Abstract
Breast cancer exhibits various epigenetic abnormalities that regulate gene expression and contribute to tumor characteristics. Epigenetic alterations play a significant role in cancer development and progression, and epigenetic-targeting drugs such as DNA methyltransferase inhibitors, histone-modifying enzymes, and mRNA regulators (such as miRNA mimics and antagomiRs) can reverse these alterations. Therefore, these epigenetic-targeting drugs are promising candidates for cancer treatment. However, there is currently no effective epi-drug monotherapy for breast cancer. Combining epigenetic drugs with conventional therapies has yielded positive outcomes and may be a promising strategy for breast cancer therapy. DNA methyltransferase inhibitors, such as azacitidine, and histone deacetylase inhibitors, such as vorinostat, have been used in combination with chemotherapy to treat breast cancer. miRNA regulators, such as miRNA mimics and antagomiRs, can alter the expression of specific genes involved in cancer development. miRNA mimics, such as miR-34, have been used to inhibit tumor growth, while antagomiRs, such as anti-miR-10b, have been used to inhibit metastasis. The development of epi-drugs that target specific epigenetic changes may lead to more effective monotherapy options in the future.
1. Introduction
Breast cancer is one of the most frequently diagnosed proliferative diseases in women worldwide, with an estimated number of 2.3 million new cases globally according to the GLOBOCAN 2020 data [1,2]. The risk of developing the disease is much higher in genetically predisposed women. There is no doubt that the genetic background is of key importance at every stage of the disease, starting with diagnosis and prognosis, monitoring the progression and choosing the right treatment protocol [3,4,5,6,7].
The vast majority (approx. 80–90%) of breast cancer cases are the so-called sporadic tumors. Up to 20% of diagnoses are hereditary cancers, associated with the presence of germline mutations, most often in the BRCA1 (17q21.31) or BRCA2 (13q13.1) genes. Approximately 5–15% of all diagnoses of the disease are cases with familial aggregation, for some of which the presence of mutations in the predisposition genes cannot be confirmed [1,8,9,10]. The most common cause of hereditary breast cancer are mutations in the BRCA1 or BRCA2 genes. Nevertheless, there are dozens of genes associated with the risk of breast cancer. Among them, genes participating in the repair of double-stranded DNA damage predominate. Mutations and polymorphisms in them can lead to abnormal cell growth, which can lead to the development of cancer. Among the genes that are associated with the development and progression of breast cancer, the following are currently listed [8,10,11,12,13,14,15,16,17,18,19,20]:
- BRCA1 and BRCA2 genes, which have the best documented association with breast cancer; having a mutation in these genes is responsible for a 50–80% risk of breast cancer and a 45% risk of ovarian cancer before the age of 85—with a mutation in the BRCA1 gene and a 31–56% risk of breast cancer and 11–27% of ovarian cancer in BRCA2 mutation [21,22,23,24,25,26,27,28,29,30];
- the PALB2 gene, which is responsible for the repair of damaged DNA; carriers of the defective gene have a 35% risk of developing breast cancer before the age of 70 [31,32,33,34,35,36];
- the CHEK2 gene, which is responsible for the production of a protein that inhibits tumor growth; women with a mutation in this gene have a twice as high a risk of developing breast cancer compared to the general population [37,38,39,40,41,42];
- the NBN gene, which encodes a protein regulating the DNA repair process and maintaining chromosome stability [43,44,45,46,47];
- CDKN2A gene, associated with the formation of proteins regulating the course of the cell cycle and inhibiting the growth of cancer cells [48,49,50,51,52];
- other, less known genes whose mutations may increase the risk of breast and other cancers, but which have not yet been precisely described, include TP53, PTEN, CASP8, CTLA4, BARD1, BRIP1, CYP19A1, ATM, FGFR2, H19, LSP1, MAP3K1, MAP2K4, MRE11A, RAD51 and TERT [53].
According to current guidelines, breast cancer risk assessment should be based primarily on the analysis of 14 genes: ATM, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, NBN, NF1, PALB2, PTEN, RAD51C, RAD51D, STK11, TP53. Genetic testing should be performed on every patient who: developed breast cancer before the age of 50, was diagnosed with triple-negative cancer, or developed ovarian cancer. In carriers of mutations in the BRCA1 and BRCA2 genes, the risk of developing breast cancer increases to even 80%. In addition, these BRCA1-associated breast tumors are usually triple-negative for estrogen receptor α (ER-), progesterone receptor (PR-) and HER2 (HER2-), which makes the development of targeted therapies difficult [21,22,23,24].
Detection of a specific mutation enables the implementation of appropriate further diagnostics and the development of individual and preventive recommendations, including imaging tests (ultrasound, magnetic resonance imaging, mammography). It has been shown that hereditary cancers often coexist in pairs: cancer appears in both breasts, it can develop in the ovaries, fallopian tubes, pancreas, and the risk of melanoma increases. Knowledge of the genetic background therefore enables effective and targeted monitoring of organs that are particularly vulnerable to the manifestation of the disease. Early detection of mutations enables the appropriate selection of diagnostic tests and providing the affected families with proper care and genetic counseling.
Genetic testing also plays an extremely important role in planning treatment. Conventional treatment protocols have greatly improved the management of BC patients, but subtype heterogeneity, the emergence of drug resistance, and disease relapse are major factors hampering the effectiveness of BC therapy. In the case of cancer patients, information on the genetic load changes the scope of surgery and adjuvant treatment. Knowledge about the genetic load is important for planning treatment, but also for the patient’s family, due to the high probability of the offspring inheriting the disease-causing mutation.
2. Epigenetic Regulations
In addition to the study of germline genetic changes, epigenetic changes affecting the modulation of predisposition gene expression, as well as causing disturbances in signaling pathways, especially those related to DNA repair, are being increasingly studied. Epigenetics is a critical mechanism for regulating transcription [54,55]. Epigenetic information is encoded in the structure and function of covalent modifications of DNA (hypomethylation or hypermethylation) and DNA-related nuclear chromatin histone proteins (such as acetylation, methylation, phosphorylation, sumoylation, ubiquitylation, or ADP-ribosylation). In the last few years, attention has been increasingly paid to the role of microRNAs as diverse expression regulators with high therapeutic potential. Epimutations can lead to the silencing of tumor suppressor genes independently and also in combination with pathogenic genetic mutations. Epimutations can also promote tumorigenesis by activating oncogenes. The events that lead to the onset of these epigenetic abnormalities are still not fully understood. Nevertheless, because epigenetic changes are inherited, they are selected in the rapidly growing population of cancer cells and provide a growth advantage for cancer cells, promoting their uncontrolled growth. For many genes of high predisposition, typical epigenetic changes have been determined, which affect the abnormal function of these genes and are one of the causes of cell transformation. Individual epimutations may act independently or together. As in the case of the BRCA1 gene (Figure 1), they can be differentiated and involve different mechanisms. BRCA1 alters epigenetics through physical interactions and transcriptional regulation of known epigenetic modifiers. In addition, as an E3 ubiquitin ligase, BRCA1 directly ubiquitylates histones.
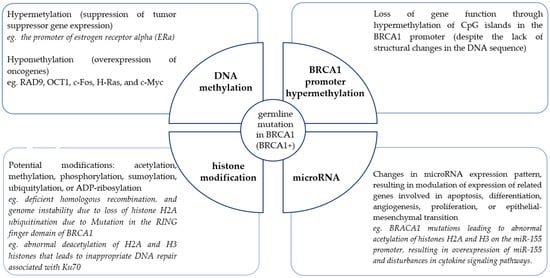
Figure 1.
Landscape of epigenetic changes in familial breast cancer associated with the BRCA1 gene (based on [56]).
Disturbances in the processes of epigenetic reprogramming of genomic DNA transcriptional activity may be the cause of neoplastic transformation. The fact that epigenetic aberrations, unlike genetic mutations, are potentially reversible and can be restored to their normal state by epigenetic therapy makes any epigenetic research promising and therapeutically relevant.
BRCA1/2-associated hereditary breast cancer is characterized by global changes in DNA methylation. In the presence of BRCA1/2 mutations, cancer cells are unable to properly repair DNA damage, leading to uncontrolled proliferation and tumor development. Changes in DNA methylation affect the expression of genes involved in DNA repair processes, cell signaling, proliferation, and differentiation, which further contributes to tumor development [57]. In the case of familial breast cancer not associated with BRCA1/2 mutations, changes in DNA methylation are also observed. It was shown that changes in DNA methylation were more correlated with familial breast cancer than with sporadic cases. These changes involved promoter regions of genes involved in cell cycle regulation, signaling, and apoptosis. In the case of sporadic breast cancer, changes in DNA methylation are more diverse and associated with the expression of different breast cancer subtypes. One study showed that DNA methylation patterns in breast cancer subtypes are so diverse that they can be used for classification [58].
It is believed that no two breast cancers are the same, and that the molecular profile of a breast tumor is different in each patient; therefore, it is very important to choose the right therapy. Unfortunately, in breast cancer, we still have few targeted therapies that directly target the changes that led to the cancer; however, the possibilities are constantly changing. The aim of this paper is to present the current state of knowledge on epigenetic factors and determining mechanisms in breast cancer, especially genetically determined, with particular emphasis on their impact on therapy. For many years, advanced research has been conducted on epigenome changes in breast cancer, and its goal is not only to understand the role of epigenetics in the development and progression of BC, but also to search for epi-drugs and develop new protocols based on epigenetic therapies.
3. Epigenetic Therapy in Breast Cancer
The reversible nature of epigenetic changes during oncogenesis has prompted the consideration of epigenetic therapy. Epigenetic therapy is a therapeutic approach that focuses on chemical modifications that affect gene activity in cancer cells. In breast cancer treatment, epigenetic therapy aims to restore the normal expression of tumor suppressor genes, which usually inhibit the growth of cancer cells. There are different types of epigenetic therapy, but the most commonly used in breast cancer treatment are histone deacetylase inhibitors (HDACi), DNA methyltransferase inhibitors (DNMTi) and RNA inhibitors [57,59]. Nevertheless, the search for effective epigenetic therapies has broadened and, in addition to these well-known inhibitors and target miRNAs, inhibition of HAT, class I, II, and IV specific HDACs, class III HDACs (sirtuins), KMT, KDM, and many kinase are being considered [60]. HDAC inhibitors inhibit the activity of enzymes that remove acetyl groups from histones, which leads to chromatin changes and activation of tumor suppressor genes. On the other hand, DNMT inhibitors inhibit the activity of enzymes that add methyl groups to DNA, which usually leads to the blocking of tumor suppressor gene expression. Inhibitors of epigenetic enzymes are referred to as “epi-drugs” [61,62,63,64,65]. RNA inhibitors, block the transcription and translation of mRNA, affecting the regulation of gene expression. This class of epigenetic regulators is most often used in the so-called replacement therapy [66].
Clinical studies have shown that epigenetic therapy can be effective in breast cancer treatment, especially in hormone-resistant breast cancer. Epigenetic drugs are being tested with increasing frequency, especially those effective in reversing DNA methylation and histone modification aberrations. In therapeutic concepts, it is important to take into account the interactions between various elements of the epigenetic machinery, and especially the synergistic effect of DNA and HDAC methylation inhibitors. In the case of microRNA expression disorders, therapeutic molecules (mimic or antagomiRs) are designed, whose task is to regulate the target miRs [66]. Combinatorial treatment strategies have been found to be more effective than individual therapeutic approaches. The first epigenetic drugs were tested for hematologic cancers.
4. DNA Methylation Aberrations
The first epimutations identified for the initial stages of tumor initiation and progression are profound changes in DNA methylation. A hallmark of the epigenome of cancer cells is genome-wide hypomethylation and promoter-specific hypermethylation of the CpG island promoter. The consequence of DNA hypomethylation is increased genomic instability, promoting chromosomal rearrangements. It also results in abnormal activation of genes (proto-oncogenes) and non-coding regions through various mechanisms that contribute to tumor development and progression.
Site-specific hypermethylation contributes to oncogenesis by silencing tumor suppressor genes. Such a silencing mechanism was confirmed for BRCA1. In addition to the direct inactivation of tumor suppressor genes, DNA hypermethylation can also indirectly lead to the silencing of various classes of genes by inactivating transcription factors and DNA repair genes [56,67]. Silencing DNA repair genes allows cells to accumulate further genetic changes leading to rapid tumor progression. The ability of DNA hypermethylation to silence predisposition genes in breast cancer is well-known, nevertheless, how the genes target this aberrant DNA methylation is not well-characterized. This mechanism is believed to provide a cell growth advantage, resulting in clonal selection and proliferation. Further understanding of how specific regions of the genome target DNA hypermethylation in cancer could potentially lead to the identification of additional therapeutic targets.
Hypermethylation most often concerns tumor suppressor genes however, it has also been confirmed for DNA repair genes, apoptosis, cell cycle regulation, cell growth, homeostasis and adhesion [62,68]. It is believed that DNA methylation status may be of value as both a diagnostic and predictive marker, including response to therapy. DNA methylation levels have been confirmed to be high at the gene loci proapoptotic genes (HOXA5, TMS1), cell cycle inhibitory genes (p16, RASSF1A) and DNA repair genes [69,70]. It has been shown that hypermethylation disorders of the genes encoding the estrogen receptor alpha and the progesterone receptor are correlated with the silencing of these genes and the development of ER- and PR-negative breast cancer. Hypermethylation of the RASSF1A gene is considered to be an important BC diagnostic marker, and the PITX2 gene is considered a marker of tamoxifen resistance [62].
The relationship between BRCA and DNA methylation is the most widely studied [71,72]. The promoter of the BRCA1 gene has more than 90 CpG islands, and the promoter of the BRCA2 gene has 70 sites that can undergo methylation [73]. The BRCA2 promoter is more often methylated; however, this gene is less frequently studied compared to BRCA2 [73]. BRCA1-associated breast tumors show less DNA methylation compared to sporadic breast tumors [71,72,74]. BRCA1 gene expression is significantly reduced or completely inhibited by the methylation of its promoter [75,76,77]. Hypermethylation of the promoter of this gene in sporadic breast cancer results in loss of function of this gene analogous to its mutation in hereditary cases [77,78]. CpG island promoter methylation is more common in young women with high-grade pathology and triple-negative breast cancer (negative estrogen receptor, negative progesterone receptor, negative ERBB2 [Her2/Neu]) [79,80,81,82]. In addition, mutated BRCA1 is correlated with a number of epigenetic modifications in carriers. BRCA1 physically interacts with the de novo methyltransferase DNMT3B, modulates heterochromatin [83], regulates the transcription of the methylation maintenance enzyme DNMT1 and prevents global DNA [67]. Genome hypermethylation in BRCA1 carriers has been shown to be significantly correlated with cancer development [84]. Strong methylation of the estrogen receptor promoter alpha is the reason for the silencing the expression of this gene in familial cases of breast cancer [85]. Loss of function by mutated BRCA1 leads to increased hypomethylation, which promotes proliferation and invasiveness, due to the increased expression of oncogenes such as RAD9, c-Fos, H-Ras and c-Myc [67,86].
Observations on the methylation status of genes involved in the initiation and progression of breast cancer have been used in attempts to improve the effectiveness of anti-cancer therapies. In vitro studies, animal models and clinical studies (Table 1) have shown the effectiveness of nucleoside analogs, e.g., cytidine analogs, i.e., azacytidine (5-aza-CR; Vidaza®, Celgene Corp., Summit, NJ, USA) and decitabine (5-aza-2′-deoxycytidine, 5-aza-CdR; Dacogen®, SuperGen, Inc., Dublin, CA, USA), in inhibiting DNA methylation in cancer cells [87]. Under culture conditions, after introducing the analogs, gene expression was induced, causing the differentiation of these cells. Cytidine chemical analogues are incorporated into the DNA of rapidly growing tumor cells during replication, and inhibit DNA methylation by forming covalent bonds with DNMT, leading to their depletion inside the cell [87]. The downregulation of DNA methylation induced by these drugs is responsible for inhibiting the growth of cancer cells by activating tumor suppressor genes that are abnormally silenced in cancer cells [88,89]. Studies have shown a reduction in tumor size in xenograft mice [90,91,92]. Tsai et al. [92] showed a correlation between the treatment of mice with azacytidine and the inhibition of tumor growth at the 5th week of therapy. Tao et al. [93] analyzed the possibility of breast cancer therapy using azacitidine. In their experiment, using the MCF7 and MDA-MB-231 cell lines, it was possible to inhibit 23 out of 26 hypermethylated genes in breast cancer. Moreover, for the CLDN6, PRA, RIN1 and VGF genes, the ability to reactivate their expression was confirmed [93]. Efforts to treat breast cancer using decitabine, which prevents DNA remethylation, are also promising. Cai et al. [94] reported that this analog is able to activate the apoptosis-inducing ligand associated with tumor necrosis factor (TRAIL) in triple-negative breast cancer cells, thereby sensitizing cancer cells to chemotherapy. In addition, reductions in tumor growth have been observed in animal models [90,92]. CTFR is highly hypermethylated in breast cancer. This change was characteristic of invasive cancers. Low levels of CFTR protein correlated with poor patient survival. Treatment with decitabine enabled high expression of CTFR and thus inhibition of cell growth [95]. Borges et al. [90] noted the importance of aberrant methylation of the PRKD1 gene promoter during tumor progression and lung metastasis. Restoring the normal methylation pattern of this gene with decitabine affected tumor aggressiveness and inhibited tumor spread in a PKD1-dependent manner. These researchers emphasized that the promoter status of the PKD1 gene may be both a marker of early diagnosis and of significant importance in therapy.
Restoring the correct epigenetic state is also important for genes for which mutations have not been found, but they are inactivated in cancer cells. Genes such as RASSF1, GSTP1, MGMT, and BRMS1 are markers of response to therapy, including difficult cases. Demethylation has also been shown to have promising effects in overcoming resistance to hormone therapy [62]. Restoration of expression of genes such as ESR1 [96] or PITX2 [97] sensitizes cancer cells to tamoxifen, and the analysis of promoter methylation status of these genes enables the identification of patients with positive expression of the hormone receptor that will not benefit from the drug administration [62].

Table 1.
Exemplary clinical studies on the role of methyltransferase inhibitors in breast cancer (based on ClinicalTrials.gov (accessed on 5 March 2023) [98]).
Table 1.
Exemplary clinical studies on the role of methyltransferase inhibitors in breast cancer (based on ClinicalTrials.gov (accessed on 5 March 2023) [98]).
| NCT Number | Study Type | Description | Outcome Measures | Study Population |
|---|---|---|---|---|
| Azacitidine | ||||
| NCT04891068 | Interventional | Determination of the effect of low-dose azacitidine therapy on tumor-infiltrating lymphocytes (TILs) in primary tumors from patients with high-risk early stage breast cancer. | Clinical response (change Ki67 and tumor size) of primary tumor following treatment with low-dose azacitidine therapy, DFS and OS measures. | Age ≥ 18 years of age at time of consent |
| NCT01349959 | Interventional | Evaluation of the response rate using RECIST criteria of the combination of azacitidine and entinostat in women with advanced breast cancer, triple-negative and hormone-refractory. | Clinical Benefit Rate, OS, PFS, change in expression of relevant genes (e.g., ER alpha and RAR beta). | Histologically or cytologically confirmed adenocarcinoma of the breast triple-negative (ER-, PR-, HER2- or hormone-positive/ HER2-, with evidence of locally advanced and inoperable or metastatic disease). |
| NCT01292083 | Interventional | Evaluation of the ability of DNA methylation inhibition using 5-azacitidine to induce expression of the ER and PR genes in solid human triple-negative invasive breast cancer. | Percentage of participants with ER/PR response after receiving 10 doses of 5-Azacitidine. | Resectable tumor measuring 2 cm or more; triple-negative invasive breast cancer |
| NCT02374099 | Interventional | Assessing the efficacy and safety of the combination of fulvestrant with CC-486 in subjects with ER+, HER2- metastatic breast cancer who have progressed after prior AI. | Percentage of participants who achieved a confirmed CR, PR or SD to treatment, estimation of DoR and TEAEs. | ≥18 years of age with metastatic breast cancer |
| NCT00748553 | Interventional | Testing whether treatment of patients with advanced or metastatic solid tumors or breast cancer with Abraxane plus Vidaza is safe and results in good tumor response. | Percentage of participants with ORR measured using RECIST 1.0 criteria, including CR, PR, SD, or PD. | Patients with advanced or metastatic HER2-negative breast cancer who have not received treatment for their metastatic disease. |
| Decitabine | ||||
| NCT03295552 | Interventional | Evaluation the effect of novel DNA demethylating agents in the treatment of metastatic TNBC (drugs: decitabine, carboplatin). | Partial response (PR) + complete response (CR) rate. | Pathologically confirmed metastatic triple-negative breast cancer, age between 18 years and 70 years. |
| NCT02957968 | Interventional | Course of immunotherapy consisting of sequential decitabine followed by pembrolizumab administered prior to a standard neoadjuvant chemotherapy regimen for patients with locally advanced HER2-negative breast cancer. | Determination of whether the immunotherapy increases the presence and percentage of tumor and/or stromal area of infiltrating lymphocytes prior to initiation of standard neoadjuvant chemotherapy. | Invasive adenocarcinoma of the breast, HER2-negative. |
| NCT03282825 | Interventional | Decitabine and paclitaxel combination therapy in treating patients with metastatic and locally advanced breast cancer. | Measure of maximum tolerant dose (MTD) and dose limiting toxicity (DLT). | Unable to operate for therapy with HER2 negative breast adenocarcinoma and metastatic breast cancer, one or more chemotherapy. |
| NCT01194908 | Interventional | Reactivation of ER using decitabine in combination with LBH589 (deacetylase inhibitor). Reactivated ER can then be targeted with agents such as tamoxifen. | Measure of MTD of Decitabine and LBH589 given in combination and determination of the safety of tamoxifen in combination with decitabine and LBH589. | ER-, PR-, HER2- metastatic or locally advanced breast cancer. |
| FdCyd | ||||
| NCT00978250 and NCT01479348 | Interventional | Testing of FdCyd (also called 5-fluoro-2′-deoxcytidine), and THU (also called tetrahydrouridine) effectiveness in treating cancer that has not responded to standard therapies. | Determination of PFS and/or the response rate (CR + PR) of FdCyd. | Individuals who were 18 years of age and older who have advanced non-small cell lung cancer, breast cancer, bladder cancer, or head and neck cancer that has progressed after receiving standard treatment. |
In the case of breast cancer, DNMTi can be used as an adjunct therapy in the treatment of advanced and hormone-resistant breast cancer. The studies that have been conducted have shown that DNMTi reduce the size of breast tumors and inhibit their growth, in addition to improving the effectiveness of hormonal therapy. In addition, DNMTi may also contribute to the inhibition of the development of metastases in other organs, which may improve the overall effectiveness of treatment. However, DNMTi therapy is still in the clinical trial stage, and its effectiveness in the treatment of breast cancer requires further research and confirmation.
5. Changes in Histone Modifications
Covalent modifications of histones most often concern the N-terminal tails, which can undergo various post-translational changes including the following: methylation, acetylation, ubiquitylation, sumoylation and phosphorylation at specific residues [99]. These are key changes in the regulation of cellular processes such as transcription, replication and genome repair. Depending on which residues are modified and the type of modifications present, histone modifications can result in activation or repression. The global consequence of HDAC-mediated loss of acetylated H4-lysine trimethylation is gene repression. In turn, alteration of the methylation pattern, especially of H3 histones, mediated by HMT, has been associated with abnormal gene silencing [100]. The most common histone modification is acetylation, a dynamic event carried out by histone acetyltransferases (HATs) and the deacetylase complex histone factor (HDAC). These enzymes, respectively, add and remove acetyl groups from the tails of [101].
BRCA1 is involved in numerous histone modifications leading to the modulation of chromatin activity. Of these, the most common are: histone H2A ubiquitination (resulting in satellite DNA overexpression, homologous recombination disorders, genomic instability) [102,103,104], deacetylation of H2A and H3, interactions with HDAC1 resulting in deacetylation of DNA repair genes (e.g., KDM5B, Ku70 pathway genes) [105,106,107,108]. In addition, BRCA1 interacts with CBP and p300, two structurally related HATs [101]. Zheng et al. [109] described the mechanism of estrogen receptor repression through the interaction of the BRCA1 gene with the catalytic subunits of the deacetylase complex histone. BRCA1-dependent ERα repression is largely restored by the HDAC inhibitor trichostatin A. Moreover, BRCA1 and HDAC2 interactions have an effect on the epigenetic silencing of mir-155 [110]. Silencing of miR-155 expression occurs upon the binding of BRCA1 to the oncomiR promoter. The consequence of the combination is the recruitment of HDAC2 to deacetylate histones H2A and H3. It has been experimentally shown that the administration of HDAC inhibitors restores the normal level of miR-155 expression. However, this is not possible in BRCA1-deficient cells because a mutation in the BRCT domain prevents interaction with HDAC2 [110]. Loss of histone acetylation may be the cause of gene silencing abnormalities. Therefore, it was considered that treatment with HDAC inhibitors, leading to the restoration of normal histone acetylation patterns, may have anti-cancer effects, following growth arrest, activation of apoptosis and induction of differentiation. Thanks to the introduction of HDAC inhibitors into cancer cells, it is possible to reactivate silenced tumor suppressor genes. Recently, HMT inhibitors (e.g., DZNep) are also being actively investigated.
There are four groups of HDACs: (1) short-chain fatty acids, e.g., sodium butyrate and valproic acid; (2) hydroxamic acids, e.g., trichostatin A, vorinostat, Panobinostat; (3) cyclic tetrapeptides, e.g., depsipetide, romidepsin / isostax; (4) benzamides, e.g., entinostat, tacedynalin [94,111]. Numerous HDAC inhibitors have also been tested to date, but only vorinostat and romidepsin (the treatment of lymphoma skin with T cells) [112,113,114,115,116,117]. Nevertheless, the clinical use of these epidrugs is acceptable in a number of solid tumor cancers, including breast cancer (Table 2), due to in vitro and in vivo antitumor activity [118,119,120,121,122]. HDAC monotherapy has been shown to have a positive effect on apoptosis induction, growth arrest, and differentiation of breast cancer [94,111,118,119,123]. Although numerous histone modifications typical of breast cancer have been described in the scientific literature, the positive effect of monotherapy with inhibitors has not yet been unequivocally proven. Much more promising are the effects of sensitizing cancer cells to radiotherapy and conventional anticancer drugs [94,124]. In overcoming drug resistance, including HER2-targeted therapies, the effectiveness has been confirmed, among others, by for valproic acid, trichostatin A and entinostat [125]. Huang et al. [125] tested the efficacy of SNDX-275, a class I HDAC inhibitor, to overcome trastuzumab resistance in erbB2 overexpressing patients. The mechanism of overcoming drug resistance involved a radical reduction of erbB3 and its phosphorylation (P-erbB3) and inhibition of Akt signaling. The combination of trastuzumab therapy and SNDX-275 significantly enhanced DNA fragmentation, induction of PARP cleavage and caspase-3 activation. The synergy effect was observed in both sensitive and resistant cells [125]. A similar effect was also observed in tamoxifen-resistant cells. Activation of ESR1 gene expression by HDACi sensitized cells to estrogen receptor-targeted therapy [126,127,128,129]. Unfortunately, the opposite effect is also observed, mainly due to the non-selective effect of HDACi on non-histone proteins. Fisk et al. [130] reported the effects of increased acetylation of heat shock proteins after the administration of verinostat. Hyperacetylation of hsp90 inhibits its protective function. As a result of this modification, the level of ERalpha decreased in cancer cells and increased ubiquitination. Munster et al. [131] analyzed the effect of vorinostat on tamoxifen therapy. In their study, they assessed histone acetylation and HDAC2 expression in peripheral blood mononuclear cells. From these, they observed significant clinical benefits for patients that correlated with histone hyperacetylation and higher baseline HDAC2 levels. Yardley et al. [132] reported the effect of an HDAC inhibitor on overcoming resistance to hormone therapies in estrogen receptor positive breast cancer. For this purpose, they conducted phase II clinical trials evaluating entinostat in combination with exemestane in patients with metastatic breast cancer. Based on their observations, they concluded that changes in the acetylation pattern may be of clinical benefit in ER-targeted therapies, as the combination of exemestane with entinostat significantly improved progression-free survival and overall survival [132]. Ramaswamy et al. [133] analyzed the efficacy of vorinostat with paclitaxel and bevacizumab in a phase I/II study involving 54 patients with metastatic breast cancer. They observed a significant effect of vorinostat on the improvement of treatment results (49% objective response rate (partial + complete remission) and 78% clinical benefit rate (objective response + disease stabilization > 6 months), with not too severe side effects. As a mechanism of tumor cell sensitization, the authors indicate the induction of histone and alpha tubulin acetylation, and epigenetic inhibition of Hsp90 function [133].
Table 2.
Exemplary clinical studies on the role of histone deacetylase inhibitors in breast cancer (based on ClinicalTrials.gov (accessed on 5 March 2023) [98]).
In the treatment of breast cancer, HDACi can be used both as stand-alone drugs and in combination with other anti-cancer therapies. Studies have shown that HDACi have the ability to induce apoptosis of breast cancer cells, which contributes to the reduction in tumor volume. They can also act synergistically with hormonal drugs and increase their effectiveness. HDACi can also be used in the therapy of breast cancer associated with mutations in the BRCA1 gene, which is involved in DNA damage repair. Research suggests that HDACi may contribute to increasing the sensitivity of cells to therapy, leading to a reduction in tumor size.
Most of the current histone deacetylase inhibitors have limited efficacy against solid tumors and can produce toxic side effects leading to drug resistance. As a result, there is a need for the development of new histone modification inhibitors with improved anti-tumor activities and reduced toxicities for breast cancer therapy, as well as further investigation into their mechanisms of action. In addition to the inhibitors described above, other promising therapeutic agents such as histone methyltransferase (KMT) and histone demethylase (KDM)are being investigated in in vivo and in vitro studies for their potential in treating various types of cancers, including breast cancer. KMT is an enzyme that transfers a methyl group to the lysine or arginine position of histones, which can lead to gene transcription activation or repression [134,135]. One KMT inhibitor that has shown efficacy in mouse breast cancer studies is GSK-J4. Yan et al. [136] demonstrated that GSK-J4 had the ability to effectively inhibit breast cancer stem cells by reducing their expansion, self-renewal capacity, and expression of stemness-related markers. The SETD8 inhibitor has been investigated as a potential therapy for breast cancer associated with the BRCA1 mutation. New findings suggest that SETD8 and the methylation of its corresponding histone H4K20 play a role in determining the choice of DNA double-strand break repair pathway [137]. KDM, on the other hand, removes methyl groups from histones, which can also affect gene activity [138]. The KDM4 family, including KDM4A-F, plays a role in oncogene activation, tumor suppressor silencing, and chromosomal instability, making them potential therapeutic targets. Inhibitors targeting KDM4 enzymes have shown anticancer effects in vitro; however, their structural similarities and active domain conservation pose challenges in discovering selective inhibitors. Despite promising results, no KDM4 inhibitors have entered clinical trials yet [139]. One KDM inhibitor that shows potential in breast cancer treatment is also GSK-J1 [140]. Wang et al. used GSK-J1, a small-molecule inhibitor of JMJD3/KDM6B to treat LPS-induced mammary gland inflammation in mice and mouse mammary epithelial cells in vivo and in vitro. The KDM1A (LSD1) inhibitor has been investigated as a therapeutic target in breast cancer because it has shown promising results in inhibiting the growth and invasion of cancer cells [141]. However, research on KMT and KDM inhibitors in breast cancer therapy is still in its early stages and requires further investigation to confirm their efficacy and safety. All of the inhibitors mentioned are being currently investigated as single therapies; however, research indicates that combinations of KMT and KDM inhibitors may prove more effective in treating breast cancer (Li et al., 2021) [142]. It is worth noting that unlike traditional chemotherapy, which kills both cancerous and healthy cells, KMT and KDM inhibitors directly affect molecular processes associated with cancer, which can lead to more targeted and effective breast cancer therapy.
6. The Role of miRNAs
miRNA expression pattern is associated with oncogenesis. MiRNAs are a special class of small (18–26 nucleotides), evolutionarily conserved non-coding RNA molecules, which, through post-transcriptional regulation of gene expression, affect the proper development and maintenance of tissue homeostasis, as well as being involved in the development of pathological processes [143,144,145]. They are thought to be responsible for regulating close to 30% of human mRNAs; one miRNA can regulate multiple target sequences, and one gene can be controlled by numerous microRNAs. In recent years, they have gained popularity as non-invasive biomarkers for assessing tumor development, as their significant role in carcinogenesis, cancer progression, cell cycle checkpoint bypass, drug response regulation, and invasion has been described [146,147,148,149,150,151] (Figure 2). It has been shown that miRNAs act as both oncogenes and tumor suppressor genes, which gives them a wide range of modulation processes in cancer cells [152,153,154]. Numerous studies have identified miRs associated with abnormal expression and function of breast cancer predisposition genes (Figure 3).
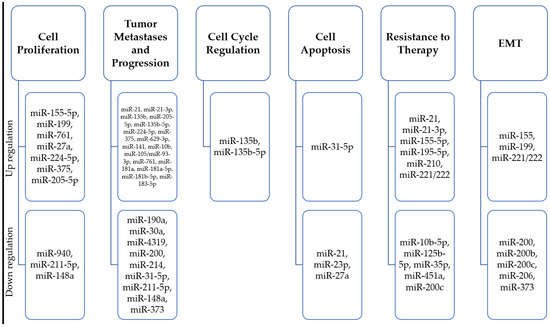
Figure 2.
Summary of microRNA molecules characteristic of different stages of progression of hereditary and familial breast cancer.
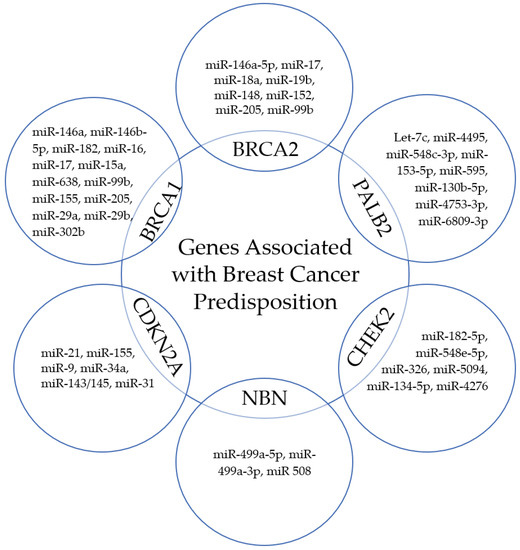
Figure 3.
Selected miRs correlated with the expression and function of breast cancer predisposition genes.
Research on the importance and potential of miRs in oncology is extremely intensive; however, most breast cancer clinical trials are still observational (Table 3). Their goal is to identify miRs that modulate the response of cancer cells to specific drugs, or to analyze the relationship between the selected miRs and the effectiveness of neoadjuvant and adjuvant chemotherapy. Both in scientific and clinical research, it was possible to identify miRs that may be markers of drug resistance or predictors of hormonal sensitivity, as well as molecules associated with the risk of developing complications of therapy.
In this review, a selective choice of presented microRNAs related to breast cancer therapy and monitoring, drug resistance, serving as targets for targeted therapy, and complementing standard breast cancer treatment was made. To maintain an appropriate scope of the review, we focused on microRNAs that can supplement standard breast cancer treatment and have the potential for use in therapeutic protocols. Their significance and prospects for application in clinical practice were presented.
MicroRNAs are currently an important area of research in cancer therapy. The use of miRNAs in breast cancer therapy aims to impact the proliferation, invasion, migration, angiogenesis, and apoptosis of cancer cells. MiRNAs can have the following goals and types of applications in oncology therapy:
- (1)
- Diagnosis: miRNAs can be used as biomarkers for assessing the risk of breast cancer development, diagnosis, and disease monitoring [155,156,157,158,159].
- (2)
- Targeted therapy: miRNAs that are expressed in cancer cells in large quantities can be inhibited by specific miRNA inhibitors (antimiRNAs). AntimiRNAs are small molecules that bind to the target miRNA and inhibit its activity, leading to the inhibition of the growth and proliferation of cancer cells [160,161,162].
- (3)
- Gene therapy: the introduction of miRNAs into cancer cells that express a low level of a particular miRNA can be a method of gene therapy. In gene therapy, miRNAs can be used for therapeutic or diagnostic purposes [163,164,165].
- (4)
- Combined therapy: miRNAs can be used in combination with other anticancer drugs to increase their efficacy. For example, miRNAs and chemo- or immunotherapeutic drugs can be used together to increase the effectiveness of therapy and reduce the toxicity of the drugs [166,167,168,169].
In the therapy of breast cancer, various miRNAs play significant roles, including the following:
- (1)
- MiR-21: this is one of the most commonly identified miRNAs associated with breast cancer. MiR-21 has been shown to contribute to breast cancer development and progression by regulating a range of genes responsible for proliferation, angiogenesis, and invasion of cancer cells [170,171,172,173,174].
- (2)
- MiR-34a: miR-34a has been shown to inhibit proliferation and induce apoptosis of breast cancer cells, making it a potential therapeutic target [175,176,177].
- (3)
- MiR-155: it has been shown that miR-155 is involved in inflammatory processes and proliferation of cancer cells in breast cancer. Therefore, miR-155 has become a subject of interest as a potential therapeutic target [178,179,180,181,182].
- (4)
- MiR-200: this is a miRNA that is involved in the invasion and metastasis of breast cancer through the regulation of EMT (epithelial-mesenchymal transition) processes. Increasing miR-200 expression has been shown to reduce the ability of cancer cells to invade and migrate, making it a potential therapeutic target [183,184].
- (5)
- MiR-10b: it has been shown that miR-10b is involved in the metastasis of breast cancer through the regulation of EMT processes. Therefore, miR-10b has become a subject of interest as a potential therapeutic target [185,186,187].
One of the most extensively studied is the suppressor miR-34a [188]. MiR-34a is one of the most important factors involved in the regulation of cell cycle and apoptosis. Laboratory studies have shown that miR-34a is often mutated or decreased in breast cancer. The use of miR-34a in breast cancer therapy involves introducing an additional dose of miR-34a into cancer cells, which leads to the inhibition of proliferation, invasion, migration, and induction of apoptosis. Studies have shown that miR-34a can also enhance the effect of chemotherapy in breast cancer. For example, in one study, breast cancer cells were treated with a combination of synthetic miR-34a and 5-fluorouracil (5-FU), a chemotherapy drug commonly used in breast cancer treatment. The results showed that the combination of miR-34a and 5-FU led to a greater inhibition of cell growth and increased apoptosis compared to treatment with 5-FU alone. Adams et al. [189] demonstrated the benefits of miR-34a replacement therapy in retarding the growth of subcutaneous and orthotopic transplanted tumors. They have experimentally proven that in triple-negative breast cancer models, restoring miR-34a expression leads to the inhibition of proliferation and invasion as well as the activation of senescence. Moreover, they described a negative feedback between miR-34a and c-SRC affecting the sensitization of cancer cells to dasatinib [189]. MiR-34a also has an effect on 5-fluorouracil chemotherapy as shown by Li et al. [190]. In their study, they noticed a significant reduction in the level of miR-34a in breast cancer cell lines and breast cancer samples, which resulted in no inhibition of cancer cell invasion and inhibition of apoptosis. The beneficial effect, including sensitization of cells to 5-FU after administration of miR-34, was due to the effect of targeting Bcl-2 and SIRT1. These studies confirmed the efficacy of the mimetic miR-34a (MXR34) as a potential therapeutic agent for patients with breast cancer [190] and other types of cancer [191,192,193,194]. Li et al. [195] observed that the expression of miR-34a in tissues and drug-resistant cell lines is significantly reduced and correlated with breast cancer multidrug resistance (MDR). As noted by the authors, patients with low miR-34a expression had worse overall survival and disease-free survival; the introduction of the mimetic miR-34a in vitro led to a partial reversal of MDR. The authors indicated Bcl-2, CCND1 and NOTCH1 as the targets of this miR, while also noting the lack of a direct effect of miR-34a on the expression of HER-2, TP53 and TOP-2a. One of the challenges in the use of miR-34a in breast cancer therapy is the problem of delivering miRNA to cancer cells. Currently, various methods of miRNA transport are being investigated, such as lipid nanoparticles, viral vectors, or vector-based nanoparticles, which aim to improve the effectiveness of delivering miRNA to cancer cells.
Xue et al. [169] identified elevated miR-621 expression associated with paclitaxel and carboplatin (PTX/CBP) sensitivity. Administration of the mimetic miR-621 made it possible to sensitize breast tumors to these drugs by inhibiting FBXO11 and enhancing p53 activity [169]. Mei et al. [196] showed that overexpression of miR-21 is associated with taxol resistance, and Chen and Bourguignon [197] showed that upregulation of this molecule has an effect on the increase in Bcl-2 activity. Shaban et al. [198] highlighted that miR-34 and miR-21 can predict the response of breast cancer patients to chemo-radiotherapy, especially by regulating Bcl-2, BRCA1, BRCA2, and p53 targets in breast cancer cells. In turn, Yadav et al. [199] observed a decrease in miR-21 expression in breast cancer patients who received neoadjuvant therapy. Differential expression of miR-125b and changes in miR-21 expression during neoadjuvant chemotherapy were associated with response to chemotherapy and disease-free survival. The correlation of downregulation of miR-125b expression with resistance to four cycles of neoadjuvant 5-fluorouracil, epirubicin and cyclophosphamide was confirmed by Wang et al. [200]. The mechanism of action involved modulation E2F2 expression. MiR-125b is involved in regulating numerous signaling pathways, including NF-κB, p53, PI3K/Akt/mTOR, ErbB2, Wnt. Hence, it is also important marker involved in controlling cell proliferation, differentiation, metabolism, apoptosis, drug resistance and tumor immunity [201]. In HER2-negative patients, the differential expression of miR-222, miR-20a and miR-451 in clinical response to neoadjuvant chemotherapy has been confirmed to be related to chemosensitivity [202].
Cardiotoxicity is one of the most significant complications among breast cancer patients as a result of the use of agents such as anthracyclines and monoclonal antibodies directed against HER2. Therefore, the search for prognostic biomarkers of this event is crucial in mitigating the risk of cardiotoxicity in vulnerable patients [203,204]. Numerous microRNAs are considered among the candidates, including hsa-miR-1273 g-3p and hsa-miR-4638-3p (regulating TGF-β and CTGF pathways, responsible for atherosclerotic plaque instability and heart failure [205]), hsa-miR-208 (associated with fibrosis and EMT progression, and specifically targeted to the BMP co-receptor, endoglin [206,207]), hsa-miR-130a (associated with cardiomyopathy [208,209,210]), hsa-miR-29a (involved in hemolysis of blood cells [211,212]) or proangiogenic hsa-miR-17-5p, hsa-miR-19a, hsa-miR-378, hsa-Let-7b [208,209] and hsa-miR-126 [207,211]. On the basis of laboratory tests, the concept of introducing antimiRs as a real cardioprotective agent in patients after chemotherapy is being considered.
Table 3.
Examples of clinical trials taking into account the importance of microRNAs in the treatment of breast cancer.
Table 3.
Examples of clinical trials taking into account the importance of microRNAs in the treatment of breast cancer.
| NCT Number | Study Type | Description | Outcome Measures | Study Population | Publications |
|---|---|---|---|---|---|
| NCT03779022 | Observational | miRNA and relevant biomarkers of BC patients undergoing neoadjuvant treatment | Clinical disease response was evaluated for every two cycles of chemotherapy till surgery with RECIST criteria. | Patients with early stage breast cancer patients, with stage II-III disease | [200,202,213,214] |
| NCT01598285 | Observational | Genome-wide association study (GWAS) and microRNA (miRNA) profiling for identification of genetic variants and blood miRNA signatures predictors of bevacizumab response cancer | To identify miRNA signatures in whole blood as bevacizumab response predictors in metastatic breast cancer patients | Patients suffering from metastatic (disseminated at the time of diagnosis) breast cancer, treated with bevacizumab. | [98] |
| NCT02656589 | Observational | A perspective study of the predictive value of microRNA in patients with HER2 positive advanced stage breast cancer who were treated with herceptin | Progression-free survival (PFS) evaluation defined as the interval from the diagnosis of advanced breast cancer with HER2 positive to disease progression, relapse, death due to any causes or last follow-up. The follow-up interval is 2 years. | Advanced breast cancer patients first diagnosis, ≥ 18yrs ages, HER2 positive: immunohistochemistry (+++) or FISH (+), stage IV, the patients have no history of chemotherapy, hormone therapy, radiotherapy or surgery after diagnosis of breast cancer. | [215,216,217] |
| NCT01612871 | Interventional | Circulating miRNAs as biomarkers of hormone sensitivity in breast cancer | Analysis of the larger-scale circulating miRNAs in plasma of these patients before and after one month of treatment with tamoxifen or anti aromatase. Correlation between the specific miRNAs initial expression and the appearance of an objective response or clinical benefit of hormone therapy and the time to progression. | Drugs: Tamoxifen, Letrozole, Anastrozole, Exemestane Women with metastatic invasive breast cancer or locally advanced (without surgical project), for which treatment with tamoxifen or anti aromatase. Cancer HER2-negative. | [98] |
| NCT05151224 | Observational | Circulating microRNA-21 expression level before and after neoadjuvant systemic therapy in breast carcinoma | Describes miRNA 21 expression level before and after neoadjuvant systemic therapy in breast cancer patient. | Invasive breast cancer, from stage IIB to stage IIIC, all subtypes are included, either HR (ER, PR)-positive or -negative, HER2-positive or -negative, eligible to neoadjuvant systemic therapy. Neoadjuvant systemic treatment composed of anthracyclines-based chemotherapy and taxanes, trastuzumab for HER2-positive patients. | [196,197,199,200,202,218,219,220] |
| NCT01722851 | Observational | Novel breast cancer biomarkers and their use for guiding and monitoring response to chemotherapy | Relationship between changes in a patients circulating miRNA expression levels over the course of their systemic therapy, and their response to that treatment. | Cohort 1: All patients with a new diagnosis of breast cancer, who are destined to undergo neoadjuvant chemotherapy. OR Cohort 2: All breast cancer patients who present with metastatic disease, disease recurrence or progression who will receive up-front chemotherapy ± hormonal therapy. OR Cohort 3: All breast cancer patient who present with metastatic disease who are commencing hormonal therapy only. | [98] |
| NCT02950207 | Observational | Prospective observational study of antitumor activity correlation between hormonal therapy and expression miRNA-100 | Mono-centric, observational, prospective study, designed for patients with diagnosis of hormone-positive breast cancer to evaluate the correlation between the response to hormonal treatment indicated by the reduction of the level of Ki67 and miRNA100 in two groups of patients | Post-menopausal hormone-positive breast cancer patients. Histological diagnosis of invasive carcinoma of the breast. X-ray evidence (mammography and / or ultrasound) strongly suggestive for the presence of invasive breast cancer (BIRADS 4c or BIRADS 5) of greater than 15mm diameter. Positivity for the estrogen receptor and / or to the progestin defined as the expression of one or both hormone receptors in ≥10% of tumor cells, negativity for HER2. | [98] |
Clinical trials using miRNA in breast cancer therapy are still in the early stages. Most of them have focused mainly on Phase I and II clinical trials aimed at evaluating the safety and tolerability of miRNA. However, the results of these trials are promising, suggesting potential therapeutic benefits and the safety of miRNA use in breast cancer treatment. Several miRNAs, such as miR-34a, miR-21, miR-155, miR-29b, and miR-16, have been identified as potential therapeutic targets in breast cancer therapy. Introducing miRNA as a new therapeutic tool may help to better understand pathological processes and result in improved therapeutic outcomes for patients with breast cancer.
7. Summary
The clinical trials conducted to date have failed to confirm the high effectiveness of monotherapy based on epigenetic drugs. According to Falahi et al. [111], anticancer efficacy of epitherapy in breast cancer is at the level of 10%. However, much better results are obtained when the therapy is combined with cytostatics or targeted therapy. Significantly higher progression-free survival and survival were observed in the phase I and II studies.
The introduction of epigenetic drugs for oncotherapy raises questions about their possible side effects. It is believed that epitherapeutics primarily affect rapidly dividing cells; therefore, their toxic effect on normal cells should be minimal. Nevertheless, an alternative approach is actively sought to develop non-nucleoside compounds that can, e.g., effectively inhibit DNA methylation without being incorporated into DNA, such as SGI-1027, RG108 and MG98 [221,222,223]. The mechanism of action of these molecules involves blocking the DNMT catalytic/ cofactor binding sites, or targeting their regulatory messenger RNA sequences. Non-nucleoside DNMTs of natural origin such as polyphenols or epigallocatechin-3-gallate are also tested [87,224,225]. It has been shown that these compounds have, e.g., inhibition effect, VEGF inhibition and apoptosis induction, and ESR1 re- expression. Nevertheless, the proliferation-inhibiting potential of drugs based on non-nucleoside compounds is not satisfactory. The advantage of miRNAs is that, as natural cellular components, they should not cause side effects and toxicity, and subsequent studies prove high efficiency and low antigenicity of such therapy. However, the clinical use of miRs in breast cancer therapy still requires a significant amount of research work, including refining methods of therapeutic manipulation, delivery to target cells, overcoming immune barriers and maintaining long-term activity of nanoconstructs [66].
Epigenome characteristics can be important in determining prognosis and can be used to stratify patients into risk categories. It is also helpful in identifying breast cancer patients who are likely to respond well to neoadjuvant and adjuvant chemotherapy, or who are sensitive or resistant to a given therapeutic agent. Monitoring individual epimutations or circulating microRNAs could improve patient response to chemotherapy and hormone therapies.
Current breast cancer therapy protocols do not allow the introduction of epigenetic drugs as monotherapy. Such therapy is in its early stages, and requires careful study of its benefits for patients, potential side effects, interactions with other drugs and the exact mechanisms of both the effect on the cancer cell and the acquisition of resistance by it. Much more promising are the possibilities of using epi-drugs in combination with chemotherapeutics and targeted therapies to increase or restore sensitivity to these drugs.
Author Contributions
Conceptualization, J.S. and A.T.; methodology, J.S. and M.S.; formal analysis, J.S., M.S. and C.C.; investigation, J.S. amd J.J.-T.; resources, J.S.; data curation, J.S.; writing—original draft preparation, J.S. and A.T.; writing—review and editing, C.C., A.T. and J.S.; visualization, J.S.; supervision, A.T., J.J-T and C.C.; project administration, J.S. and A.T.; funding acquisition, A.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Rojas, K.; Stuckey, A. Breast Cancer Epidemiology and Risk Factors. Clin. Obstet. Gynecol. 2016, 59, 651–672. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, A. Breast cancer: Epidemiology and risk factors. Clin. Obstet. Gynecol. 2011, 54, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Lukasiewicz, S.; Czeczelewski, M.; Forma, A.; Baj, J.; Sitarz, R.; Stanislawek, A. Breast Cancer-Epidemiology, Risk Factors, Classification, Prognostic Markers, and Current Treatment Strategies—An Updated Review. Cancers 2021, 13, 4287. [Google Scholar] [CrossRef]
- Powell, C.B.; Laurent, C.; Garcia, C.; Hoodfar, E.; Karlea, A.; Kobelka, C.; Lee, J.; Roh, J.; Kushi, L.H. Factors influencing genetic counseling and testing for hereditary breast and ovarian cancer syndrome in a large US health care system. Clin. Genet. 2022, 101, 324–334. [Google Scholar] [CrossRef]
- Bredart, A.; De Pauw, A.; Anota, A.; Tuchler, A.; Dick, J.; Muller, A.; Kop, J.L.; Rhiem, K.; Schmutzler, R.; Devilee, P.; et al. Information needs on breast cancer genetic and non-genetic risk factors in relatives of women with a BRCA1/2 or PALB2 pathogenic variant. Breast 2021, 60, 38–44. [Google Scholar] [CrossRef]
- van der Groep, P.; Bouter, A.; van der Zanden, R.; Siccama, I.; Menko, F.H.; Gille, J.J.; van Kalken, C.; van der Wall, E.; Verheijen, R.H.; van Diest, P.J. Distinction between hereditary and sporadic breast cancer on the basis of clinicopathological data. J. Clin. Pathol. 2006, 59, 611–617. [Google Scholar] [CrossRef]
- Anderson, D.E. Familial versus sporadic breast cancer. Cancer 1992, 70, 1740–1746. [Google Scholar] [CrossRef]
- Arpino, G.; Pensabene, M.; Condello, C.; Ruocco, R.; Cerillo, I.; Lauria, R.; Forestieri, V.; Giuliano, M.; De Angelis, C.; Montella, M.; et al. Tumor characteristics and prognosis in familial breast cancer. BMC Cancer 2016, 16, 924. [Google Scholar] [CrossRef]
- Fleisher, B.; Clarke, C.; Ait-Oudhia, S. Current advances in biomarkers for targeted therapy in triple-negative breast cancer. Breast Cancer 2016, 8, 183–197. [Google Scholar] [CrossRef]
- Haffty, B.G.; Euhus, D.M.; Pierce, L.J. Genetic Factors in the Locoregional Management of Breast Cancer. J. Clin. Oncol. 2020, 38, 2220–2229. [Google Scholar] [CrossRef]
- Lee, A.; Mavaddat, N.; Wilcox, A.N.; Cunningham, A.P.; Carver, T.; Hartley, S.; Babb de Villiers, C.; Izquierdo, A.; Simard, J.; Schmidt, M.K.; et al. BOADICEA: A comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet. Med. 2019, 21, 1708–1718. [Google Scholar] [CrossRef]
- Blaes, A.H.; Jewett, P.I.; McKay, K.; Riley, D.; Jatoi, I.; Trentham-Dietz, A.; Chrischilles, E.; Klemp, J.R. Factors associated with genetic testing in a cohort of breast cancer survivors. Breast J. 2019, 25, 1241–1244. [Google Scholar] [CrossRef]
- Paz, M.; de Alencar, M.; Gomes Junior, A.L.; da Conceicao Machado, K.; Islam, M.T.; Ali, E.S.; Shill, M.C.; Ahmed, M.I.; Uddin, S.J.; da Mata, A.; et al. Correlations between Risk Factors for Breast Cancer and Genetic Instability in Cancer Patients—A Clinical Perspective Study. Front. Genet. 2017, 8, 236. [Google Scholar] [CrossRef]
- Harro, C.M.; Monteiro, A.N. Dissecting genetic risk factors in breast cancer. Oncotarget 2017, 8, 12540–12541. [Google Scholar] [CrossRef]
- O’Brien, K.M.; Cole, S.R.; Engel, L.S.; Bensen, J.T.; Poole, C.; Herring, A.H.; Millikan, R.C. Breast cancer subtypes and previously established genetic risk factors: A bayesian approach. Cancer Epidemiol. Biomark. Prev. 2014, 23, 84–97. [Google Scholar] [CrossRef]
- Garcia-Closas, M.; Gunsoy, N.B.; Chatterjee, N. Combined associations of genetic and environmental risk factors: Implications for prevention of breast cancer. J. Natl. Cancer. Inst. 2014, 106, dju305. [Google Scholar] [CrossRef]
- Erturk, E.; Cecener, G.; Polatkan, V.; Gokgoz, S.; Egeli, U.; Tunca, B.; Tezcan, G.; Demirdogen, E.; Ak, S.; Tasdelen, I. Evaluation of genetic variations in miRNA-binding sites of BRCA1 and BRCA2 genes as risk factors for the development of early-onset and/or familial breast cancer. Asian Pac. J. Cancer Prev. 2014, 15, 8319–8324. [Google Scholar] [CrossRef]
- Amer, M.H. Genetic factors and breast cancer laterality. Cancer Manag. Res. 2014, 6, 191–203. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Stefansson, I.M.; Chappuis, P.O.; Begin, L.R.; Goffin, J.R.; Wong, N.; Trudel, M.; Akslen, L.A. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003, 95, 1482–1485. [Google Scholar] [CrossRef] [PubMed]
- Laakso, M.; Loman, N.; Borg, A.; Isola, J. Cytokeratin 5/14-positive breast cancer: True basal phenotype confined to BRCA1 tumors. Mod. Pathol. 2005, 18, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.R.; Reis-Filho, J.S.; Fulford, L.; Penault-Llorca, F.; van der Vijver, M.; Parry, S.; Bishop, T.; Benitez, J.; Rivas, C.; Bignon, Y.J.; et al. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin. Cancer Res. 2005, 11, 5175–5180. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Girardi, A.; Calvello, M.; Gandini, S.; Gaeta, A.; Marabelli, M.; Magnoni, F.; Veronesi, P.; Guerrieri-Gonzaga, A.; Bonanni, B. Prognostic impact of germline BRCA1/2 pathogenic variants in breast cancer. Breast Cancer Res. Treat. 2023, 197, 103–112. [Google Scholar] [CrossRef]
- Menezes, M.C.S.; Raheem, F.; Mina, L.; Ernst, B.; Batalini, F. PARP Inhibitors for Breast Cancer: Germline BRCA1/2 and Beyond. Cancers 2022, 14, 4332. [Google Scholar] [CrossRef]
- Paik, H.J.; Jung, Y.J.; Kim, D.I.; Lee, S.; Jung, C.S.; Kang, S.K.; Kim, J.J.; Oh, S.Y.; Joo, J.H.; Kim, H.Y. Clinicopathological Features of BRCA1/2 Mutation-Positive Breast Cancer. Oncology 2021, 99, 499–506. [Google Scholar] [CrossRef]
- Solano, A.R.; Mele, P.G.; Jalil, F.S.; Liria, N.C.; Podesta, E.J.; Gutierrez, L.G. Study of the Genetic Variants in BRCA1/2 and Non-BRCA Genes in a Population-Based Cohort of 2155 Breast/Ovary Cancer Patients, Including 443 Triple-Negative Breast Cancer Patients, in Argentina. Cancers 2021, 13, 2711. [Google Scholar] [CrossRef]
- Miklikova, S.; Trnkova, L.; Plava, J.; Bohac, M.; Kuniakova, M.; Cihova, M. The Role of BRCA1/2-Mutated Tumor Microenvironment in Breast Cancer. Cancers 2021, 13, 575. [Google Scholar] [CrossRef]
- Vasconcelos de Matos, L.; Fernandes, L.; Louro, P.; Placido, A.; Barros, M.; Vaz, F. Challenges and Considerations on Risk-Reducing Surgery in BRCA1/2 Patients with Advanced Breast Cancer. Curr. Oncol. 2021, 28, 10050. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, H.; Fu, F.; Li, Z.; Feng, Q.; Wu, W.; Liu, Y.; Wang, C.; Chen, Y. Spectrum of PALB2 germline mutations and characteristics of PALB2-related breast cancer: Screening of 16,501 unselected patients with breast cancer and 5890 controls by next-generation sequencing. Cancer 2020, 126, 3202–3208. [Google Scholar] [CrossRef]
- Behl, S.; Hamel, N.; de Ladurantaye, M.; Lepage, S.; Lapointe, R.; Mes-Masson, A.M.; Foulkes, W.D. Founder BRCA1/BRCA2/PALB2 pathogenic variants in French-Canadian breast cancer cases and controls. Sci. Rep. 2020, 10, 6491. [Google Scholar] [CrossRef]
- Cybulski, C.; Kluzniak, W.; Huzarski, T.; Wokolorczyk, D.; Kashyap, A.; Jakubowska, A.; Szwiec, M.; Byrski, T.; Debniak, T.; Gorski, B.; et al. Clinical outcomes in women with breast cancer and a PALB2 mutation: A prospective cohort analysis. Lancet Oncol. 2015, 16, 638–644. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkas, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [CrossRef]
- Balia, C.; Sensi, E.; Lombardi, G.; Roncella, M.; Bevilacqua, G.; Caligo, M.A. PALB2: A novel inactivating mutation in a Italian breast cancer family. Fam. Cancer 2010, 9, 531–536. [Google Scholar] [CrossRef]
- Ameziane, N.; van den Ouweland, A.M.; Adank, M.A.; Vijzelaar, R.N.; Errami, A.; Dorsman, J.C.; Joenje, H.; Meijers-Heijboer, H.; Waisfisz, Q. Lack of large genomic deletions in BRIP1, PALB2, and FANCD2 genes in BRCA1/2 negative familial breast cancer. Breast Cancer Res. Treat. 2009, 118, 651–653. [Google Scholar] [CrossRef]
- Soleimani, T.; Bourdon, C.; Davis, J.; Fortes, T. A case report of biallelic CHEK2 heterozygous variant presenting with breast cancer. Clin. Case Rep. 2023, 11, e6820. [Google Scholar] [CrossRef]
- Yadav, S.; Boddicker, N.J.; Na, J.; Polley, E.C.; Hu, C.; Hart, S.N.; Gnanaolivu, R.D.; Larson, N.; Holtegaard, S.; Huang, H.; et al. Contralateral Breast Cancer Risk Among Carriers of Germline Pathogenic Variants in ATM, BRCA1, BRCA2, CHEK2, and PALB2. J. Clin. Oncol. 2023, 41, 1703–1713. [Google Scholar] [CrossRef]
- Graffeo, R.; Rana, H.Q.; Conforti, F.; Bonanni, B.; Cardoso, M.J.; Paluch-Shimon, S.; Pagani, O.; Goldhirsch, A.; Partridge, A.H.; Lambertini, M.; et al. Moderate penetrance genes complicate genetic testing for breast cancer diagnosis: ATM, CHEK2, BARD1 and RAD51D. Breast 2022, 65, 32–40. [Google Scholar] [CrossRef]
- Lowry, K.P.; Geuzinge, H.A.; Stout, N.K.; Alagoz, O.; Hampton, J.; Kerlikowske, K.; de Koning, H.J.; Miglioretti, D.L.; van Ravesteyn, N.T.; Schechter, C.; et al. Breast Cancer Screening Strategies for Women With ATM, CHEK2, and PALB2 Pathogenic Variants: A Comparative Modeling Analysis. JAMA Oncol. 2022, 8, 587–596. [Google Scholar] [CrossRef]
- Ghunaim, H.; Laenen, A.; De Keyzer, F.; Soens, J.; Keupers, M.; Postema, S.; Neven, P.; Van Ongeval, C. Comparing breast cancer imaging characteristics of CHEK2 with BRCA1 and BRCA2 gene mutation carriers. Eur. J. Radiol. 2022, 146, 110074. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Tenedini, E.; Piombino, C.; Venturelli, M.; Marchi, I.; Gasparini, E.; Barbieri, E.; Razzaboni, E.; Domati, F.; Caggia, F.; et al. Clinicopathologic Profile of Breast Cancer in Germline ATM and CHEK2 Mutation Carriers. Genes 2021, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Goricar, K.; Dugar, F.; Dolzan, V.; Marinko, T. NBN, RAD51 and XRCC3 Polymorphisms as Potential Predictive Biomarkers of Adjuvant Radiotherapy Toxicity in Early HER2-Positive Breast Cancer. Cancers 2022, 14, 4365. [Google Scholar] [CrossRef] [PubMed]
- Zuntini, R.; Bonora, E.; Pradella, L.M.; Amato, L.B.; Vidone, M.; De Fanti, S.; Catucci, I.; Cortesi, L.; Medici, V.; Ferrari, S.; et al. Detecting Variants in the NBN Gene While Testing for Hereditary Breast Cancer: What to Do Next? Int. J. Mol. Sci. 2021, 22, 5832. [Google Scholar] [CrossRef]
- Nithya, P.; ChandraSekar, A. NBN Gene Analysis and it’s Impact on Breast Cancer. J. Med. Syst. 2019, 43, 270. [Google Scholar] [CrossRef]
- Uzunoglu, H.; Korak, T.; Ergul, E.; Uren, N.; Sazci, A.; Utkan, N.Z.; Kargi, E.; Triyaki, C.; Yirmibesoglu, O. Association of the nibrin gene (NBN) variants with breast cancer. Biomed. Rep. 2016, 4, 369–373. [Google Scholar] [CrossRef]
- Sokolenko, A.P.; Bogdanova, N.; Kluzniak, W.; Preobrazhenskaya, E.V.; Kuligina, E.S.; Iyevleva, A.G.; Aleksakhina, S.N.; Mitiushkina, N.V.; Gorodnova, T.V.; Bessonov, A.A.; et al. Double heterozygotes among breast cancer patients analyzed for BRCA1, CHEK2, ATM, NBN/NBS1, and BLM germ-line mutations. Breast Cancer Res. Treat. 2014, 145, 553–562. [Google Scholar] [CrossRef]
- Bartels, S.; van Luttikhuizen, J.L.; Christgen, M.; Magel, L.; Luft, A.; Hanzelmann, S.; Lehmann, U.; Schlegelberger, B.; Leo, F.; Steinemann, D.; et al. CDKN2A loss and PIK3CA mutation in myoepithelial-like metaplastic breast cancer. J. Pathol. 2018, 245, 373–383. [Google Scholar] [CrossRef]
- ShahidSales, S.; Mehramiz, M.; Ghasemi, F.; Aledavood, A.; Shamsi, M.; Hassanian, S.M.; Ghayour-Mobarhan, M.; Avan, A. A genetic variant in CDKN2A/B gene is associated with the increased risk of breast cancer. J. Clin. Lab. Anal. 2018, 32, e22190. [Google Scholar] [CrossRef]
- Nagore, E.; Montoro, A.; Garcia-Casado, Z.; Botella-Estrada, R.; Insa, A.; Lluch, A.; Lopez-Guerrero, J.A.; Guillen, C. Germline mutations in CDKN2A are infrequent in female patients with melanoma and breast cancer. Melanoma Res. 2009, 19, 211–214. [Google Scholar] [CrossRef]
- Knappskog, S. New doctorial cancer research: Germline genetic alterations affecting CDKN2A, MDM2, and CDKN1A in melanoma and breast cancer patients. Crit. Rev. Oncog. 2007, 13, 261–262, discussion 263. [Google Scholar] [CrossRef]
- Debniak, T.; Gorski, B.; Huzarski, T.; Byrski, T.; Cybulski, C.; Mackiewicz, A.; Gozdecka-Grodecka, S.; Gronwald, J.; Kowalska, E.; Haus, O.; et al. A common variant of CDKN2A (p16) predisposes to breast cancer. J. Med. Genet. 2005, 42, 763–765. [Google Scholar] [CrossRef]
- Rajendran, B.K.; Deng, C.X. Characterization of potential driver mutations involved in human breast cancer by computational approaches. Oncotarget 2017, 8, 50252–50272. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Grewal, S.I.; Moazed, D. Heterochromatin and epigenetic control of gene expression. Science 2003, 301, 798–802. [Google Scholar] [CrossRef]
- Downs, B.; Wang, S.M. Epigenetic changes in BRCA1-mutated familial breast cancer. Cancer Genet. 2015, 208, 237–240. [Google Scholar] [CrossRef][Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Yang, X.; Lay, F.; Han, H.; Jones, P.A. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol. Sci. 2010, 31, 536–546. [Google Scholar] [CrossRef]
- Ascoët, S.; De Waard, M. Diagnostic and Therapeutic Value of Aptamers in Envenomation Cases. Int. J. Mol. Sci. 2020, 21, 3565. [Google Scholar] [CrossRef]
- Shukla, S.; Penta, D.; Mondal, P.; Meeran, S.M. Epigenetics of Breast Cancer: Clinical Status of Epi-drugs and Phytochemicals. Adv. Exp. Med. Biol. 2019, 1152, 293–310. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell. 2010, 19, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, O.A.; Esteller, M. Epigenetic modifications in breast cancer and their role in personalized medicine. Am. J. Pathol. 2013, 183, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J. Targeting epigenetic readers in cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef]
- Szczepanek, J.; Skorupa, M.; Tretyn, A. MicroRNA as a Potential Therapeutic Molecule in Cancer. Cells 2022, 11, 1008. [Google Scholar] [CrossRef]
- Shukla, V.; Coumoul, X.; Lahusen, T.; Wang, R.H.; Xu, X.; Vassilopoulos, A.; Xiao, C.; Lee, M.H.; Man, Y.G.; Ouchi, M.; et al. BRCA1 affects global DNA methylation through regulation of DNMT1. Cell Res. 2010, 20, 1201–1215. [Google Scholar] [CrossRef]
- Locke, W.J.; Clark, S.J. Epigenome remodelling in breast cancer: Insights from an early in vitro model of carcinogenesis. Breast Cancer Res. 2012, 14, 215. [Google Scholar] [CrossRef]
- Radpour, R.; Barekati, Z.; Kohler, C.; Lv, Q.; Burki, N.; Diesch, C.; Bitzer, J.; Zheng, H.; Schmid, S.; Zhong, X.Y. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS ONE 2011, 6, e16080. [Google Scholar] [CrossRef]
- Nam, H.J.; Baek, S.H. Epigenetic Regulation in Breast Cancer. In Translational Research in Breast Cancer; Springer: Singapore, 2021; pp. 103–119. [Google Scholar] [CrossRef]
- Suijkerbuijk, K.P.M.; Fackler, M.J.; Sukumar, S.; van Gils, C.H.; van Laar, T.; van der Wall, E.; Vooijs, M.; van Diest, P.J. Methylation is less abundant in BRCA1-associated compared with sporadic breast cancer. Ann. Oncol. 2008, 19, 1870–1874. [Google Scholar] [CrossRef]
- Vasilatos, S.N.; Broadwater, G.; Barry, W.T.; Baker, J.C., Jr.; Lem, S.; Dietze, E.C.; Bean, G.R.; Bryson, A.D.; Pilie, P.G.; Goldenberg, V.; et al. CpG island tumor suppressor promoter methylation in non-BRCA-associated early mammary carcinogenesis. Cancer Epidemiol. Biomark. Prev. 2009, 18, 901–914. [Google Scholar] [CrossRef]
- Vos, S.; Moelans, C.B.; van Diest, P.J. BRCA promoter methylation in sporadic versus BRCA germline mutation-related breast cancers. Breast Cancer Res. 2017, 19, 64. [Google Scholar] [CrossRef]
- Bernardino, J.; Roux, C.; Almeida, A.; Vogt, N.; Gibaud, A.; Gerbault-Seureau, M.; Magdelenat, H.; Bourgeois, C.A.; Malfoy, B.; Dutrillaux, B. DNA hypomethylation in breast cancer: An independent parameter of tumor progression? Cancer Genet. Cytogenet. 1997, 97, 83–89. [Google Scholar] [CrossRef]
- Wei, M.; Grushko, T.A.; Dignam, J.; Hagos, F.; Nanda, R.; Sveen, L.; Xu, J.; Fackenthal, J.; Tretiakova, M.; Das, S.; et al. BRCA1 promoter methylation in sporadic breast cancer is associated with reduced BRCA1 copy number and chromosome 17 aneusomy. Cancer Res. 2005, 65, 10692–10699. [Google Scholar] [CrossRef]
- Wu, L.; Wang, F.; Xu, R.; Zhang, S.; Peng, X.; Feng, Y.; Wang, J.; Lu, C. Promoter methylation of BRCA1 in the prognosis of breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2013, 142, 619–627. [Google Scholar] [CrossRef]
- Pang, D.; Zhao, Y.; Xue, W.; Shan, M.; Chen, Y.; Zhang, Y.; Zhang, G.; Liu, F.; Li, D.; Yang, Y. Methylation profiles of the BRCA1 promoter in hereditary and sporadic breast cancer among Han Chinese. Med. Oncol. 2012, 29, 1561–1568. [Google Scholar] [CrossRef]
- Turner, N.C.; Reis-Filho, J.S.; Russell, A.M.; Springall, R.J.; Ryder, K.; Steele, D.; Savage, K.; Gillett, C.E.; Schmitt, F.C.; Ashworth, A.; et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene 2007, 26, 2126–2132. [Google Scholar] [CrossRef]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar]
- Lips, E.H.; Mulder, L.; Oonk, A.; van der Kolk, L.E.; Hogervorst, F.B.; Imholz, A.L.; Wesseling, J.; Rodenhuis, S.; Nederlof, P.M. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br. J. Cancer 2013, 108, 2172–2177. [Google Scholar] [CrossRef]
- Toffoli, S.; Bar, I.; Abdel-Sater, F.; Delree, P.; Hilbert, P.; Cavallin, F.; Moreau, F.; Van Criekinge, W.; Lacroix-Triki, M.; Campone, M.; et al. Identification by array comparative genomic hybridization of a new amplicon on chromosome 17q highly recurrent in BRCA1 mutated triple negative breast cancer. Breast Cancer Res. 2014, 16, 466. [Google Scholar] [CrossRef]
- Severson, T.M.; Peeters, J.; Majewski, I.; Michaut, M.; Bosma, A.; Schouten, P.C.; Chin, S.F.; Pereira, B.; Goldgraben, M.A.; Bismeijer, T.; et al. BRCA1-like signature in triple negative breast cancer: Molecular and clinical characterization reveals subgroups with therapeutic potential. Mol. Oncol. 2015, 9, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Filipponi, D.; Muller, J.; Emelyanov, A.; Bulavin, D.V. Wip1 controls global heterochromatin silencing via ATM/BRCA1-dependent DNA methylation. Cancer Cell 2013, 24, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Anjum, S.; Fourkala, E.O.; Zikan, M.; Wong, A.; Gentry-Maharaj, A.; Jones, A.; Hardy, R.; Cibula, D.; Kuh, D.; Jacobs, I.J.; et al. A BRCA1-mutation associated DNA methylation signature in blood cells predicts sporadic breast cancer incidence and survival. Genome Med. 2014, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Archey, W.B.; McEachern, K.A.; Robson, M.; Offit, K.; Vaziri, S.A.; Casey, G.; Borg, A.; Arrick, B.A. Increased CpG methylation of the estrogen receptor gene in BRCA1-linked estrogen receptor-negative breast cancers. Oncogene 2002, 21, 7034–7041. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheng, C.K.; Chow, L.W.; Loo, W.T.; Chan, T.K.; Chan, V. The cell cycle checkpoint gene Rad9 is a novel oncogene activated by 11q13 amplification and DNA methylation in breast cancer. Cancer Res. 2005, 65, 8646–8654. [Google Scholar] [CrossRef]
- Martinet, N.; Michel, B.Y.; Bertrand, P.; Benhida, R. Small molecules DNA methyl transferases inhibitors. MedChemComm 2012, 3, 263–273. [Google Scholar] [CrossRef]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef]
- Ramos, M.-P.; Wijetunga, N.A.; McLellan, A.S.; Suzuki, M.; Greally, J.M. DNA demethylation by 5-aza-2′-deoxycytidine is imprinted, targeted to euchromatin, and has limited transcriptional consequences. Epigenetics Chromatin 2015, 8, 11. [Google Scholar] [CrossRef]
- Borges, S.; Doppler, H.; Perez, E.A.; Andorfer, C.A.; Sun, Z.; Anastasiadis, P.Z.; Thompson, E.; Geiger, X.J.; Storz, P. Pharmacologic reversion of epigenetic silencing of the PRKD1 promoter blocks breast tumor cell invasion and metastasis. Breast Cancer Res. 2013, 15, R66. [Google Scholar] [CrossRef]
- Derissen, E.J.; Beijnen, J.H.; Schellens, J.H. Concise drug review: Azacitidine and decitabine. Oncologist 2013, 18, 619–624. [Google Scholar] [CrossRef]
- Tsai, H.C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef]
- Tao, Y.; Liu, S.; Briones, V.; Geiman, T.M.; Muegge, K. Treatment of breast cancer cells with DNA demethylating agents leads to a release of Pol II stalling at genes with DNA-hypermethylated regions upstream of TSS. Nucleic Acids Res. 2011, 39, 9508–9520. [Google Scholar] [CrossRef]
- Cai, F.-F.; Kohler, C.; Zhang, B.; Wang, M.-H.; Chen, W.-J.; Zhong, X.-Y. Epigenetic Therapy for Breast Cancer. Int. J. Mol. Sci. 2011, 12, 4465–4476. [Google Scholar] [CrossRef]
- Liu, K.; Dong, F.; Gao, H.; Guo, Y.; Li, H.; Yang, F.; Zhao, P.; Dai, Y.; Wang, J.; Zhou, W.; et al. Promoter hypermethylation of the CFTR gene as a novel diagnostic and prognostic marker of breast cancer. Cell Biol. Int. 2020, 44, 603–609. [Google Scholar] [CrossRef]
- Widschwendter, M.; Siegmund, K.D.; Muller, H.M.; Fiegl, H.; Marth, C.; Muller-Holzner, E.; Jones, P.A.; Laird, P.W. Association of breast cancer DNA methylation profiles with hormone receptor status and response to tamoxifen. Cancer Res. 2004, 64, 3807–3813. [Google Scholar] [CrossRef]
- Maier, S.; Nimmrich, I.; Koenig, T.; Eppenberger-Castori, S.; Bohlmann, I.; Paradiso, A.; Spyratos, F.; Thomssen, C.; Mueller, V.; Nahrig, J.; et al. DNA-methylation of the homeodomain transcription factor PITX2 reliably predicts risk of distant disease recurrence in tamoxifen-treated, node-negative breast cancer patients—Technical and clinical validation in a multi-centre setting in collaboration with the European Organisation for Research and Treatment of Cancer (EORTC) PathoBiology group. Eur. J. Cancer 2007, 43, 1679–1686. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ (accessed on 21 January 2023).
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef]
- Pao, G.M.; Janknecht, R.; Ruffner, H.; Hunter, T.; Verma, I.M. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc. Natl. Acad. Sci. USA 2000, 97, 1020–1025. [Google Scholar] [CrossRef]
- Yarden, R.I.; Brody, L.C. BRCA1 interacts with components of the histone deacetylase complex. Proc. Natl. Acad. Sci. USA 1999, 96, 4983–4988. [Google Scholar] [CrossRef]
- Witus, S.R.; Burrell, A.L.; Farrell, D.P.; Kang, J.; Wang, M.; Hansen, J.M.; Pravat, A.; Tuttle, L.M.; Stewart, M.D.; Brzovic, P.S.; et al. BRCA1/BARD1 site-specific ubiquitylation of nucleosomal H2A is directed by BARD1. Nat. Struct. Mol. Biol. 2021, 28, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Kalb, R.; Mallery, D.L.; Larkin, C.; Huang, J.T.; Hiom, K. BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 2014, 8, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Chu, Y.; Ma, H.; Zhang, Y.; Zhang, X.; Zhao, D.; Li, Z.; Wang, J.; Gao, Y.E.; Xiao, L.; et al. Epigenetic interventions increase the radiation sensitivity of cancer cells. Curr. Pharm. Des. 2014, 20, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, L.; Yang, S.; Song, N.; Zhou, X.; Gao, J.; Yu, N.; Shan, L.; Wang, Q.; Liang, J.; et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc. Natl. Acad. Sci. USA 2014, 111, 7096–7101. [Google Scholar] [CrossRef]
- Veeck, J.; Esteller, M. Breast cancer epigenetics: From DNA methylation to microRNAs. J. Mammary Gland. Biol. Neoplasia 2010, 15, 5–17. [Google Scholar] [CrossRef]
- Zhu, Q.; Pao, G.M.; Huynh, A.M.; Suh, H.; Tonnu, N.; Nederlof, P.M.; Gage, F.H.; Verma, I.M. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 2011, 477, 179–184. [Google Scholar] [CrossRef]
- Zheng, L.; Annab, L.A.; Afshari, C.A.; Lee, W.H.; Boyer, T.G. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 9587–9592. [Google Scholar] [CrossRef]
- Chang, S.; Wang, R.H.; Akagi, K.; Kim, K.A.; Martin, B.K.; Cavallone, L.; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer; Haines, D.C.; Basik, M.; Mai, P.; et al. Tumor suppressor BRCA1 epigenetically controls oncogenic microRNA-155. Nat. Med. 2011, 17, 1275–1282. [Google Scholar] [CrossRef]
- Falahi, F.; van Kruchten, M.; Martinet, N.; Hospers, G.A.; Rots, M.G. Current and upcoming approaches to exploit the reversibility of epigenetic mutations in breast cancer. Breast Cancer Res. 2014, 16, 412. [Google Scholar] [CrossRef]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef]
- Piekarz, R.L.; Frye, R.; Turner, M.; Wright, J.J.; Allen, S.L.; Kirschbaum, M.H.; Zain, J.; Prince, H.M.; Leonard, J.P.; Geskin, L.J.; et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2009, 27, 5410–5417. [Google Scholar] [CrossRef]
- Kim, M.; Thompson, L.A.; Wenger, S.D.; O’Bryant, C.L. Romidepsin: A histone deacetylase inhibitor for refractory cutaneous T-cell lymphoma. Ann. Pharmacother. 2012, 46, 1340–1348. [Google Scholar] [CrossRef]
- Prince, H.M.; Dickinson, M. Romidepsin for cutaneous T-cell lymphoma. Clin. Cancer Res. 2012, 18, 3509–3515. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; He, K.; Sridhara, R.; Abraham, S.; Booth, B.P.; Verbois, L.; Morse, D.E.; Jee, J.M.; Pope, S.; et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin. Cancer Res. 2007, 13, 2318–2322. [Google Scholar] [CrossRef]
- Beckers, T.; Burkhardt, C.; Wieland, H.; Gimmnich, P.; Ciossek, T.; Maier, T.; Sanders, K. Distinct pharmacological properties of second generation HDAC inhibitors with the benzamide or hydroxamate head group. Int. J. Cancer 2007, 121, 1138–1148. [Google Scholar] [CrossRef]
- Kim, Y.J.; Greer, C.B.; Cecchini, K.R.; Harris, L.N.; Tuck, D.P.; Kim, T.H. HDAC inhibitors induce transcriptional repression of high copy number genes in breast cancer through elongation blockade. Oncogene 2013, 32, 2828–2835. [Google Scholar] [CrossRef]
- Hsu, K.W.; Huang, C.Y.; Tam, K.W.; Lin, C.Y.; Huang, L.C.; Lin, C.L.; Hsieh, W.S.; Chi, W.M.; Chang, Y.J.; Wei, P.L.; et al. The Application of Non-Invasive Apoptosis Detection Sensor (NIADS) on Histone Deacetylation Inhibitor (HDACi)-Induced Breast Cancer Cell Death. Int. J. Mol. Sci. 2018, 19, 452. [Google Scholar] [CrossRef]
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [Google Scholar] [CrossRef]
- Lane, A.A.; Chabner, B.A. Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 2009, 27, 5459–5468. [Google Scholar] [CrossRef]
- Cooper, S.J.; von Roemeling, C.A.; Kang, K.H.; Marlow, L.A.; Grebe, S.K.; Menefee, M.E.; Tun, H.W.; Colon-Otero, G.; Perez, E.A.; Copland, J.A. Reexpression of Tumor Suppressor, sFRP1, Leads to Antitumor Synergy of Combined HDAC and Methyltransferase Inhibitors in Chemoresistant Cancers. Mol. Cancer Ther. 2012, 11, 2105–2115. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Davis, R.; Singh, K.P.; Kurzrock, R.; Ross, D.D.; Srivastava, R.K. Suberoylanilide hydroxamic acid (Zolinza/vorinostat) sensitizes TRAIL-resistant breast cancer cells orthotopically implanted in BALB/c nude mice. Mol. Cancer Ther. 2009, 8, 1596–1605. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, X.; Wang, S.; Lee, C.-K.; Yang, X.; Liu, B. HDAC inhibitor SNDX-275 enhances efficacy of trastuzumab in erbB2-overexpressing breast cancer cells and exhibits potential to overcome trastuzumab resistance. Cancer Lett. 2011, 307, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Tate, C.R.; Rhodes, L.V.; Segar, H.C.; Driver, J.L.; Pounder, F.N.; Burow, M.E.; Collins-Burow, B.M. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res. 2012, 14, R79. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Cartron, P.F.; Jouvenot, M.; Delage-Mourroux, R. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics 2013, 8, 237–245. [Google Scholar] [CrossRef]
- Sappok, A.; Mahlknecht, U. Ribavirin restores ESR1 gene expression and tamoxifen sensitivity in ESR1 negative breast cancer cell lines. Clin. Epigenetics 2011, 3, 8. [Google Scholar] [CrossRef]
- Thomas, S.; Munster, P.N. Histone deacetylase inhibitor induced modulation of anti-estrogen therapy. Cancer Lett. 2009, 280, 184–191. [Google Scholar] [CrossRef]
- Fiskus, W.; Ren, Y.; Mohapatra, A.; Bali, P.; Mandawat, A.; Rao, R.; Herger, B.; Yang, Y.; Atadja, P.; Wu, J.; et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-alpha levels and transcriptional activity: A result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin. Cancer Res. 2007, 13, 4882–4890. [Google Scholar] [CrossRef]
- Munster, P.N.; Thurn, K.T.; Thomas, S.; Raha, P.; Lacevic, M.; Miller, A.; Melisko, M.; Ismail-Khan, R.; Rugo, H.; Moasser, M.; et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br. J. Cancer 2011, 104, 1828–1835. [Google Scholar] [CrossRef]
- Yardley, D.A.; Ismail-Khan, R.R.; Melichar, B.; Lichinitser, M.; Munster, P.N.; Klein, P.M.; Cruickshank, S.; Miller, K.D.; Lee, M.J.; Trepel, J.B. Randomized phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J. Clin. Oncol. 2013, 31, 2128–2135. [Google Scholar] [CrossRef]
- Ramaswamy, B.; Fiskus, W.; Cohen, B.; Pellegrino, C.; Hershman, D.L.; Chuang, E.; Luu, T.; Somlo, G.; Goetz, M.; Swaby, R.; et al. Phase I-II study of vorinostat plus paclitaxel and bevacizumab in metastatic breast cancer: Evidence for vorinostat-induced tubulin acetylation and Hsp90 inhibition in vivo. Breast Cancer Res. Treat. 2012, 132, 1063–1072. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef]
- Tian, X.; Zhang, S.; Liu, H.-M.; Zhang, Y.-B.; Blair, C.; Mercola, D.; Sassone-Corsi, P.; Zi, X. Histone Lysine-Specific Methyltransferases and Demethylases in Carcinogenesis: New Targets for Cancer Therapy and Prevention. Curr. Cancer Drug Targets 2013, 13, 558–579. [Google Scholar] [CrossRef]
- Yan, N.; Xu, L.; Wu, X.; Zhang, L.; Fei, X.; Cao, Y.; Zhang, F. GSKJ4, an H3K27me3 demethylase inhibitor, effectively suppresses the breast cancer stem cells. Exp. Cell Res. 2017, 359, 405–414. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, L.; Sun, J.; Hu, X.; Kalvakolanu, D.V.; Ren, H.; Guo, B. Roles for the methyltransferase SETD8 in DNA damage repair. Clin. Epigenetics 2022, 14, 34. [Google Scholar] [CrossRef]
- Shen, A.; Yu, X.-Y. Histone lysine demethylase inhibitor (HDMi) as chemo-sensitizing agent. In Epigenetic Regulation in Overcoming Chemoresistance; Academic Press: Cambridge, MA, USA, 2021; pp. 41–55. [Google Scholar] [CrossRef]
- Varghese, B.; Del Gaudio, N.; Cobellis, G.; Altucci, L.; Nebbioso, A. KDM4 Involvement in Breast Cancer and Possible Therapeutic Approaches. Front. Oncol. 2021, 11, 750315. [Google Scholar] [CrossRef]
- Wang, J.-J.; Wang, X.; Xian, Y.-E.; Chen, Z.-Q.; Sun, Y.-P.; Fu, Y.-W.; Wu, Z.-K.; Li, P.-X.; Zhou, E.-S.; Yang, Z.-T. The JMJD3 histone demethylase inhibitor GSK-J1 ameliorates lipopolysaccharide-induced inflammation in a mastitis model. J. Biol. Chem. 2022, 298, 102017. [Google Scholar] [CrossRef]
- Yang, G.-J.; Liu, Y.-J.; Ding, L.-J.; Tao, F.; Zhu, M.-H.; Shi, Z.-Y.; Wen, J.-M.; Niu, M.-Y.; Li, X.; Xu, Z.-S.; et al. A state-of-the-art review on LSD1 and its inhibitors in breast cancer: Molecular mechanisms and therapeutic significance. Front. Pharmacol. 2022, 13, 98957. [Google Scholar] [CrossRef]
- Li, W.; Wu, H.; Sui, S.; Wang, Q.; Xu, S.; Pang, D. Targeting Histone Modifications in Breast Cancer: A Precise Weapon on the Way. Front Cell Dev Biol. 2021, 14, 736935. [Google Scholar] [CrossRef]
- Rajewsky, N. L(ou)sy miRNA targets? Nat. Struct. Mol. Biol. 2006, 13, 754–755. [Google Scholar] [CrossRef]
- Liu, W.; Mao, S.Y.; Zhu, W.Y. Impact of tiny miRNAs on cancers. World J. Gastroenterol. 2007, 13, 497–502. [Google Scholar] [CrossRef]
- Berezikov, E.; Guryev, V.; van de Belt, J.; Wienholds, E.; Plasterk, R.H.; Cuppen, E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell 2005, 120, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H. Clinical Relevance of Circulating, Cell-Free and Exosomal microRNAs in Plasma and Serum of Breast Cancer Patients. Oncol. Res. Treat. 2017, 40, 423–429. [Google Scholar] [CrossRef]
- Davey, M.G.; Davies, M.; Lowery, A.J.; Miller, N.; Kerin, M.J. The Role of MicroRNA as Clinical Biomarkers for Breast Cancer Surgery and Treatment. Int. J. Mol. Sci. 2021, 22, 8290. [Google Scholar] [CrossRef] [PubMed]
- Al-Othman, N.; Ahram, M.; Alqaraleh, M. Role of androgen and microRNA in triple-negative breast cancer. Breast Dis. 2020, 39, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.M.; Yigit, M.V. The role of microRNA in resistance to breast cancer therapy. Wiley Interdiscip. Rev. RNA 2014, 5, 823–833. [Google Scholar] [CrossRef]
- Tekiner, T.A.; Basaga, H. Role of microRNA deregulation in breast cancer cell chemoresistance and stemness. Curr. Med. Chem. 2013, 20, 3358–3369. [Google Scholar] [CrossRef]
- Teo, M.T.; Landi, D.; Taylor, C.F.; Elliott, F.; Vaslin, L.; Cox, D.G.; Hall, J.; Landi, S.; Bishop, D.T.; Kiltie, A.E. The role of microRNA-binding site polymorphisms in DNA repair genes as risk factors for bladder cancer and breast cancer and their impact on radiotherapy outcomes. Carcinogenesis 2012, 33, 581–586. [Google Scholar] [CrossRef]
- Cho, W.C. MicroRNAs: Potential biomarkers for cancer diagnosis, prognosis and targets for therapy. Int. J. Biochem. Cell Biol. 2010, 42, 1273–1281. [Google Scholar] [CrossRef]
- Cho, W.C. Great potential of miRNAs as predictive and prognostic markers for cancer. Expert Rev. Mol. Diagn. 2012, 12, 315–318. [Google Scholar] [CrossRef]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. microRNAs as oncogenes and tumor suppressors. Dev. Biol. 2007, 302, 1–12. [Google Scholar] [CrossRef]
- Uyisenga, J.P.; Debit, A.; Poulet, C.; Freres, P.; Poncin, A.; Thiry, J.; Mutesa, L.; Jerusalem, G.; Bours, V.; Josse, C. Differences in plasma microRNA content impair microRNA-based signature for breast cancer diagnosis in cohorts recruited from heterogeneous environmental sites. Sci. Rep. 2021, 11, 11698. [Google Scholar] [CrossRef]
- Zou, X.; Xia, T.; Li, M.; Wang, T.; Liu, P.; Zhou, X.; Huang, Z.; Zhu, W. MicroRNA profiling in serum: Potential signatures for breast cancer diagnosis. Cancer Biomark. 2021, 30, 41–53. [Google Scholar] [CrossRef]
- Braga, E.A.; Burdennyy, A.M.; Pronina, I.V.; Filippova, E.A.; Kazubskaya, T.P.; Fridman, M.V.; Khodyrev, D.S.; Karpukhin, A.V.; Loginov, V.I.; Kushlinskii, N.E. System of Markers Based on the Methylation of a Group of Proapoptotic Genes in Combination with MicroRNA in the Diagnosis of Breast Cancer. Bull. Exp. Biol. Med. 2020, 168, 366–370. [Google Scholar] [CrossRef]
- Kahraman, M.; Roske, A.; Laufer, T.; Fehlmann, T.; Backes, C.; Kern, F.; Kohlhaas, J.; Schrors, H.; Saiz, A.; Zabler, C.; et al. MicroRNA in diagnosis and therapy monitoring of early-stage triple-negative breast cancer. Sci. Rep. 2018, 8, 11584. [Google Scholar] [CrossRef]
- Andorfer, C.A.; Necela, B.M.; Thompson, E.A.; Perez, E.A. MicroRNA signatures: Clinical biomarkers for the diagnosis and treatment of breast cancer. Trends Mol. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef]
- Sun, X.; Xu, H.; Huang, T.; Zhang, C.; Wu, J.; Luo, S. Simultaneous delivery of anti-miRNA and docetaxel with supramolecular self-assembled “chitosome” for improving chemosensitivity of triple negative breast cancer cells. Drug Deliv. Transl. Res. 2021, 11, 192–204. [Google Scholar] [CrossRef]
- Yin, H.; Xiong, G.; Guo, S.; Xu, C.; Xu, R.; Guo, P.; Shu, D. Delivery of Anti-miRNA for Triple-Negative Breast Cancer Therapy Using RNA Nanoparticles Targeting Stem Cell Marker CD133. Mol. Ther. 2019, 27, 1252–1261. [Google Scholar] [CrossRef]
- Shu, D.; Li, H.; Shu, Y.; Xiong, G.; Carson, W.E., 3rd; Haque, F.; Xu, R.; Guo, P. Systemic Delivery of Anti-miRNA for Suppression of Triple Negative Breast Cancer Utilizing RNA Nanotechnology. ACS Nano 2015, 9, 9731–9740. [Google Scholar] [CrossRef]
- Gorur, A.; Bayraktar, R.; Ivan, C.; Mokhlis, H.A.; Bayraktar, E.; Kahraman, N.; Karakas, D.; Karamil, S.; Kabil, N.N.; Kanlikilicer, P.; et al. ncRNA therapy with miRNA-22-3p suppresses the growth of triple-negative breast cancer. Mol. Ther. Nucleic Acids 2021, 23, 930–943. [Google Scholar] [CrossRef]
- Qattan, A. Novel miRNA Targets and Therapies in the Triple-Negative Breast Cancer Microenvironment: An Emerging Hope for a Challenging Disease. Int. J. Mol. Sci. 2020, 21, 8905. [Google Scholar] [CrossRef] [PubMed]
- Valcourt, D.M.; Day, E.S. Dual Regulation of miR-34a and Notch Signaling in Triple-Negative Breast Cancer by Antibody/miRNA Nanocarriers. Mol. Ther. Nucleic Acids 2020, 21, 290–298. [Google Scholar] [CrossRef]
- Dwivedi, S.; Pareek, P.; Vishnoi, J.R.; Sharma, P.; Misra, S. Downregulation of miRNA-21 and cancer stem cells after chemotherapy results in better outcome in breast cancer patients. World J. Stem Cells 2022, 14, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Cheng, Z.; Yang, Y.; Shen, J.; Zhu, Y.; Ling, L.; Lin, W.; Yu, Z.; Li, Z.; Tan, W.; et al. A 10-miRNA risk score-based prediction model for pathological complete response to neoadjuvant chemotherapy in hormone receptor-positive breast cancer. Sci. China Life Sci. 2022, 65, 2205–2217. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, E.M.; Ragle Aure, M.; Haugen, M.H.; Kleivi Sahlberg, K.; Kristensen, V.N.; Nebdal, D.; Borresen-Dale, A.L.; Lingjaerde, O.C.; Engebraaten, O. miRNA expression changes during the course of neoadjuvant bevacizumab and chemotherapy treatment in breast cancer. Mol. Oncol. 2019, 13, 2278–2296. [Google Scholar] [CrossRef]
- Xue, J.; Chi, Y.; Chen, Y.; Huang, S.; Ye, X.; Niu, J.; Wang, W.; Pfeffer, L.M.; Shao, Z.M.; Wu, Z.H.; et al. MiRNA-621 sensitizes breast cancer to chemotherapy by suppressing FBXO11 and enhancing p53 activity. Oncogene 2016, 35, 448–458. [Google Scholar] [CrossRef]
- Rama, A.R.; Lara, P.; Mesas, C.; Quinonero, F.; Velez, C.; Melguizo, C.; Prados, J. Circular Sponge against miR-21 Enhances the Antitumor Activity of Doxorubicin against Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 14803. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Xue, J.; Liang, W.; Zhang, Z.; Yang, X.; Qiao, Z.; Jiang, Y.; Wang, J.; Cao, X.; et al. Co-treatment with miR-21-5p inhibitor and Aurora kinase inhibitor reversine suppresses breast cancer progression by targeting sprouty RTK signaling antagonist 2. Bioengineered 2022, 13, 455–468. [Google Scholar] [CrossRef]
- Wu, H.; Chen, S.; Liu, C.; Li, J.; Wei, X.; Jia, M.; Guo, J.; Jin, J.; Meng, D.; Zhi, X. SPTBN1 inhibits growth and epithelial-mesenchymal transition in breast cancer by downregulating miR-21. Eur. J. Pharmacol. 2021, 909, 174401. [Google Scholar] [CrossRef]
- Arisan, E.D.; Rencuzogullari, O.; Cieza-Borrella, C.; Miralles Arenas, F.; Dwek, M.; Lange, S.; Uysal-Onganer, P. MiR-21 Is Required for the Epithelial-Mesenchymal Transition in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 1557. [Google Scholar] [CrossRef]
- Anwar, S.L.; Sari, D.N.I.; Kartika, A.I.; Fitria, M.S.; Tanjung, D.S.; Rakhmina, D.; Wardana, T.; Astuti, I.; Haryana, S.M.; Aryandono, T. Upregulation of Circulating MiR-21 Expression as a Potential Biomarker for Therapeutic Monitoring and Clinical Outcome in Breast Cancer. Asian Pac. J. Cancer Prev. 2019, 20, 1223–1228. [Google Scholar] [CrossRef]
- Mohammady, M.; Ghetmiri, S.I.; Baharizade, M.; Morowvat, M.H.; Torabi, S. Expanding the Biotherapeutics Realm via miR-34a: “Potent Clever Little” Agent in Breast Cancer Therapy. Curr. Pharm. Biotechnol. 2019, 20, 665–673. [Google Scholar] [CrossRef]
- Xia, Y.; Deng, X.; Cao, M.; Liu, S.; Zhang, X.; Xiao, X.; Shen, S.; Hu, Q.; Sheng, W. Nanodiamond-based layer-by-layer nanohybrids mediate targeted delivery of miR-34a for triple negative breast cancer therapy. RSC Adv. 2018, 8, 13789–13797. [Google Scholar] [CrossRef]
- Wu, M.Y.; Fu, J.; Xiao, X.; Wu, J.; Wu, R.C. MiR-34a regulates therapy resistance by targeting HDAC1 and HDAC7 in breast cancer. Cancer Lett. 2014, 354, 311–319. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, Y.; Yang, R.; Mu, J.; Zhou, Z.; Sun, M. Poly-antioxidants for enhanced anti-miR-155 delivery and synergistic therapy of metastatic breast cancer. Biomater. Sci. 2022, 10, 3637–3646. [Google Scholar] [CrossRef]
- Shang, M.; Wu, Y.; Wang, Y.; Cai, Y.; Jin, J.; Yang, Z. Dual antisense oligonucleotide targeting miR-21/miR-155 synergize photodynamic therapy to treat triple-negative breast cancer and inhibit metastasis. Biomed. Pharmacother. 2022, 146, 112564. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Yu, G.; Sun, Z.; Wang, T.; Tian, X.; Duan, X.; Zhang, C. Exosomal miR-1246 and miR-155 as predictive and prognostic biomarkers for trastuzumab-based therapy resistance in HER2-positive breast cancer. Cancer Chemother. Pharmacol. 2020, 86, 761–772. [Google Scholar] [CrossRef]
- Abtin, M.; Alivand, M.R.; Khaniani, M.S.; Bastami, M.; Zaeifizadeh, M.; Derakhshan, S.M. Simultaneous downregulation of miR-21 and miR-155 through oleuropein for breast cancer prevention and therapy. J. Cell Biochem. 2018, 119, 7151–7165. [Google Scholar] [CrossRef]
- Zaleska, K.; Przybyla, A.; Kulcenty, K.; Wichtowski, M.; Mackiewicz, A.; Suchorska, W.; Murawa, D. Wound fluids affect miR-21, miR-155 and miR-221 expression in breast cancer cell lines, and this effect is partially abrogated by intraoperative radiation therapy treatment. Oncol. Lett. 2017, 14, 4029–4036. [Google Scholar] [CrossRef]
- Fischer, C.; Deutsch, T.M.; Feisst, M.; Rippinger, N.; Riedel, F.; Hartkopf, A.D.; Brucker, S.Y.; Domschke, C.; Fremd, C.; Michel, L.; et al. Circulating miR-200 family as predictive markers during systemic therapy of metastatic breast cancer. Arch. Gynecol. Obstet. 2022, 306, 875–885. [Google Scholar] [CrossRef]
- Mekala, J.R.; Naushad, S.M.; Ponnusamy, L.; Arivazhagan, G.; Sakthiprasad, V.; Pal-Bhadra, M. Epigenetic regulation of miR-200 as the potential strategy for the therapy against triple-negative breast cancer. Gene 2018, 641, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Khalighfard, S.; Alizadeh, A.M.; Irani, S.; Omranipour, R. Plasma miR-21, miR-155, miR-10b, and Let-7a as the potential biomarkers for the monitoring of breast cancer patients. Sci. Rep. 2018, 8, 17981. [Google Scholar] [CrossRef] [PubMed]
- Fkih M’hamed, I.; Privat, M.; Trimeche, M.; Penault-Llorca, F.; Bignon, Y.J.; Kenani, A. miR-10b, miR-26a, miR-146a and miR-153 Expression in Triple Negative Vs Non Triple Negative Breast Cancer: Potential Biomarkers. Pathol. Oncol. Res. 2017, 23, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.; Kavishwar, A.; Ross, A.; Wang, P.; Tabassum, D.P.; Polyak, K.; Barteneva, N.; Petkova, V.; Pantazopoulos, P.; Tena, A.; et al. Combining miR-10b-Targeted Nanotherapy with Low-Dose Doxorubicin Elicits Durable Regressions of Metastatic Breast Cancer. Cancer Res. 2015, 75, 4407–4415. [Google Scholar] [CrossRef]
- Imani, S.; Zhang, X.; Hosseinifard, H.; Fu, S.; Fu, J. The diagnostic role of microRNA-34a in breast cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 23177–23187. [Google Scholar] [CrossRef]
- Adams, B.D.; Wali, V.B.; Cheng, C.J.; Inukai, S.; Booth, C.J.; Agarwal, S.; Rimm, D.L.; Gyorffy, B.; Santarpia, L.; Pusztai, L.; et al. miR-34a Silences c-SRC to Attenuate Tumor Growth in Triple-Negative Breast Cancer. Cancer Res. 2016, 76, 927–939. [Google Scholar] [CrossRef]
- Li, L.; Yuan, L.; Luo, J.; Gao, J.; Guo, J.; Xie, X. MiR-34a inhibits proliferation and migration of breast cancer through down-regulation of Bcl-2 and SIRT1. Clin. Exp. Med. 2013, 13, 109–117. [Google Scholar] [CrossRef]
- Pichler, M.; Calin, G.A. MicroRNAs in cancer: From developmental genes in worms to their clinical application in patients. Br. J. Cancer 2015, 113, 569–573. [Google Scholar] [CrossRef]
- Smith, B.; Agarwal, P.; Bhowmick, N.A. MicroRNA applications for prostate, ovarian and breast cancer in the era of precision medicine. Endocr. Relat. Cancer 2017, 24, R157–R172. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- Lucibello, G.; Mograbi, B.; Milano, G.; Hofman, P.; Brest, P. PD-L1 regulation revisited: Impact on immunotherapeutic strategies. Trends Mol. Med. 2021, 27, 868–881. [Google Scholar] [CrossRef]
- Li, Z.H.; Weng, X.; Xiong, Q.Y.; Tu, J.H.; Xiao, A.; Qiu, W.; Gong, Y.; Hu, E.W.; Huang, S.; Cao, Y.L. miR-34a expression in human breast cancer is associated with drug resistance. Oncotarget 2017, 8, 106270–106282. [Google Scholar] [CrossRef]
- Mei, M.; Ren, Y.; Zhou, X.; Yuan, X.B.; Han, L.; Wang, G.X.; Jia, Z.; Pu, P.Y.; Kang, C.S.; Yao, Z. Downregulation of miR-21 enhances chemotherapeutic effect of taxol in breast carcinoma cells. Technol. Cancer Res. Treat. 2010, 9, 77–86. [Google Scholar] [CrossRef]
- Chen, L.; Bourguignon, L.Y.W. Hyaluronan-CD44 interaction promotes c-Jun signaling and miRNA21 expression leading to Bcl-2 expression and chemoresistance in breast cancer cells. Mol. Cancer 2014, 13, 52. [Google Scholar] [CrossRef]
- Shaban, N.Z.; Ibrahim, N.K.; Saada, H.N.; El-Rashidy, F.H.; Shaaban, H.M.; Farrag, M.A.; ElDebaiky, K.; Kodous, A.S. miR-34a and miR-21 as biomarkers in evaluating the response of chemo-radiotherapy in Egyptian breast cancer patients. J. Radiat. Res. Appl. Sci. 2022, 15, 285–292. [Google Scholar] [CrossRef]
- Yadav, P.; Mirza, M.; Nandi, K.; Jain, S.K.; Kaza, R.C.; Khurana, N.; Ray, P.C.; Saxena, A. Serum microRNA-21 expression as a prognostic and therapeutic biomarker for breast cancer patients. Tumour Biol. 2016, 37, 15275–15282. [Google Scholar] [CrossRef]
- Wang, H.; Tan, G.; Dong, L.; Cheng, L.; Li, K.; Wang, Z.; Luo, H. Circulating MiR-125b as a marker predicting chemoresistance in breast cancer. PLoS ONE 2012, 7, e34210. [Google Scholar] [CrossRef]
- Wang, Y.; Zeng, G.; Jiang, Y. The Emerging Roles of miR-125b in Cancers. Cancer Manag. Res. 2020, 12, 1079–1088. [Google Scholar] [CrossRef]
- Zhu, W.; Liu, M.; Fan, Y.; Ma, F.; Xu, N.; Xu, B. Dynamics of circulating microRNAs as a novel indicator of clinical response to neoadjuvant chemotherapy in breast cancer. Cancer Med. 2018, 7, 4420–4433. [Google Scholar] [CrossRef]
- Cardinale, D.; Biasillo, G.; Cipolla, C.M. Curing Cancer, Saving the Heart: A Challenge That Cardioncology Should Not Miss. Curr. Cardiol. Rep. 2016, 18, 51. [Google Scholar] [CrossRef]
- Brown, C.; Mantzaris, M.; Nicolaou, E.; Karanasiou, G.; Papageorgiou, E.; Curigliano, G.; Cardinale, D.; Filippatos, G.; Memos, N.; Naka, K.K.; et al. A systematic review of miRNAs as biomarkers for chemotherapy-induced cardiotoxicity in breast cancer patients reveals potentially clinically informative panels as well as key challenges in miRNA research. Cardiooncology 2022, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Yadi, W.; Shurui, C.; Tong, Z.; Suxian, C.; Qing, T.; Dongning, H. Bioinformatic analysis of peripheral blood miRNA of breast cancer patients in relation with anthracycline cardiotoxicity. BMC Cardiovasc. Disord. 2020, 20, 43. [Google Scholar] [CrossRef] [PubMed]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Investig. 2009, 119, 2772–2786. [Google Scholar] [CrossRef] [PubMed]
- Freres, P.; Bouznad, N.; Servais, L.; Josse, C.; Wenric, S.; Poncin, A.; Thiry, J.; Moonen, M.; Oury, C.; Lancellotti, P.; et al. Variations of circulating cardiac biomarkers during and after anthracycline-containing chemotherapy in breast cancer patients. BMC Cancer 2018, 18, 102. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, X.; Dong, H.; Ke, S.; Zheng, W.H. Let-7f and miRNA-126 correlate with reduced cardiotoxicity risk in triple-negative breast cancer patients who underwent neoadjuvant chemotherapy. Int. J. Clin. Exp. Pathol. 2018, 11, 4987–4995. [Google Scholar]
- Qin, X.; Chang, F.; Wang, Z.; Jiang, W. Correlation of circulating pro-angiogenic miRNAs with cardiotoxicity induced by epirubicin/cyclophosphamide followed by docetaxel in patients with breast cancer. Cancer Biomark. 2018, 23, 473–484. [Google Scholar] [CrossRef]
- Feng, Q.; Ren, Y.; Hou, A.; Guo, J.; Mao, Z.; Liu, S.; Wang, B.; Bai, Z.; Hou, X. MicroRNA-130a Increases and Predicts Cardiotoxicity during Adjuvant Chemotherapy in Human Epidermal Growth Factor Receptor-2-Positive Breast Cancer. J. Breast Cancer 2021, 24, 153–163. [Google Scholar] [CrossRef]
- Lakhani, H.V.; Pillai, S.S.; Zehra, M.; Dao, B.; Tirona, M.T.; Thompson, E.; Sodhi, K. Detecting early onset of anthracyclines-induced cardiotoxicity using a novel panel of biomarkers in West-Virginian population with breast cancer. Sci. Rep. 2021, 11, 7954. [Google Scholar] [CrossRef]
- Ntelios, D.; Meditskou, S.; Efthimiadis, G.; Pitsis, A.; Nikolakaki, E.; Girtovitis, F.; Parcharidou, D.; Zegkos, T.; Kouidou, S.; Karvounis, H.; et al. Elevated plasma levels of miR-29a are associated with hemolysis in patients with hypertrophic cardiomyopathy. Clin. Chim. Acta 2017, 471, 321–326. [Google Scholar] [CrossRef]
- Weisz, A.; Li, Q.; Liu, M.; Ma, F.; Luo, Y.; Cai, R.; Wang, L.; Xu, N.; Xu, B. Circulating miR-19a and miR-205 in Serum May Predict the Sensitivity of Luminal A Subtype of Breast Cancer Patients to Neoadjuvant Chemotherapy with Epirubicin Plus Paclitaxel. PLoS ONE 2014, 9, e104870. [Google Scholar] [CrossRef]
- Zhao, R.; Wu, J.; Jia, W.; Gong, C.; Yu, F.; Ren, Z.; Chen, K.; He, J.; Su, F. Plasma miR-221 as a Predictive Biomarker for Chemoresistance in Breast Cancer Patients who Previously Received Neoadjuvant Chemotherapy. Onkologie 2011, 34, 675–680. [Google Scholar] [CrossRef]
- Kleivi Sahlberg, K.; Bottai, G.; Naume, B.; Burwinkel, B.; Calin, G.A.; Borresen-Dale, A.L.; Santarpia, L. A serum microRNA signature predicts tumor relapse and survival in triple-negative breast cancer patients. Clin. Cancer Res. 2015, 21, 1207–1214. [Google Scholar] [CrossRef]
- Chan, M.; Liaw, C.S.; Ji, S.M.; Tan, H.H.; Wong, C.Y.; Thike, A.A.; Tan, P.H.; Ho, G.H.; Lee, A.S.-G. Identification of Circulating MicroRNA Signatures for Breast Cancer Detection. Clin. Cancer Res. 2013, 19, 4477–4487. [Google Scholar] [CrossRef]
- Roth, C.; Rack, B.; Müller, V.; Janni, W.; Pantel, K.; Schwarzenbach, H. Circulating microRNAs as blood-based markers for patients with primary and metastatic breast cancer. Breast Cancer Res. 2010, 12, R90. [Google Scholar] [CrossRef]
- Petrović, N. miR-21 Might be Involved in Breast Cancer Promotion and Invasion Rather than in Initial Events of Breast Cancer Development. Mol. Diagn. Ther. 2016, 20, 97–110. [Google Scholar] [CrossRef]
- Yan, L.X.; Wu, Q.N.; Zhang, Y.; Li, Y.Y.; Liao, D.Z.; Hou, J.H.; Fu, J.; Zeng, M.S.; Yun, J.P.; Wu, Q.L.; et al. Knockdown of miR-21 in human breast cancer cell lines inhibits proliferation, in vitro migration and in vivotumor growth. Breast Cancer Res. 2011, 13, R2. [Google Scholar] [CrossRef]
- Liu, B.; Su, F.; Chen, M.; Li, Y.; Qi, X.; Xiao, J.; Li, X.; Liu, X.; Liang, W.; Zhang, Y.; et al. Serum miR-21 and miR-125b as markers predicting neoadjuvant chemotherapy response and prognosis in stage II/III breast cancer. Hum. Pathol. 2017, 64, 44–52. [Google Scholar] [CrossRef]
- Amato, R.J. Inhibition of DNA methylation by antisense oligonucleotide MG98 as cancer therapy. Clin. Genitourin Cancer 2007, 5, 422–426. [Google Scholar] [CrossRef]
- Hu, C.; Liu, X.; Zeng, Y.; Liu, J.; Wu, F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: Mechanism and clinical application. Clin. Epigenetics 2021, 13, 166. [Google Scholar] [CrossRef]
- Roy, D.M.; Walsh, L.A.; Chan, T.A. Driver mutations of cancer epigenomes. Protein Cell 2014, 5, 265–296. [Google Scholar] [CrossRef]
- Braicu, C.; Gherman, C.D.; Irimie, A.; Berindan-Neagoe, I. Epigallocatechin-3-Gallate (EGCG) inhibits cell proliferation and migratory behaviour of triple negative breast cancer cells. J. Nanosci. Nanotechnol. 2013, 13, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, Y.Y.; Meeran, S.M.; Tollefsbol, T.O. Synergistic epigenetic reactivation of estrogen receptor-alpha (ERalpha) by combined green tea polyphenol and histone deacetylase inhibitor in ERalpha-negative breast cancer cells. Mol. Cancer 2010, 9, 274. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).