
Severe Trauma-Induced Coagulopathy: Molecular Mechanisms Underlying Critical Illness


 ,
,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
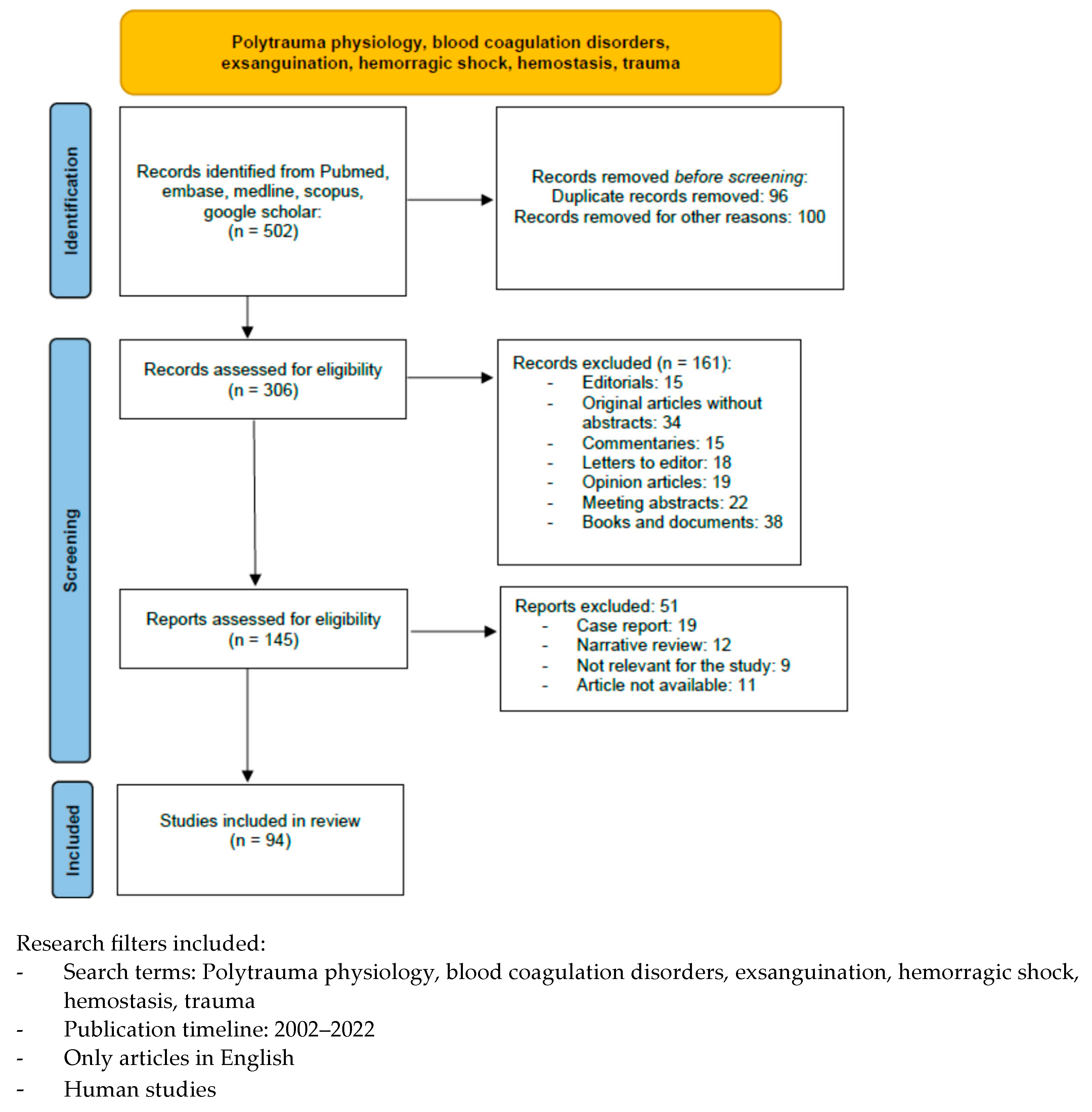
2. Materials and Methods
3. Results
4. Discussion
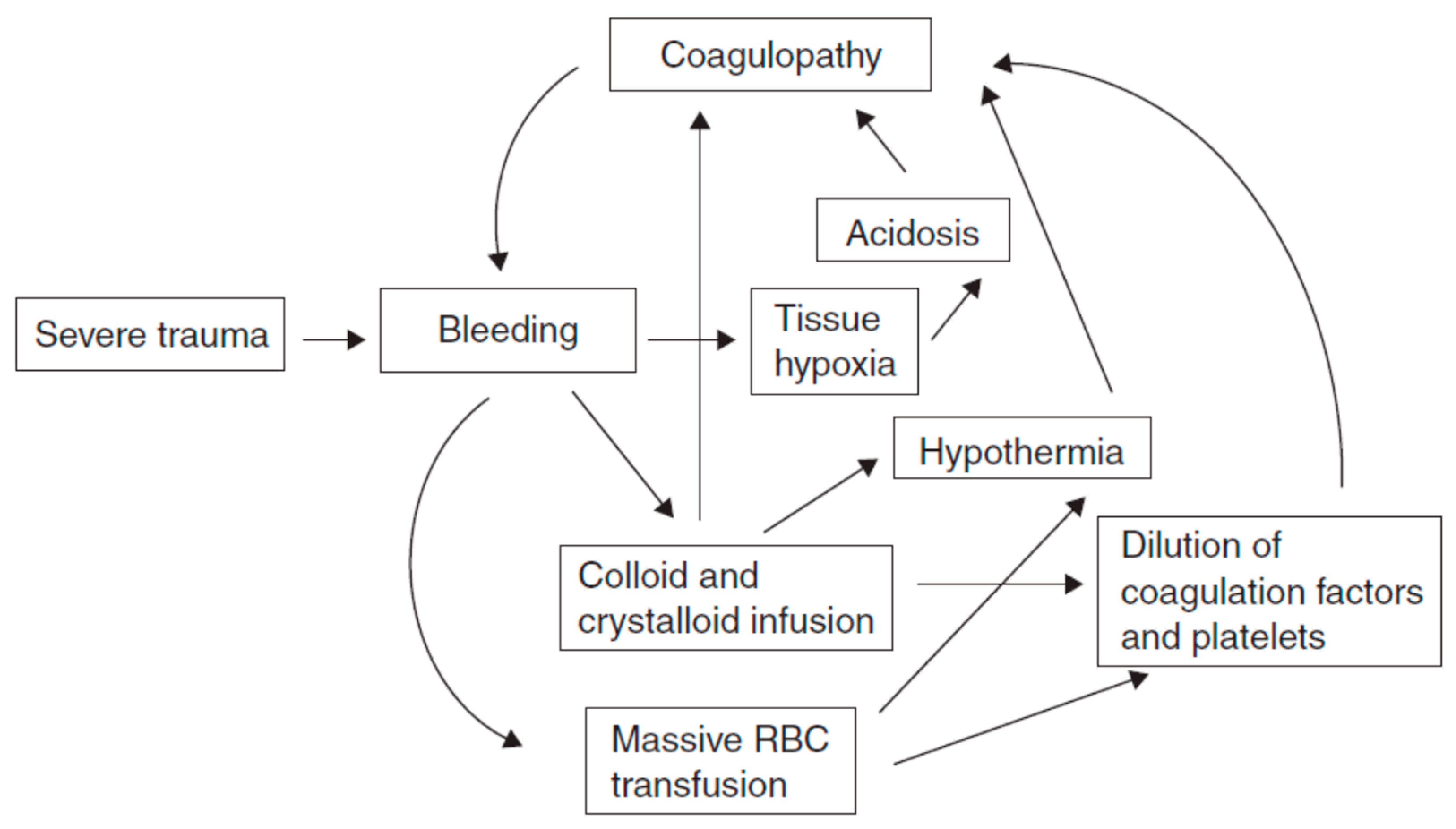
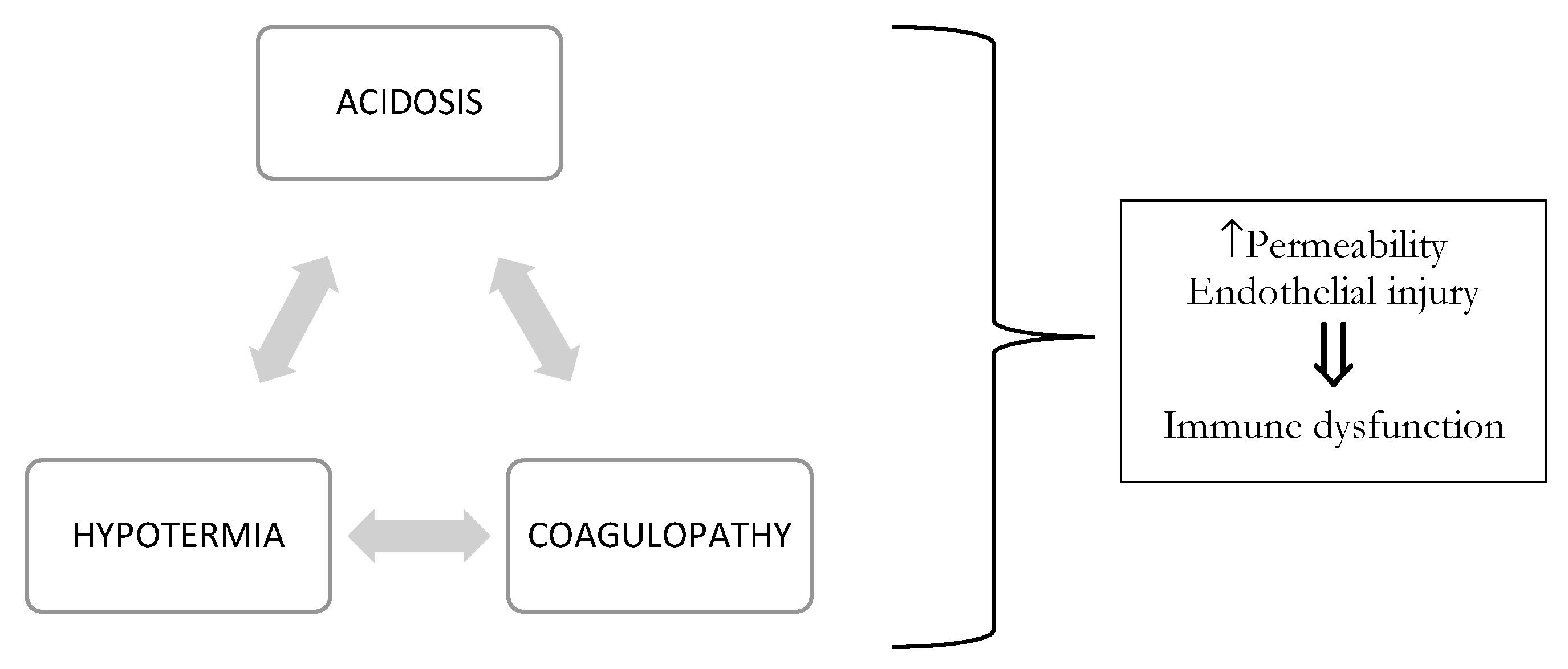
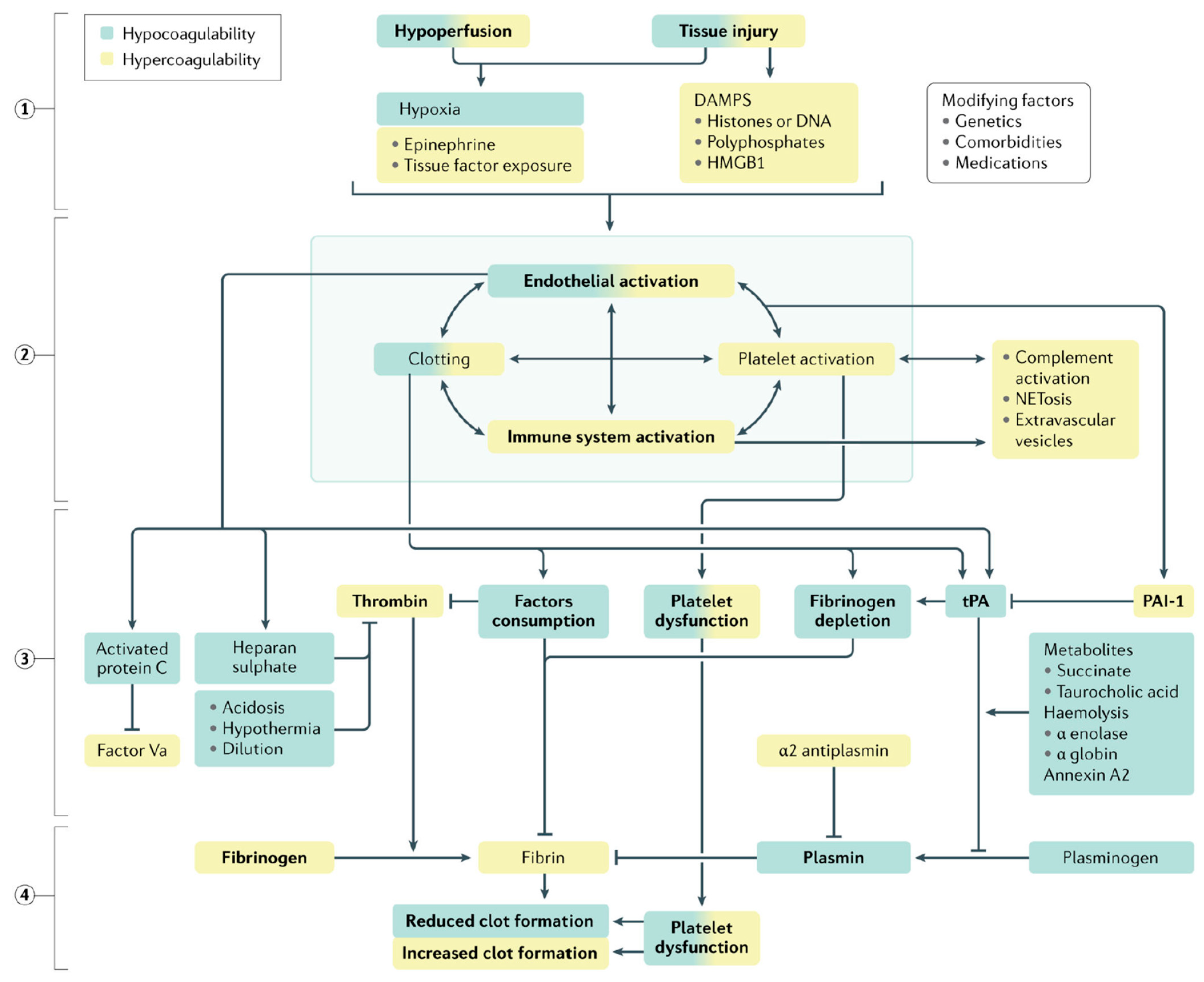
4.1. TIC Pathophysiology
4.1.1. Inflammatory Responses to Injury
4.1.2. Systemic Endothelial Dysfunction
4.1.3. Dysregulated Coagulation
4.1.4. Fibrinogen Depletion and Alterations in Fibrinolysis
4.1.5. Platelet Dysfunction
4.2. Diagnosis
4.3. Treatment
4.3.1. Goal-Directed Transfusion
4.3.2. Thromboelastography-Based Transfusion
4.3.3. Fibrinogen Concentrates
4.3.4. Pharmaceutical Hemostatic Agents
5. Conclusions
6. Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brohi, K.; Singh, J.; Heron, M.; Coats, T. Acute traumatic coagulopathy. J. Trauma 2003, 54, 1127–1130. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, J.B.; Lynn, M.; McKenney, M.G.; Cohn, S.M.; Murtha, M. Early coagulopathy predicts mortality in trauma. J. Trauma 2003, 55, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Maegele, M.; Lefering, R.; Yucel, N. Early coagulopathy in multiple injury: An analysis from the German Trauma Registry on 8724 patients. Injury 2007, 38, 298–304. [Google Scholar] [CrossRef]
- Saviano, A.; Ojetti, V.; Zanza, C.; Franceschi, F.; Longhitano, Y.; Martuscelli, E.; Maiese, A.; Volonnino, G.; Bertozzi, G.; Ferrara, M.; et al. Liver Trauma: Management in the Emergency Setting and Medico-Legal Implications. Diagnostics 2022, 12, 1456. [Google Scholar] [CrossRef] [PubMed]
- Lier, H. Preconditions of hemostasis in trauma: A review. The influence of acidosis, hypocalcemia, anemia, and hypothermia on functional hemostasis in trauma. J. Trauma 2008, 65, 951–960. [Google Scholar] [CrossRef]
- Zanza, C. Lactic Acidosis Related to Pharmacotherapy and Human Diseases. Pharmaceuticals 2022, 15, 1496. [Google Scholar] [CrossRef]
- Meng, Z.H.; Wolberg, A.S.; Hoffman, M. The effect of temperature and pH on the activity of factor VIIa: Implications for the efficacy of high-dose factor VIIa in hypothermic and acidotic patients. J. Trauma 2003, 55, 886. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, A.S.; Meng, Z.H.; Hoffman, M. A systematic evaluation of the effect of temperature on coagulation enzyme activity and platelet function. J. Trauma 2004, 56, 1221. [Google Scholar] [CrossRef]
- Tsuei, B.J.; Paul, A. Hypothermia in the trauma patient. Injury 2004, 35, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Farkash, U.; Lynn, M.; Scope, A. Does prehospital fluid administration impact core body temperature and coagulation functions in combat casualties? Injury 2002, 33, 103. [Google Scholar] [CrossRef]
- Chang, R.; Cardenas, J.C.; Wade, C.E.; Holcomb, J.B. Advances in the understanding of trauma-induced coagulopathy. Blood 2016, 128, 1043–1049. [Google Scholar] [CrossRef] [Green Version]
- Spivey, M.; Parr, M.J. Therapeutic approaches in trauma-induced coagulopathy. Minerva Anestesiol. 2005, 71, 281–289. [Google Scholar] [PubMed]
- Hess, J.R.; Brohi, K.; Dutton, R.P. The coagulopathy of trauma: A review of mechanisms. J. Trauma 2008, 65, 748–754. [Google Scholar] [CrossRef] [Green Version]
- Moore, H.B.; Gando, S.; Iba, T. Defining trauma-induced coagulopathy with respect to future implications for patient management: Communication from the SSC of the ISTH. J. Thromb. Haemost. 2020, 18, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Gando, S.; Levi, M.; Toh, C.H. Disseminated intravascular coagulation. Nat. Rev. Dis. Prim. 2016, 2, 16037. [Google Scholar] [CrossRef]
- Zanza, C.; Racca, F.; Longhitano, Y.; Piccioni, A.; Franceschi, F.; Artico, M.; Abenavoli, L.; Maiese, A.; Passaro, G.; Volonnino, G.; et al. Risk Management and Treatment of Coagulation Disorders Related to COVID-19 Infection. Int. J. Environ. Res. Public Health 2021, 18, 1268. [Google Scholar] [CrossRef]
- Sarah, E. Increased mortality associated with the early coagulopathy of trauma in combat casualties. J. Trauma 2008, 64, 1459–1463; discussion 1463–1465. [Google Scholar]
- Coleman, J.J. Whole blood thrombin generation is impaired in injured patients requiring a massive transfusion. J. Am. Coll. Surg. 2021, 232, 709–716. [Google Scholar] [CrossRef]
- Savioli, G.; Ceresa, I.F.; Macedonio, S.; Gerosa, S.; Belliato, M.; Luzzi, S.; Lucifero, A.G.; Manzoni, F.; Ricevuti, G.; Bressan, M.A. Major Trauma in Elderly Patients: Worse Mortality and Outcomes in an Italian Trauma Center. J. Emerg. Trauma Shock 2021, 14, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Ditzel, R.M., Jr. A review of transfusion- and trauma-induced hypocalcemia: Is it time to change the lethal triad to the lethal diamond? J. Trauma Acute Care Surg. 2020, 88, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Sloos, P.H.; Vulliamy, P.; van’t Veer, C. Platelet dysfunction after trauma: From mechanisms to targeted treatment. Transfusion 2022, 62 (Suppl. 1), S281–S300. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M. Pathophysiology of trauma-induced coagulopathy: Disseminated intravascular coagulation with the fibrinolytic phenotype. J. Intensive Care 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Longhitano, Y.; Racca, F.; Zanza, C.; Muncinelli, M.; Guagliano, A.; Peretti, E.; Minerba, A.C.; Mari, M.; Boverio, R.; Salio, M.; et al. Venous Thrombo-Embolism in Hospitalized SARS-CoV-2 Patients Treated with Three Different Anticoagulation Protocols: Prospective Observational Study. Biology 2020, 9, 310. [Google Scholar] [CrossRef]
- Pape, H.-C. Pathophysiology in patients with polytrauma. Injury 2022, 53, 2400–2412. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.J.; Call, M.; Nelson, M. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann. Surg. 2012, 255, 379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanza, C.; Romenskaya, T.; Manetti, A.C.; Franceschi, F.; La Russa, R.; Bertozzi, G.; Maiese, A.; Savioli, G.; Volonnino, G.; Longhitano, Y. Cytokine Storm in COVID-19: Immunopathogenesis and Therapy. Medicina 2022, 58, 144. [Google Scholar] [CrossRef]
- Chesebro, B.B.; Rahn, P.; Carles, M. Increase in activated protein C mediates acute traumatic coagulopathy in mice. Shock 2009, 32, 659. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Ji, Y.; Zhang, X. Endogenous activated protein C signaling is critical to protection of mice from lipopolysaccaride-induced septic shock. J. Thromb. Haemost. 2009, 7, 851. [Google Scholar] [CrossRef]
- Finigan, J.H.; Dudek, S.M.; Singleton, P.A. Activated protein C mediates novel lung endothelial barrier enhancement: Role of sphingosine 1-phosphate receptor transactivation. J. Biol. Chem. 2005, 280, 17286. [Google Scholar] [CrossRef] [Green Version]
- Finigan, J.H.; Boueiz, A.; Wilkinson, E. Activated protein C protects against ventilator-induced pulmonary capillary leak. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L1002–L1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.J.; Bir, N.; Rahn, P. Protein C depletion early after trauma increases the risk of ventilator-associated pneumonia. J. Trauma 2009, 67, 1176. [Google Scholar] [CrossRef]
- Ostrowski, S.R.; Johansson, P.I. Endothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathy. J. Trauma Acute Care Surg. 2012, 73, 60. [Google Scholar] [CrossRef]
- Johansson, P.I.; Stensballe, J.; Rasmussen, L.S.; Ostrowski, S.R. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann. Surg. 2011, 254, 194. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.G.; Ostrowski, S.R.; Cardenas, J.C.; Baer, L.A.; Tomasek, J.S.; Henriksen, H.H.; Stensballe, J.; Cotton, B.A.; Holcomb, J.B.; Johansson, P.I.; et al. Syndecan-1, A Quantitative Marker for the Endotheliopathy of Trauma. J. Am. Coll. Surg. 2017, 225, 419–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosnier, L.O.; Zlokovic, B.V.; Griffin, J.H. The cytoprotective protein C pathway. Blood 2007, 109, 3161. [Google Scholar] [CrossRef] [PubMed]
- Matijevic, N.; Wang, Y.W.; Wade, C.E. Cellular microparticle and thrombogram phenotypes in the Prospective Observational Multicenter Major Trauma Transfusion (PROMMTT) study: Correlation with coagulopathy. Thromb. Res. 2014, 134, 652. [Google Scholar] [CrossRef] [Green Version]
- Nekludov, M.; Mobarrez, F.; Gryth, D. Formation of microparticles in the injured brain of patients with severe isolated traumatic brain injury. J. Neurotrauma 2014, 31, 1927. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppenheim, J.J.; Yang, D. Alarmins: Chemotactic activators of immune responses. Curr. Opin. Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef]
- Cadroy, Y.; Diquélou, A.; Dupouy, D. The thrombomodulin/protein C/protein S anticoagulant pathway modulates the thrombogenic properties of the normal resting and stimulated endothelium. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 520. [Google Scholar] [CrossRef]
- Mosesson, M.W. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 2005, 3, 1894–1904. [Google Scholar] [CrossRef]
- Schlimp, C.J.; Schochl, H. The role of fibrinogen in trauma-induced coagulopathy. Hamostaseologie 2014, 34, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Raza, I. The incidence and magnitude of fibrinolytic activation in trauma patients. J. Thromb. Haemost. 2013, 11, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, A.R. Vitronectin functions as a cofactor for rapid inhibition of activated protein C by plasminogen activator inhibitor-1. Implications for the mechanism of profibrinolytic action of activated protein C. J. Biol. Chem. 2001, 276, 15567. [Google Scholar] [CrossRef] [Green Version]
- Bajzar, L.; Jain, N.; Wang, P.; Walker, J.B. Thrombin activatable fibrinolysis inhibitor: Not just an inhibitor of fibrinolysis. Crit. Care Med. 2004, 32, S320. [Google Scholar] [CrossRef]
- Rourke, C. Fibrinogen levels during trauma hemorrhage, response to replacement therapy, and association with patient outcomes. J. Thromb. Haemost. 2012, 10, 1342–1351. [Google Scholar] [CrossRef]
- McQuilten, Z.K.; Wood, E.M.; Bailey, M.; Cameron, P.A.; Cooper, D.J. Fibrinogen is an independent predictor of mortality in major trauma patients: A five-year statewide cohort study. Injury 2017, 48, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K. Impact of fibrinogen levels on outcomes after acute injury in patients requiring a massive transfusion. J. Am. Coll. Surg. 2013, 216, 290–297. [Google Scholar] [CrossRef]
- Davenport, R.A.; Brohi, K. Coagulopathy in trauma patients: Importance of thrombocyte function? Curr. Opin. Anesthesiol. 2009, 22, 261. [Google Scholar] [CrossRef]
- Rondina, M.T.; Weyrich, A.S.; Zimmerman, G.A. Platelets as cellular effectors of inflammation in vascular diseases. Circ. Res. 2013, 112, 1506–1519. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, A.S.; Zimmerman, G.A. Platelets: Signaling cells in the immune continuum. Trends Immunol. 2004, 25, 489–495. [Google Scholar] [CrossRef]
- Kutcher, M.E. Characterization of platelet dysfunction after trauma. J. Trauma Acute Care Surg. 2012, 73, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Brown, L.M.; Call, M.S.; Knudson, M.M. A normal platelet count may not be enough: The impact of admission platelet count on mortality and transfusion in severely injured trauma patients. J. Trauma 2011, 71, S337. [Google Scholar] [CrossRef] [Green Version]
- Savioli, G.; Zanza, C.; Longhitano, Y.; Nardone, A.; Varesi, A.; Ceresa, I.F.; Manetti, A.C.; Volonnino, G.; Maiese, A.; La Russa, R. Heat-Related Illness in Emergency and Critical Care: Recommendations for Recognition and Management with Medico-Legal Considerations. Biomedicines 2022, 10, 2542. [Google Scholar] [CrossRef]
- Starr, N.E.; Matthay, Z.A.; Fields, A.T. Identification of injury and shock driven effects on ex vivo platelet aggregometry: A cautionary tale of phenotyping. J. Trauma Acute Care Surg. 2020, 89, 20–28. [Google Scholar] [CrossRef]
- Kornblith, L.Z. Perhaps it’s not the platelet: Ristocetin uncovers the potential role of von Willebrand factor in impaired platelet aggregation following traumatic brain injury. J. Trauma Acute Care Surg. 2018, 85, 873–880. [Google Scholar] [CrossRef]
- Moore, H.B. Shock induced systemic hyperfibrinolysis is attenuated by plasma first resuscitation. J. Trauma Acute Care Surg. 2015, 79, 897–903; discussion 903–904. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [Green Version]
- Ding, N. Toll-like receptor 4 regulates platelet function and contributes to coagulation abnormality and organ injury in hemorrhagic shock and resuscitation. Circ.-Cardiovasc. Genet. 2014, 7, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Zipperle, J. Potential role of platelet-leukocyte aggregation in trauma-induced coagulopathy: Ex vivo findings. J. Trauma Acute Care Surg. 2017, 82, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.E.; Moore, H.B.; Kornblith, L.Z.; Neal, M.D.; Hoffman, M.; Mutch, N.J.; Schöchl, H.; Hunt, B.J.; Sauaia, A. Trauma-induced coagulopathy. Nat. Rev. Dis. Prim. 2021, 7, 30, Author Correction in Nat. Rev. Dis. Prim. 2022, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Gall, L.S.; Vulliamy, P.; Gillespie, S. The S100A10 Pathway Mediates an Occult Hyperfibrinolytic Subtype in Trauma Patients. Ann. Surg. 2019, 269, 1184. [Google Scholar] [CrossRef] [PubMed]
- Kornblith, L.Z.; Kutcher, M.E.; Redick, B.J. Fibrinogen and platelet contributions to clot formation: Implications for trauma resuscitation and thromboprophylaxis. J. Trauma Acute Care Surg. 2014, 76, 255. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.J.; Kutcher, M.; Redick, B. Clinical and mechanistic drivers of acute traumatic coagulopathy. J. Trauma Acute Care Surg. 2013, 75, S40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, E.; Moore, E.E.; Moore, H.B. Management of trauma-induced coagulopathy with thrombelastography. Crit. Care Clin. 2017, 33, 119–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veigas, P.V.; Callum, J.; Rizoli, S.; Nascimento, B. A systematic review on the rotational thrombelastometry (ROTEM®) values for the diagnosis of coagulopathy, prediction and guidance of blood transfusion and prediction of mortality in trauma patients. Scand. J. Trauma Resusc. Emerg. Med. 2016, 24, 114. [Google Scholar] [CrossRef] [Green Version]
- Martini, W.Z.; Chinkes, D.L.; Sondeen, J.; Dubick, M.A. Effects of hemorrhage and lactated Ringer’s resuscitation on coagulation and fibrinogen metabolism in swine. Shock 2006, 26, 396–401. [Google Scholar] [CrossRef]
- Mardel, S.N.; Saunders, F.M.; Allen, H.; Ibbotson, R.M. Reduced quality of clot formation with gelatin-based plasma substitutes. Br. J. Anaesth. 1998, 80, 204–207. [Google Scholar] [CrossRef] [Green Version]
- Mittermayr, M.; Streif, W.; Haas, T.; Fries, D.; Velik-Salchner, C.; Innerhofer, P. Hemostatic changes after crystalloid or colloid fluid administration during major orthopedic surgery: The role of fibrinogen administration. Anesth. Analg. 2007, 105, 905–917. [Google Scholar] [CrossRef]
- Wang, K.; Santiago, R. Tranexamic acid—A narrative review for the emergency medicine clinician. Am. J. Emerg. Med. 2022, 56, 33–44, Erratum in Am. J. Emerg. Med. 2022, 58, 353. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.J.; Dubose, J.J.; Rasmussen, T.E. Military Application of Tranexamic Acid in Trauma Emergency Resuscitation (MATTERs) Study. Arch. Surg. 2012, 147, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CRASH-2 Collaborators; Roberts, I.; Shakur, H. The importance of early treatment with tranexamic acid in bleeding trauma patients: An exploratory analysis of the CRASH-2 randomised controlled trial. Lancet 2011, 377, 1096–1101, 1101.e1–1101.e2. [Google Scholar]
- Spahn, D.R.; Bouillon, B.; Cerny, V. The European guideline on management of major bleeding and coagulopathy following trauma: Fifth edition. Crit. Care 2019, 23, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taeuber, I.; Weibel, S.; Herrmann, E.; Neef, V.; Schlesinger, T.; Kranke, P.; Meybohm, P. Association of Intravenous Tranexamic Acid With Thromboembolic Events and Mortality: A Systematic Review, Meta-analysis, and Meta-regression. JAMA Surg. 2021, 156, e210884. [Google Scholar] [CrossRef]
- Holcomb, J.B.; del Junco, D.J.; Fox, E.E. The prospective, observational, multicenter, major trauma transfusion (PROMMTT) study: Comparative effectiveness of a time-varying treatment with competing risks. JAMA Surg. 2013, 148, 127. [Google Scholar] [CrossRef]
- Holcomb, J.B.; Tilley, B.C.; Baraniuk, S. Transfusion of plasma, platelets, and red blood cells in a 1, 1, 1 vs a 1, 1, 2 ratio and mortality in patients with severe trauma: The PROPPR randomized clinical trial. JAMA 2015, 313, 471. [Google Scholar] [CrossRef]
- Moore, H.B.; Moore, E.E.; Chapman, M.P. Plasma-first resuscitation to treat haemorrhagic shock during emergency ground transportation in an urban area: A randomised trial. Lancet 2018, 392, 283. [Google Scholar] [CrossRef]
- del Junco, D.J.; Holcomb, J.B.; Fox, E.E. Resuscitate early with plasma and platelets or balance blood products gradually: Findings from the PROMMTT study. J. Trauma Acute Care Surg. 2013, 75, S24. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, E.; Moore, E.E.; Moore, H.B. Goal-directed hemostatic resuscitation of trauma-induced coagulopathy: A pragmatic randomized clinical trial comparing a viscoelastic assay to conventional coagulation assays. Ann. Surg. 2016, 263, 1051–1059. [Google Scholar] [CrossRef]
- Nascimento, B.; Callum, J.; Tien, H. Fibrinogen in the initial resuscitation of severe trauma (FiiRST): A randomized feasibility trial. Br. J. Anaesth. 2016, 117, 775–782. [Google Scholar] [CrossRef] [Green Version]
- Rizoli, S.B.; Boffard, K.D.; Riou, B. Recombinant activated factor VII as an adjunctive therapy for bleeding control in severe trauma patients with coagulopathy: Subgroup analysis from two randomized trials. Crit. Care 2006, 10, R178. [Google Scholar] [CrossRef] [Green Version]
- Dickneite, G.; Dörr, B.; Kaspereit, F.; Tanaka, K.A. Prothrombin complex concentrate versus recombinant factor VIIa for reversal of hemodilutional coagulopathy in a porcine trauma model. J. Trauma 2010, 68, 1151. [Google Scholar] [CrossRef]
- Ansell, J.; Hirsh, J.; Poller, L. The pharmacology and management of the vitamin K antagonists: The Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004, 126, 204S. [Google Scholar] [CrossRef] [Green Version]
- Joseph, B.; Amini, A.; Friese, R.S.; Houdek, M.; Hays, D.; Kulvatunyou, N.; Wynne, J.; O’Keeffe, T.; Latifi, R.; Rhee, P. Factor IX complex for the correction of traumatic coagulopathy. J. Trauma Acute Care Surg. 2012, 72, 828–834. [Google Scholar] [CrossRef]
- Joseph, B.; Hadjizacharia, P.; Aziz, H.; Kulvatunyou, N.; Tang, A.; Pandit, V.; Wynne, J.; O’Keeffe, T.; Friese, R.S.; Rhee, P. Prothrombin complex concentrate: An effective therapy in reversing the coagulopathy of traumatic brain injury. J. Trauma 2013, 74, 248–253. [Google Scholar] [CrossRef]
- Spinella, P.C.; Holcomb, J.B. Resuscitation and transfusion principles for traumatic hemorrhagic shock. Blood Rev. 2009, 23, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Brohi, K. Acute traumatic coagulopathy: Initiated by hypoperfusion: Modulated through the protein C pathway? Ann. Surg. 2007, 245, 812. [Google Scholar] [CrossRef]
- Joseph, B.; Aziz, H.; Pandit, V.; Hays, D.; Kulvatunyou, N.; Yousuf, Z.; Tang, A.; O’Keeffe, T.; Green, D.; Friese, R.S.; et al. Prothrombin complex concentrate versus fresh-frozen plasma for reversal of coagulopathy of trauma: Is there a difference? World J. Surg. 2014, 38, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, P.M.; Levi, M. Prevention and treatment of major blood loss. N. Engl. J. Med. 2007, 356, 2301. [Google Scholar] [CrossRef]
- Hanke, A.A.; Dellweg, C.; Kienbaum, P. Effects of desmopressin on platelet function under conditions of hypothermia and acidosis: An in vitro study using multiple electrode aggregometry. Anaesthesia 2010, 65, 688. [Google Scholar] [CrossRef] [PubMed]
- Ying, C.L.; Tsang, S.F.; Ng, K.F. The potential use of desmopressin to correct hypothermia-induced impairment of primary haemostasis-an in vitro study using PFA-100. Resuscitation 2008, 76, 129. [Google Scholar] [CrossRef] [PubMed]
- Savioli, G.; Ceresa, I.F.; Gri, N.; Bavestrello Piccini, G.; Longhitano, Y.; Zanza, C.; Piccioni, A.; Esposito, C.; Ricevuti, G.; Bressan, M.A. Emergency Department Overcrowding: Understanding the Factors to Find Corresponding Solutions. J. Pers. Med. 2022, 12, 279. [Google Scholar] [CrossRef] [PubMed]
- Zanza, C.; Piccolella, F.; Racca, F.; Romenskaya, T.; Longhitano, Y.; Franceschi, F.; Savioli, G.; Bertozzi, G.; De Simone, S.; Cipolloni, L.; et al. Ketamine in Acute Brain Injury: Current Opinion Following Cerebral Circulation and Electrical Activity. Healthcare 2022, 10, 566. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zanza, C.; Romenskaya, T.; Racca, F.; Rocca, E.; Piccolella, F.; Piccioni, A.; Saviano, A.; Formenti-Ujlaki, G.; Savioli, G.; Franceschi, F.; et al. Severe Trauma-Induced Coagulopathy: Molecular Mechanisms Underlying Critical Illness. Int. J. Mol. Sci. 2023, 24, 7118. https://doi.org/10.3390/ijms24087118
Zanza C, Romenskaya T, Racca F, Rocca E, Piccolella F, Piccioni A, Saviano A, Formenti-Ujlaki G, Savioli G, Franceschi F, et al. Severe Trauma-Induced Coagulopathy: Molecular Mechanisms Underlying Critical Illness. International Journal of Molecular Sciences. 2023; 24(8):7118. https://doi.org/10.3390/ijms24087118
Chicago/Turabian StyleZanza, Christian, Tatsiana Romenskaya, Fabrizio Racca, Eduardo Rocca, Fabio Piccolella, Andrea Piccioni, Angela Saviano, George Formenti-Ujlaki, Gabriele Savioli, Francesco Franceschi, and et al. 2023. "Severe Trauma-Induced Coagulopathy: Molecular Mechanisms Underlying Critical Illness" International Journal of Molecular Sciences 24, no. 8: 7118. https://doi.org/10.3390/ijms24087118
APA StyleZanza, C., Romenskaya, T., Racca, F., Rocca, E., Piccolella, F., Piccioni, A., Saviano, A., Formenti-Ujlaki, G., Savioli, G., Franceschi, F., & Longhitano, Y. (2023). Severe Trauma-Induced Coagulopathy: Molecular Mechanisms Underlying Critical Illness. International Journal of Molecular Sciences, 24(8), 7118. https://doi.org/10.3390/ijms24087118