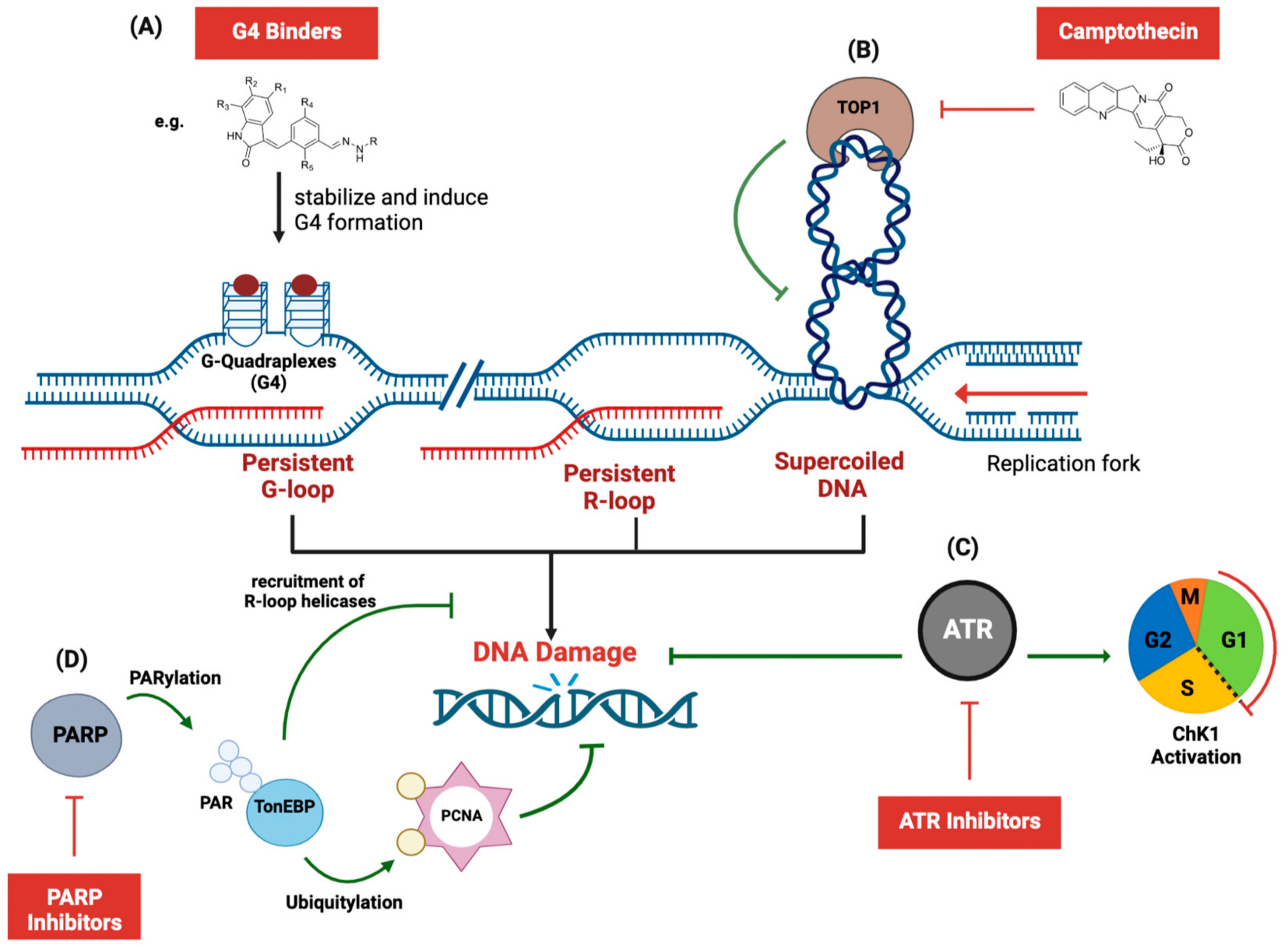
- Miglietta, G.; Russo, M.; Capranico, G. G-quadruplex–R-loop interactions and the mechanism of anticancer G-quadruplex binders. Nucleic Acids Res. 2020, 48, 11942–11957. [Google Scholar] [CrossRef]
- Camarillo, R.; Jimeno, S.; Huertas, P. The Effect of Atypical Nucleic Acids Structures in DNA Double Strand Break Repair: A Tale of R-loops and G-Quadruplexes. Front. Genet. 2021, 12, 742434. [Google Scholar] [CrossRef]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-quadruplexes: A promising target for cancer therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef]
- Amato, J.; Miglietta, G.; Morigi, R.; Iaccarino, N.; Locatelli, A.; Leoni, A.; Novellino, E.; Pagano, B.; Capranico, G.; Randazzo, A. Monohydrazone Based G-Quadruplex Selective Ligands Induce DNA Damage and Genome Instability in Human Cancer Cells. J. Med. Chem. 2020, 63, 3090–3103. [Google Scholar] [CrossRef]
- De Magis, A.; Manzo, S.G.; Russo, M.; Marinello, J.; Morigi, R.; Sordet, O.; Capranico, G. DNA damage and genome instability by G-quadruplex ligands are mediated by R loops in human cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Haensel-Hertsch, R.; Simeone, A.; Shea, A.; Hui, W.W.I.; Zyner, K.G.; Marsico, G.; Rueda, O.M.; Bruna, A.; Martin, A.; Zhang, X.; et al. Landscape of G-quadruplex DNA structural regions in breast cancer. Nat. Genet. 2020, 52, 878–883. [Google Scholar] [CrossRef]
- Ye, B.J.; Kang, H.J.; Lee-Kwon, W.; Kwon, H.M.; Choi, S.Y. PARP1-mediated PARylation of TonEBP prevents R-loop–associated DNA damage. DNA Repair 2021, 104, 103132. [Google Scholar] [CrossRef]
- Safari, M.; Litman, T.; Robey, R.W.; Aguilera, A.; Chakraborty, A.R.; Reinhold, W.C.; Basseville, A.; Petrukhin, L.; Scotto, L.; O’Connor, O.A.; et al. R-Loop–Mediated ssDNA Breaks Accumulate Following Short-Term Exposure to the HDAC Inhibitor Romidepsin. Mol. Cancer Res. 2021, 19, 1361–1374. [Google Scholar] [CrossRef]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef] [Green Version]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [Green Version]
- Moukharskaya, J.; Verschraegen, C. Topoisomerase 1 Inhibitors and Cancer Therapy. Hematol. Clin. N. Am. 2012, 26, 507–525. [Google Scholar] [CrossRef]
- Ando, K.; Shah, A.K.; Sachdev, V.; Kleinstiver, B.P.; Taylor-Parker, J.; Welch, M.M.; Hu, Y.; Salgia, R.; White, F.M.; Parvin, J.D.; et al. Camptothecin resistance is determined by the regulation of topoisomerase I degradation mediated by ubiquitin proteasome pathway. Oncotarget 2017, 8, 43733–43751. [Google Scholar] [CrossRef] [Green Version]
- Rubin, E.H.; Li, T.-K.; Duann, P.; Liu, L.F. Cellular Resistance to Topoisomerase Poisons. Cancer Treat. Res. 1996, 87, 243–260. [Google Scholar] [CrossRef]
- Hammond, E.; Asselin, M.-C.; Forster, D.; O’Connor, J.; Senra, J.; Williams, K. The Meaning, Measurement and Modification of Hypoxia in the Laboratory and the Clinic. Clin. Oncol. 2014, 26, 277–288. [Google Scholar] [CrossRef]
- Ramachandran, S.; Ma, T.S.; Griffin, J.; Ng, N.; Foskolou, I.P.; Hwang, M.-S.; Victori, P.; Cheng, W.-C.; Buffa, F.M.; Leszczynska, K.B.; et al. Hypoxia-induced SETX links replication stress with the unfolded protein response. Nat. Commun. 2021, 12, 3686. [Google Scholar] [CrossRef]
- Kumar, A.; Fournier, L.-A.; Stirling, P.C. Integrative analysis and prediction of human R-loop binding proteins. G3 Genes Genomes Genet. 2022, 12, jkac142. [Google Scholar] [CrossRef]
- Yan, Q.; Wulfridge, P.; Doherty, J.; Fernandez-Luna, J.L.; Real, P.J.; Tang, H.-Y.; Sarma, K. Proximity labeling identifies a repertoire of site-specific R-loop modulators. Nat. Commun. 2022, 13, 53. [Google Scholar] [CrossRef]
- Meghani, K.; Fuchs, W.; Detappe, A.; Drané, P.; Gogola, E.; Rottenberg, S.; Jonkers, J.; Matulonis, U.; Swisher, E.M.; Konstantinopoulos, P.A.; et al. Multifaceted Impact of MicroRNA 493-5p on Genome-Stabilizing Pathways Induces Platinum and PARP Inhibitor Resistance in BRCA2-Mutated Carcinomas. Cell Rep. 2018, 23, 100–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockhart, A.; Pires, V.B.; Bento, F.; Kellner, V.; Luke-Glaser, S.; Yakoub, G.; Ulrich, H.D.; Luke, B. RNase H1 and H2 Are Differentially Regulated to Process RNA-DNA Hybrids. Cell Rep. 2019, 29, 2890–2900.e5. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Kumari, S.; Raghavan, S.C. Depletion of RNASEH2 Activity Leads to Accumulation of DNA Double-strand Breaks and Reduced Cellular Survivability in T Cell Leukemia. J. Mol. Biol. 2022, 434, 167617. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Kahila, M.M.; Zhou, Q.; Yu, J.; Kalari, K.R.; Wang, L.; Harmsen, W.S.; Yuan, J.; Boughey, J.C.; Matthew, P.; et al. ATR Inhibition Is a Promising Radiosensitizing Strategy for Triple-Negative Breast Cancer. Mol. Cancer Ther. 2018, 17, 2462–2472. [Google Scholar] [CrossRef] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsakrmy, N.; Cui, H. R-Loops and R-Loop-Binding Proteins in Cancer Progression and Drug Resistance. Int. J. Mol. Sci. 2023, 24, 7064. https://doi.org/10.3390/ijms24087064
Elsakrmy N, Cui H. R-Loops and R-Loop-Binding Proteins in Cancer Progression and Drug Resistance. International Journal of Molecular Sciences. 2023; 24(8):7064. https://doi.org/10.3390/ijms24087064
Chicago/Turabian StyleElsakrmy, Noha, and Haissi Cui. 2023. "R-Loops and R-Loop-Binding Proteins in Cancer Progression and Drug Resistance" International Journal of Molecular Sciences 24, no. 8: 7064. https://doi.org/10.3390/ijms24087064