


Transcriptome Analysis of Roots from Wheat (Triticum aestivum L.) Varieties in Response to Drought Stress

Abstract
:1. Introduction
2. Results
2.1. Generation of RNA-Seq Data
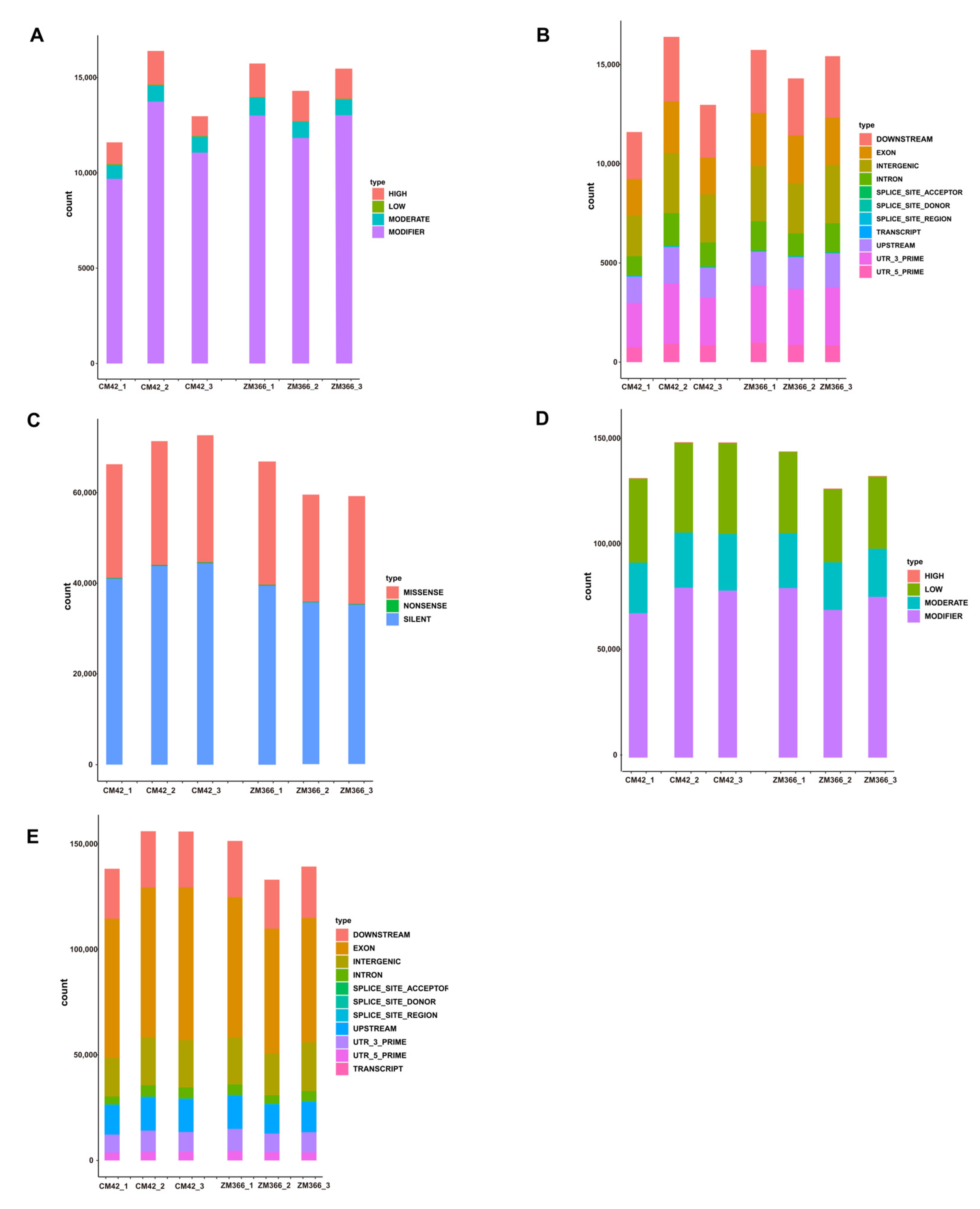
2.2. Variant Site Detection and Analysis
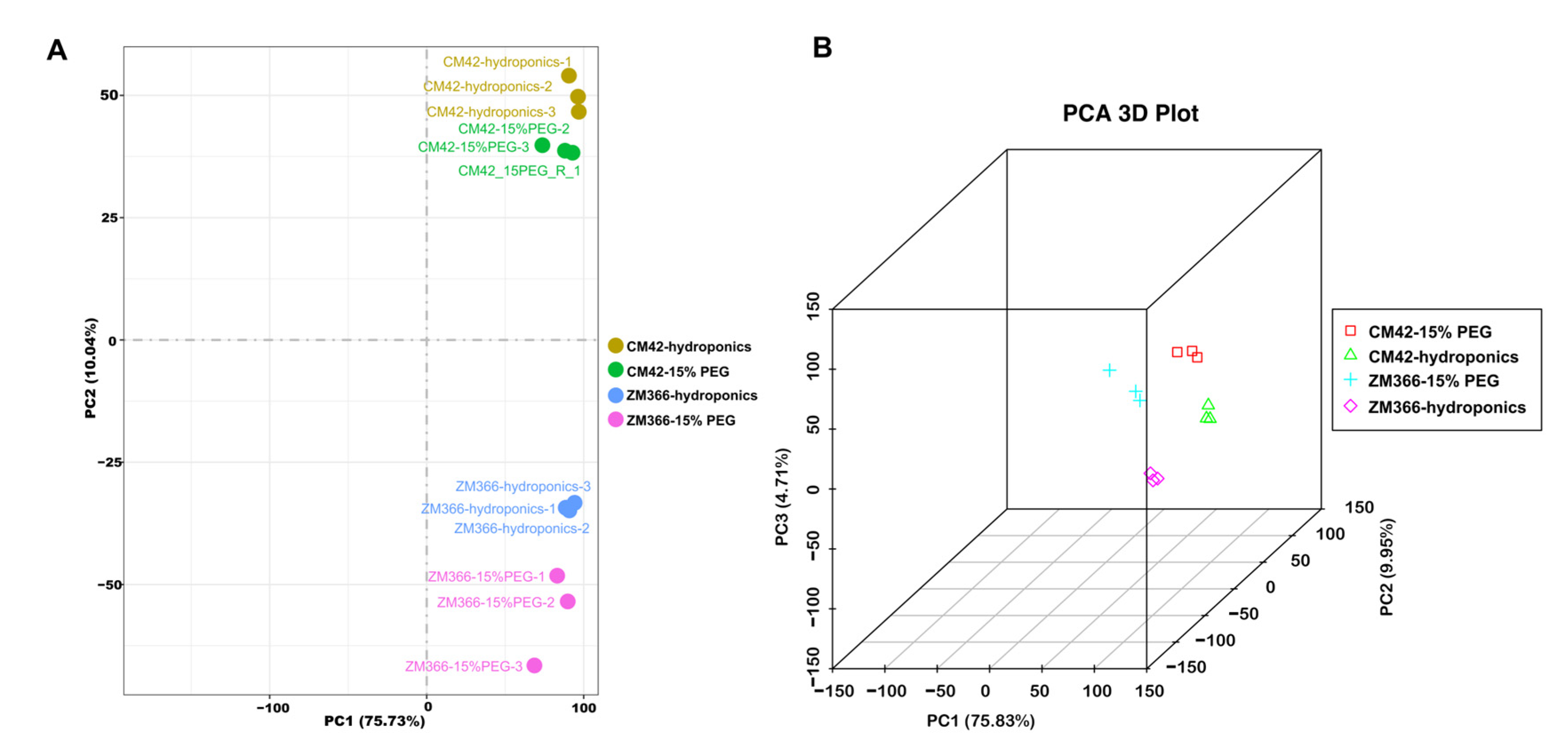
2.3. Principle Component Analysis (PCA)
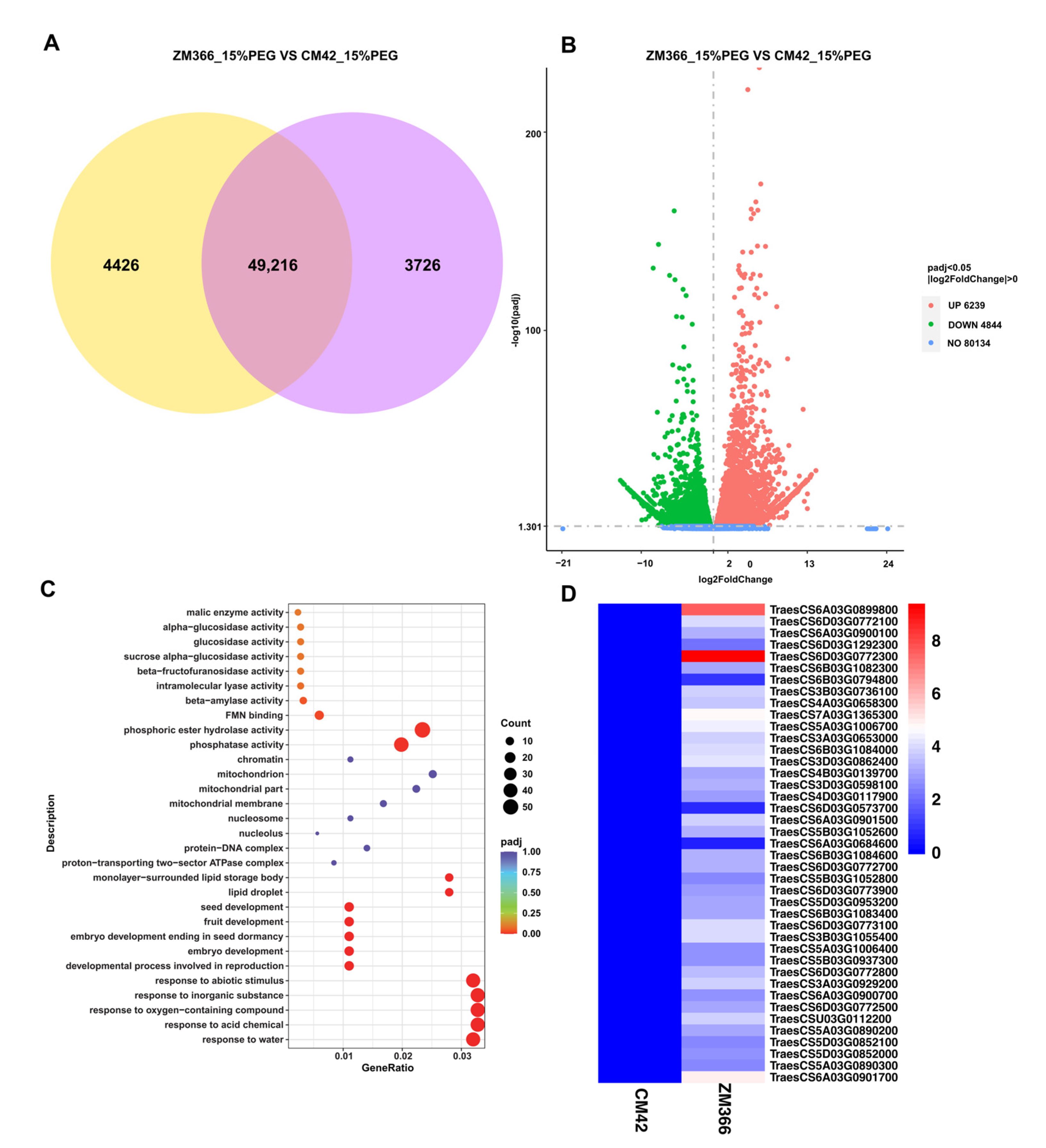
2.4. Identification of Differentially Expressed Genes
2.5. GO Enrichment Analysis of Differentially Expressed Genes (DEGs)
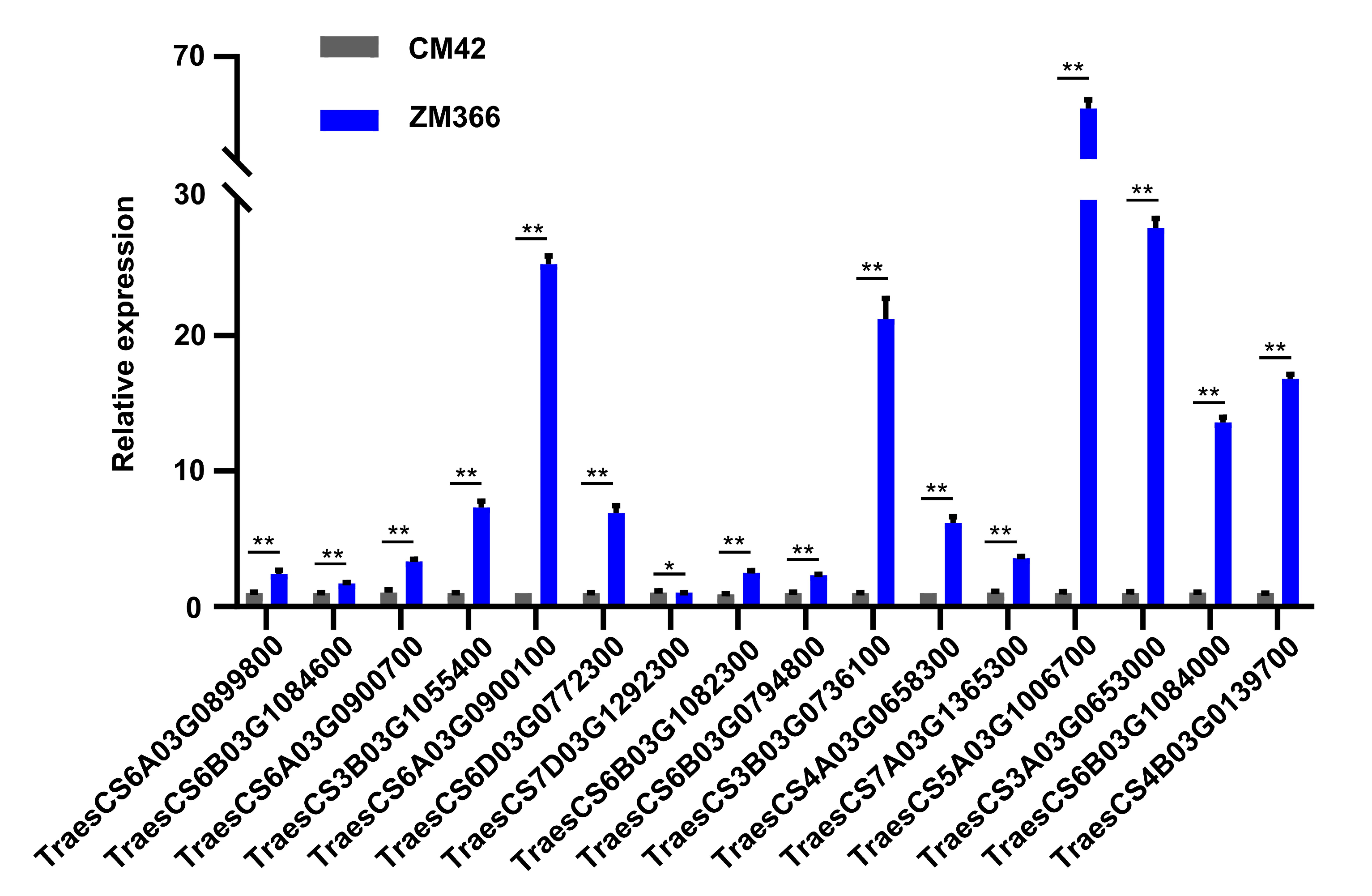
2.6. Quantitative Real-Time PCR Analysis and Functional Verification of Candidate Genes
3. Discussion
3.1. Importance of Drought Resistance Genes in Wheat According to Multi-Omics
3.2. Sixteen Key Candidate Drought Tolerance Genes Identified in the Response to 15% PEG-6000 Treatment
3.3. Genomic Era Genome Data Integration and Establishment of a Stress Gene Resource Exploration Platform
4. Materials and Methods
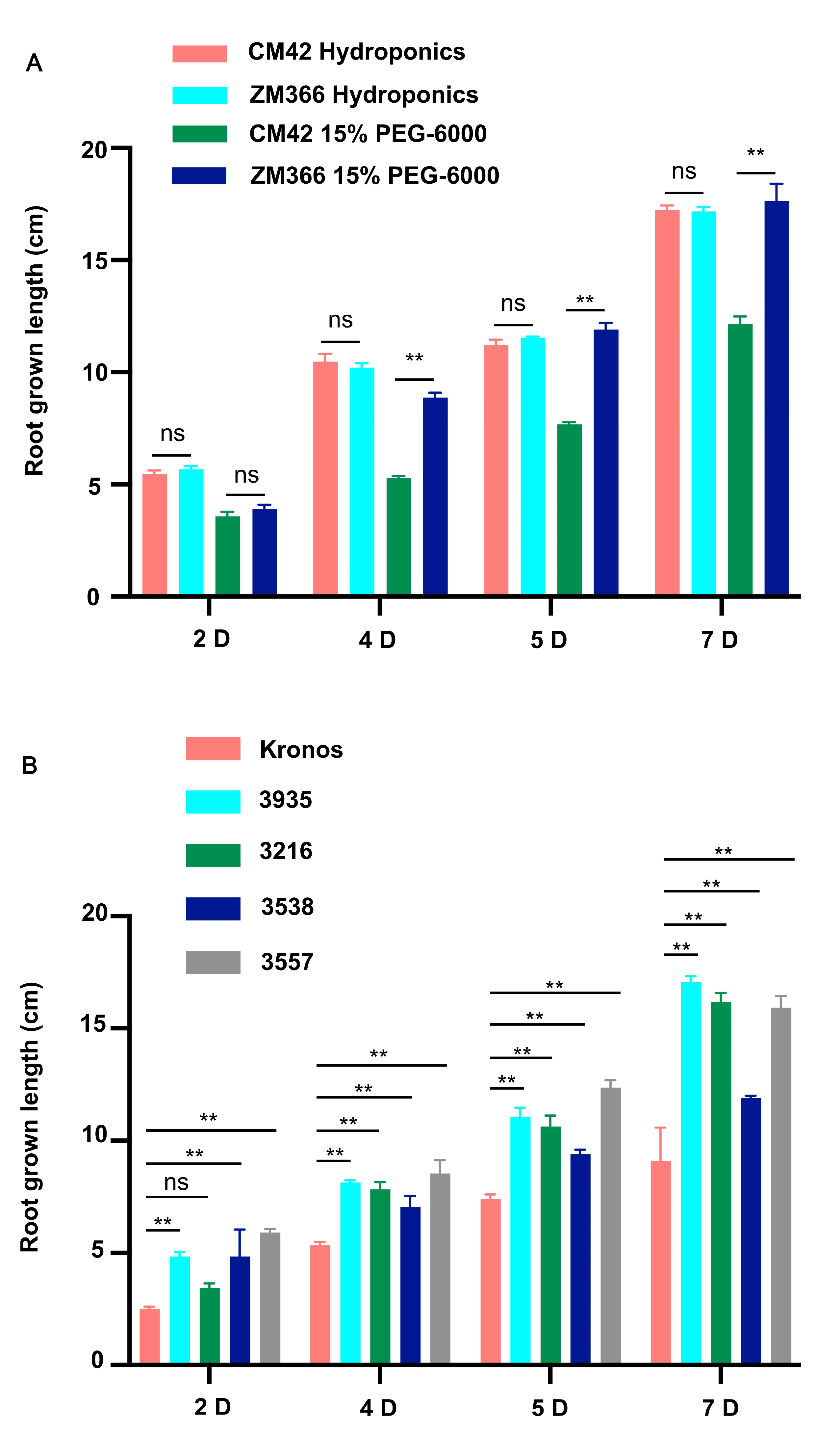
4.1. Materials and Growth Conditions
4.2. RNA Extraction, Library Construction, Sequencing, and Quality Control
4.3. Read Mapping to the Reference Genome and Novel Transcript Prediction
4.4. SNP Analysis
4.5. Quantification of Gene Expression Levels
4.6. Differential Expression Analysis (DEGs)
4.7. GO Enrichment Analysis of Differentially Expressed Genes
4.8. Quantitative Real-Time PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, Y.C. Genepools of Common Wheat. J. Triticeae Crops 2000, 20, 78–81. [Google Scholar]
- Fu, X.D.; Liu, Q.; Li, Z.S.; Zhang, A.M.; Ling, H.Q.; Tong, Y.P.; Liu, Z.Y. Research achievement and prospect development on wheat genome. Bull. Chin. Acad. Sci. 2018, 33, 909–914. [Google Scholar]
- Wang, M.; Xia, G.M. The landscape of molecular mechanisms for salt tolerance in wheat. Crop J. 2018, 6, 42–47. [Google Scholar] [CrossRef]
- Raza, A.; Mubarik, M.S.; Sharif, R.; Habib, M.; Jabeen, W.; Zhang, C.; Chen, H.; Chen, Z.H.; Siddique, K.H.M.; Zhuang, W.; et al. Developing drought-smart, ready-to-grow future crops. Plant Genome 2023, 16, e20279. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avni, R.; Nave, M.; Barad, O.; Baruch, K.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K.; et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.Z.; Zhao, S.C.; Kong, X.Y.; Li, Y.R.; Zhao, G.Y.; He, W.M.; Appels, R.; Pfeifer, M.; Tao, Y.; Zhang, X.Y.; et al. Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 2013, 496, 91–95. [Google Scholar] [CrossRef] [Green Version]
- International Wheat Genome Sequencing Consortium (IWGSC). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, 6403. [Google Scholar]
- Ling, H.Q.; Ma, B.; Shi, X.L.; Liu, H.; Dong, L.L.; Sun, H.; Cao, Y.H.; Gao, Q.; Zheng, S.S.; Li, Y.; et al. Genome sequence of the progenitor of wheat A subgenome Triticum urartu. Nature 2018, 557, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Maccaferri, M.; Harris, N.S.; Twardziok, S.O.; Pasam, R.K.; Gundlach, H.; Spannagl, M.; Ormanbekova, D.; Lux, T.; MPrade, V.; GMilner, S.; et al. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019, 51, 885–895. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Q.; Zhang, H.D.; Dong, F.Y.; Zou, J.; Juan, Z.; Gao, C.B.; Zhu, Z.W.; Liu, Y.K. Multiple roles of wheat calmodulin genes during stress treatment and TaCAM2-D as a positive regulator in response to drought and salt tolerance. Int. J. Biol. Macromol. 2022, 220, 985–997. [Google Scholar] [CrossRef]
- Sharma, E.; Borah, P.; Kaur, A.; Kaur, A.; Bhatnagar, A.; Mohapatra, T.; Kapoor, S.; Khurana, J. A comprehensive transcriptome analysis of contrasting rice cultivars highlights the role of auxin and ABA responsive genes in heat stress response. Genomics 2021, 113, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Han, S.C.; Sun, X.M.; Khan, N.U.; Zhong, Q.; Zhang, Z.Y.; Zhang, H.L.; Ming, F.; Li, Z.C.; Li, J.J. Variations in OsSPL10 confer drought tolerance by directly regulating OsNAC2 expression and ROS production in rice. J. Integr. Plant. Biol. 2022, 65, 918–933. [Google Scholar] [CrossRef]
- Yu, T.F.; Liu, Y.; Fu, J.D.; Ma, J.; Fang, Z.W.; Chen, J.; Zheng, L.; Lu, Z.W.; Zhou, Y.B.; Chen, M.; et al. The NF-Y-PYR module integrates the abscisic acid signal pathway to regulate plant stress tolerance. Plant Biotechnol. J. 2021, 19, 2589–2605. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.J.; Furtado, A.; Rangan, P. Wheat seed transcriptome reveals genes controlling key traits for human preference and crop adaptation. Curr. Opin. Plant Biol. 2018, 45, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.D.; Li, S.M.; Wang, Z.X.; Cheng, X.X.; Li, F.F.; Mei, F.M.; Chen, N.; Kang, Z.S. Regulatory changes in TaSNAC8-6A are associated with drought tolerance in wheat seedlings. Plant Biotechnol J. 2020, 18, 1078–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinowska, M.; Donnison, I.; Robson, P. Morphological and Physiological Traits that Explain Yield Response to Drought Stress in Miscanthus. Agronomy 2020, 10, 1194. [Google Scholar] [CrossRef]
- Soriano, J.M.; Alvaro, F. Discovering consensus genomic regions in wheat for root-related traits by QTL meta-analysis. Sci. Rep. 2019, 9, 10537. [Google Scholar] [CrossRef] [Green Version]
- Ansari, W.A.; Atri, N.; Pandey, M.; Singh, A.K.; Singh, B.; Pandey, S. Influence of drought stress on morphological, physiologicaland biochemical attributes of plants: A review. Biosci. Biotech-Nology Res. Asia 2019, 16, 697–709. [Google Scholar] [CrossRef]
- Mubarik, M.S.; Khan, S.H.; Sajjad, M.; Raza, A.; Hafeez, M.B.; Yasmeen, T.; Rizwan, M.; Ali, S.; Arif, M.S. Amanipulative interplay between positive and negative regulators ofphytohormones: A way forward for improving drought tolerance inplants. Physiol. Plant. 2021, 172, 1269–1290. [Google Scholar] [CrossRef]
- Mei, F.M.; Chen, B.; Du, L.Y.; Li, S.M.; Zhu, D.H.; Chen, N.; Zhang, Y.F.; Li, F.F.; Wang, Z.X.; Cheng, X.X.; et al. A gain-of-function allele of a DREB transcription factor gene ameliorates drought tolerance in wheat. Plant Cell 2022, 34, 4472–4494. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.D.; Li, S.M.; Chen, B.; Jian, C.; Mei, F.M.; Zhang, Y.F.; Li, F.F.; Chen, N.; Li, T.; Du, L.Y.; et al. Variation in cis-regulation of a NAC transcription factor contributes to drought tolerance in wheat. Mol. Plant 2022, 15, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Song, T.Q.; Wang, Y.K.; Zhang, M.F.; Li, N.; Yu, M.; Zhang, S.X.; Zhou, H.W.; Guo, S.H.; Bu, Y.N.; et al. The wheat WRKY transcription factor TaWRKY1-2D confers drought resistance in transgenic Arabidopsis and wheat (Triticum aestivum L.). Int. J. Biol. Macromol. 2023, 31, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Iquebal, M.A.; Sharma, P.; Jasrotia, R.S.; Jaiswal, S.; Kaur, A.; Saroha, M.; Angadi, U.B.; Sheoran, S.; Singh, R.; Singh, G.P.; et al. RNAseq analysis reveals drought-responsive molecular pathways with candidate genes and putative molecular markers in root tissue of wheat. Sci. Rep. 2019, 9, 13917. [Google Scholar] [CrossRef] [Green Version]
- Du, L.Y.; Huang, X.L.; Ding, L.; Wang, Z.X.; Tang, D.L.; Chen, B.; Ao, L.J.A.; Liu, Y.L.; Kang, Z.S.; Mao, H.D. TaERF87 and TaAKS1 synergistically regulate TaP5CS1/TaP5CR1-mediated proline biosynthesis to enhance drought tolerance in wheat. New Phytol. 2023, 237, 232–250. [Google Scholar] [CrossRef]
- Liu, R.X.; Wu, F.K.; Yi, X.; Lin, Y.; Wang, Z.Q.; Liu, S.H.; Deng, M.; Ma, J.; Wei, Y.M.; Zheng, Y.L.; et al. Quantitative trait loci analysis for root traits in synthetic hexaploid wheat under drought stress conditions. J. Integr. Agric. 2020, 19, 1947–1960. [Google Scholar] [CrossRef]
- Su, P.S.; Sui, C.; Li, J.Y.; Wan, K.; Sun, H.N.; Wang, S.H.; Liu, X.Q.; Guo, S.J. The Aux/IAA protein TaIAA15-1A confers drought tolerance in Brachypodium by regulating abscisic acid signal pathway. Plant Cell Rep. 2022, 12, 25. [Google Scholar] [CrossRef]
- Jing, F.L.; Miao, Y.P.; Zhang, P.P.; Chen, T.; Liu, Y.; Ma, J.F.; Li, M.F.; Yang, D.L. Characterization of TaSPP-5A gene associated with sucrose content in wheat (Triticum aestivum L.). BMC Plant Biol. 2022, 22, 58. [Google Scholar] [CrossRef]
- Tiedge, K.; Li, X.; Merrill, A.T.; Davisson, D.; Chen, Y.; Yu, P.; Tantillo, D.J.; Last, R.L.; Zerbe, P. Comparative transcriptomics and metabolomics reveal specialized metabolite drought stress responses in switchgrass (Panicum virgatum). New Phytol. 2022, 236, 1393–1408. [Google Scholar] [CrossRef]
- Kirungu, J.N.; Magwanga, R.O.; Pu, L.; Cai, X.; Xu, Y.; Hou, Y.; Zhou, Y.; Cai, Y.; Hao, F.; Zhou, Z.; et al. Knockdown of Gh_A05G1554 (GhDHN_03) and Gh_D05G1729 (GhDHN_04) Dehydrin genes, Reveals their potential role in enhancing osmotic and salt tolerance in cotton. Genomics 2020, 112, 1902–1915. [Google Scholar] [CrossRef]
- Li, X.; Liu, Q.; Feng, H.; Deng, J.; Zhang, R.; Wen, J.; Dong, J.; Wang, T. Dehydrin MtCAS31 promotes autophagic degradation under drought stress. Autophagy 2020, 16, 862–877. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, Y.; Zhang, L. Identification of upstream transcription factors and an interacting PP2C protein of dehydrin WZY2 gene in wheat. Plant Signal Behav. 2019, 14, 1678370. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.W.; Wang, M.; Wu, J.H.; Guo, W.L.; Chen, Y.M.; Li, G.W.; Wang, Y.P.; Shi, W.M.; Xia, G.M.; Fu, D.L.; et al. WheatOmics: A platform combining multiple omics data to accelerate functional genomics studies in wheat. Mol. Plant 2021, 14, 1965–1968. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.C.; Dong, C.H.; Chen, Z.X.; Gui, L.X.; Chen, C.; Li, D.P.; Xie, Z.C.; Zhang, Q.; Zhang, X.Y.; Xia, C.; et al. WheatGmap: A comprehensive platform for wheat gene mapping and genomic studies. Mol. Plant 2021, 14, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Blake, V.C.; Woodhouse, M.R.; Lazo, G.R.; Odell, S.G.; Wight, C.P.; Tinker, N.A.; Wang, Y.; Gu, Y.Q.; Birkett, C.L.; Jannink, J.L.; et al. GrainGenes: Centralized small grain resources and digital platform for geneticists and breeders. Database 2019, 2019, baz065. [Google Scholar]
- Ran, X.J.; Tang, T.F.; Wang, M.Y.; Ye, L.H.; Zhuang, Y.L.; Zhao, F.; Zhang, Y.J. CSCS: A chromatin state interface for Chinese Spring bread wheat. aBIOTECH 2021, 2, 357–364. [Google Scholar] [CrossRef]
- Duncan, O.; Trosch, J.; Fenske, R.; Taylor, N.L.; Millar, A.H. Resource: Mapping the Triticum aestivum proteome. Plant J. 2017, 89, 601–616. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.X.; Wang, Z.H.; Li, X.T.; Ni, Z.F.; Hu, Z.R.; Xin, M.M.; Peng, H.R.; Yao, Y.Y.; Sun, Q.X.; Guo, W.L. SnpHub: An easy-to-set-up web server framework for exploring large-scale genomic variation data in the post-genomic era with applications in wheat. GigaScience 2020, 9, 1–8. [Google Scholar] [CrossRef]
- Pearce, S.; Vazquez-Gross, H.; Herin, S.Y.; Hane, D.; Wang, Y.; Gu, Y.Q.; Dubcovsky, J. WheatExp: An RNA-seq expression database for polyploid wheat. BMC Plant Biol. 2015, 15, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.M.; Song, W.J.; Xie, X.M.; Wang, Z.H.; Guan, P.F.; Peng, H.R.; Jiao, Y.N.; Ni, Z.F.; Sun, Q.X.; Guo, W.L. A collinearity-incorporating homology inference strategy for connecting emerging assemblies in the Triticeae Tribe as a pilot practice in the plant pangenomic era. Mol. Plant. 2020, 13, 1694–1708. [Google Scholar] [CrossRef]
- Wang, J.R.; Fu, W.W.; Wang, R.; Hu, D.X.; Cheng, H.; Zhao, J.; Jiang, Y.; Kang, Z.S. WGVD: An integrated web-database for wheat genome variation and selective signatures. Database J. Biol. Databases Curation 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, C.C.; Chain, P.S.G. Rapid evaluation and quality control of next generation sequencing data with FaQCs. BMC Bioinform. 2014, 15, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protocol 2016, 11, 1650. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Hao, C.Y.; Jiao, C.Z.; Hou, J.; Li, T.; Liu, H.X.; Wang, Y.Q.; Zheng, J.; Liu, H.; Bi, Z.H.; Xu, F.F.; et al. Resequencing of 145 landmark cultivars reveals asymmetric sub-genome selection and strong founder genotype effects on wheat breeding in China. Mol. Plant 2020, 13, 1733–1751. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomicfeatures. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with highthroughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Method Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Candidate Gene ID | Description | Log2(FC) | p Value |
|---|---|---|---|
| TraesCS6A03G0899800 | Dehydrin, BP: response to water, abiotic stimulus | 7.60 | 2.92 × 10−45 |
| TraesCS6B03G1084600 | Dehydrin, BP: response to water, abiotic stimulus | 3.40 | 1.47 × 10−41 |
| TraesCS6A03G0900700 | Dehydrin, BP: response to water, acid chemical | 2.77 | 2.92 × 10−45 |
| TraesCS3B03G1055400 | Dehydrin, BP: response to water, abscisic acid | 4.12 | 2.92 × 10−45 |
| TraesCS6A03G0900100 | Dehydrin, BP: response to water, abscisic acid | 3.22 | 1.38 × 10−27 |
| TraesCS6D03G0772300 | Dehydrin, BP: response to water, inorganic substance | 2.24 | 5.15 × 10−17 |
| TraesCS7D03G1292300 | Dehydrin, BP: response to water, oxygen-containing compound | 9.46 | 8.39 × 10−15 |
| TraesCS6B03G1082300 | Dehydrin, BP: response to water, inorganic substance | 3.13 | 1.23 × 10−12 |
| TraesCS6B03G0794800 | Dehydrin, BP: response to water | 0.99 | 1.80 × 10−37 |
| TraesCS3B03G0736100 | Dehydrin, BP: response to water, abiotic stimulus | 3.92 | 2.19 × 10−36 |
| TraesCS4A03G0658300 | Dehydrin, BP: response to water | 3.76 | 4.84 × 10−8 |
| TraesCS7A03G1365300 | Dehydrin, BP: response to water, abiotic stimulus | 4.81 | 1.47 × 10−41 |
| TraesCS5A03G1006700 | Dehydrin, BP: response to water, oxygen-containing compound | 4.47 | 3.94 × 10−7 |
| TraesCS3A03G0653000 | Dehydrin, BP: response to water, abscisic acid | 3.94 | 3.96 × 10−7 |
| TraesCS6B03G1084000 | Dehydrin, BP: response to water, inorganic substance | 4.00 | 1.8 × 10−37 |
| TraesCS4B03G0139700 | Dehydrin, BP: response to water, abiotic stimulus | 3.12 | 2.19 × 10−36 |
| Mutation Name | Most Severe Consequence | Alleles | Location of IWGSC RefSeq v2.0 | IWGSC RefSeq v2.1 (IWGSC RefSeq v2.0) |
|---|---|---|---|---|
| Kronos3216.chr6A.581983177 | splice region variant | G/A | Chromosome 6A:581983177 | TraesCS6A03G0899800 (TraesCS6A02G350100) |
| Kronos3935.chr6A.582265082 | splice region variant | C/T | Chromosome 6A:582265082 | TraesCS6A03G0900700 (TraesCS6A02G350500) |
| Kronos3538.chr3B.667112943 | splice region variant | G/A | Chromosome 3B:667112943 | TraesCS3B03G1055400 (TraesCS3B02G428200) |
| Kronos3557.chr6B.658578115 | splice region variant | C/T | Chromosome 6B:658578115 | TraesCS6B03G1084600 (TraesCS6B02G383800) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xi, W.; Hao, C.; Li, T.; Wang, H.; Zhang, X. Transcriptome Analysis of Roots from Wheat (Triticum aestivum L.) Varieties in Response to Drought Stress. Int. J. Mol. Sci. 2023, 24, 7245. https://doi.org/10.3390/ijms24087245
Xi W, Hao C, Li T, Wang H, Zhang X. Transcriptome Analysis of Roots from Wheat (Triticum aestivum L.) Varieties in Response to Drought Stress. International Journal of Molecular Sciences. 2023; 24(8):7245. https://doi.org/10.3390/ijms24087245
Chicago/Turabian StyleXi, Wei, Chenyang Hao, Tian Li, Huajun Wang, and Xueyong Zhang. 2023. "Transcriptome Analysis of Roots from Wheat (Triticum aestivum L.) Varieties in Response to Drought Stress" International Journal of Molecular Sciences 24, no. 8: 7245. https://doi.org/10.3390/ijms24087245
APA StyleXi, W., Hao, C., Li, T., Wang, H., & Zhang, X. (2023). Transcriptome Analysis of Roots from Wheat (Triticum aestivum L.) Varieties in Response to Drought Stress. International Journal of Molecular Sciences, 24(8), 7245. https://doi.org/10.3390/ijms24087245