


Hypothalamic Reactivity and Connectivity following Intravenous Glucose Administration
 , ,
, ,
Abstract
1. Introduction
2. Results
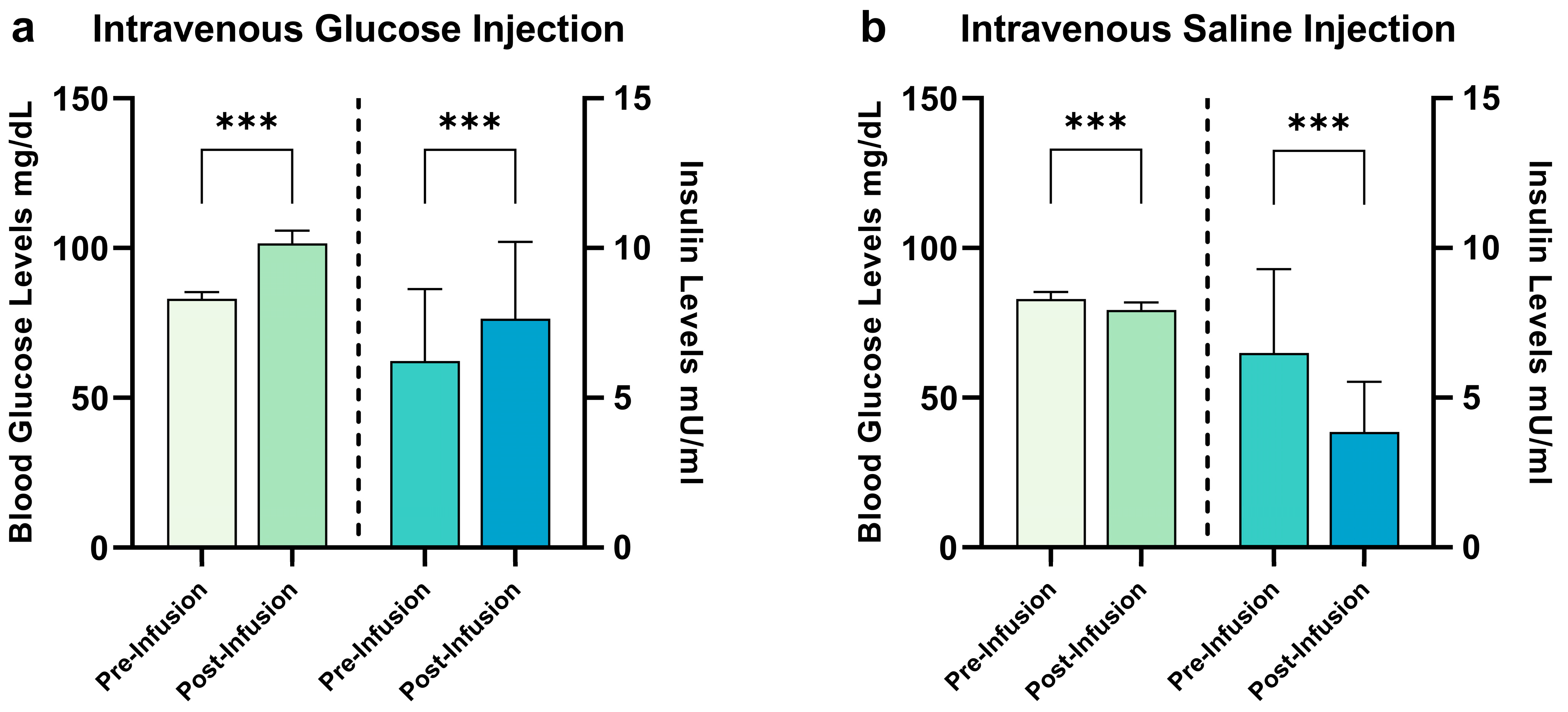
2.1. Hormonal Satiety Parameters
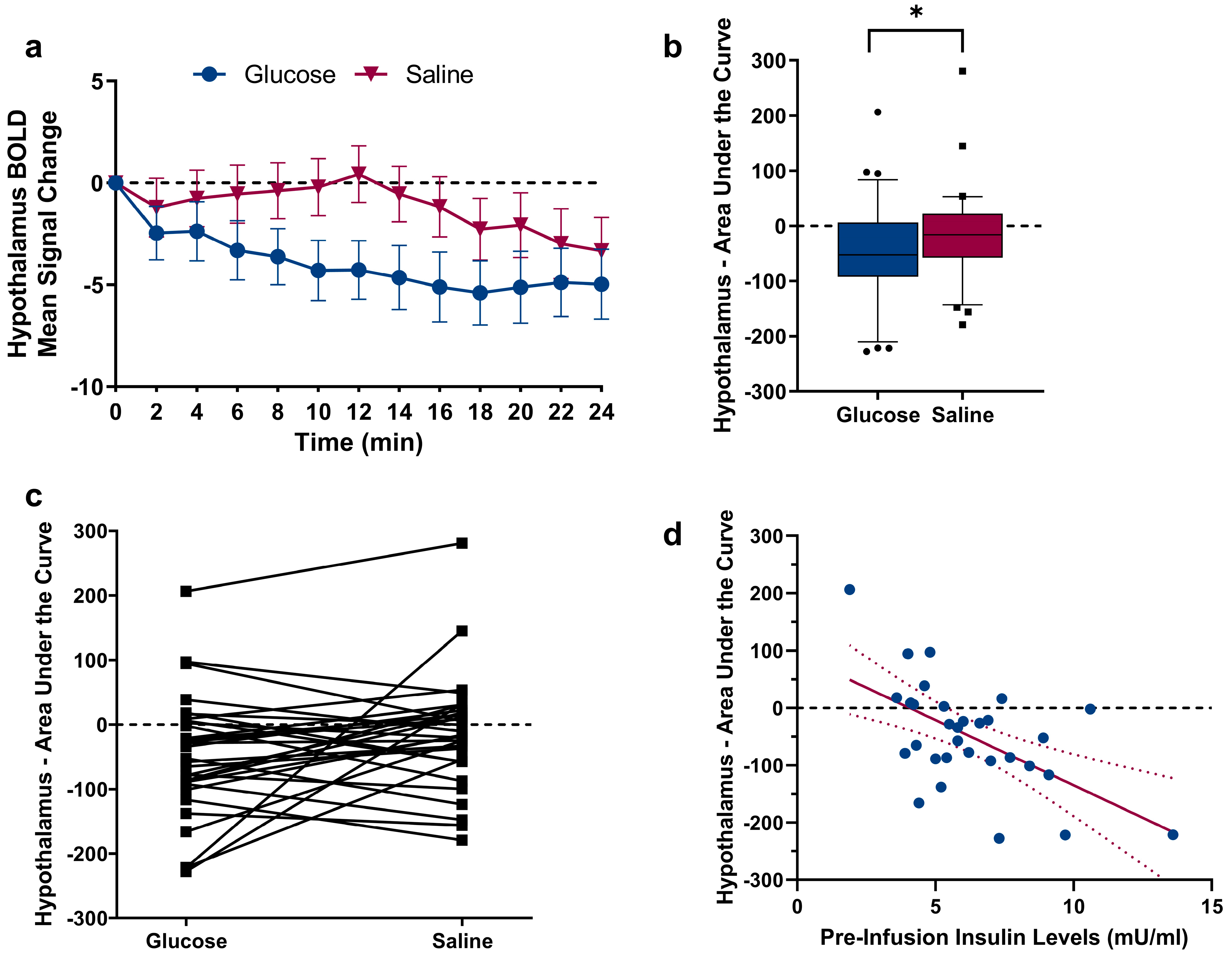
2.2. Changes in Hypothalamic Activation Induced by Glucose and Saline
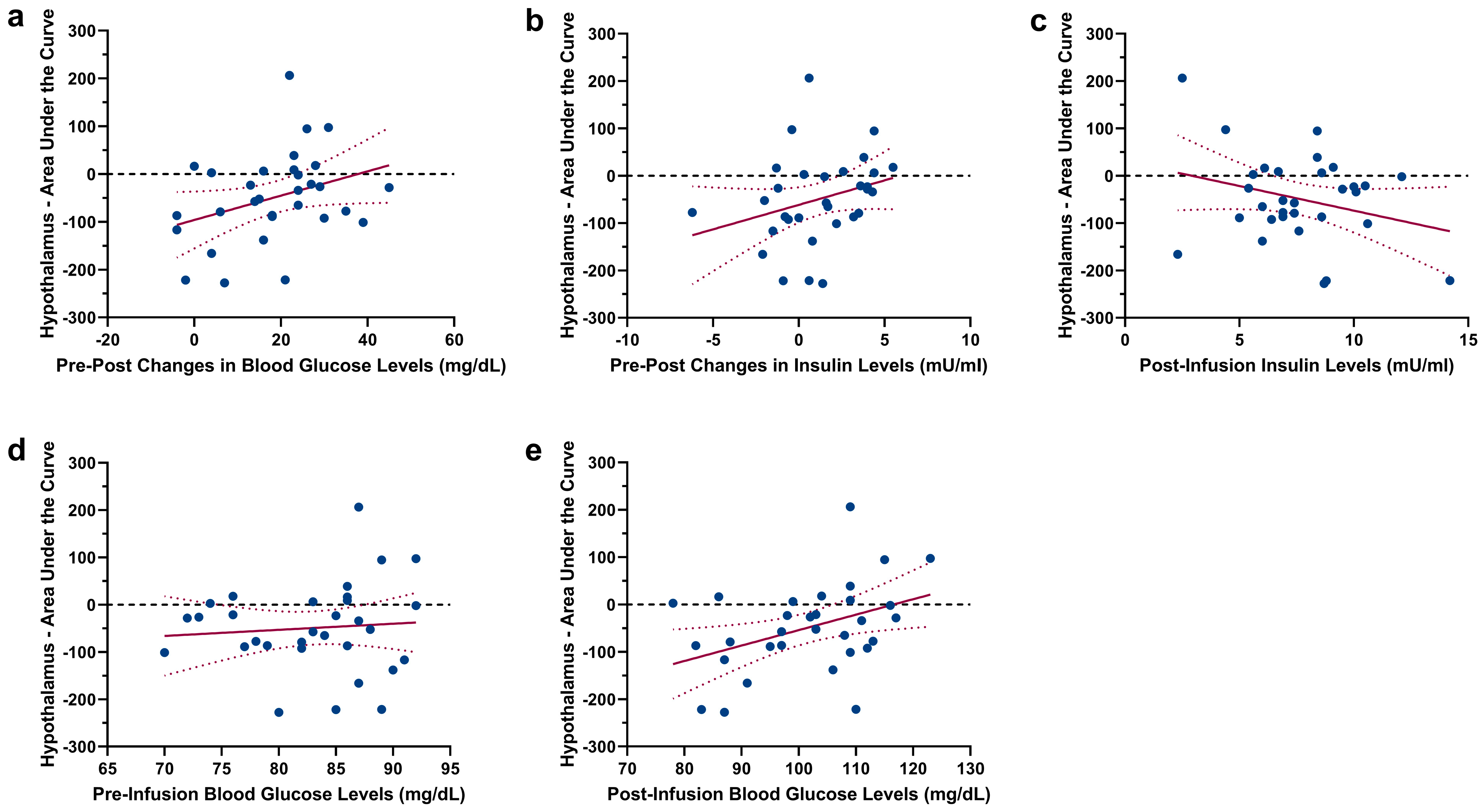
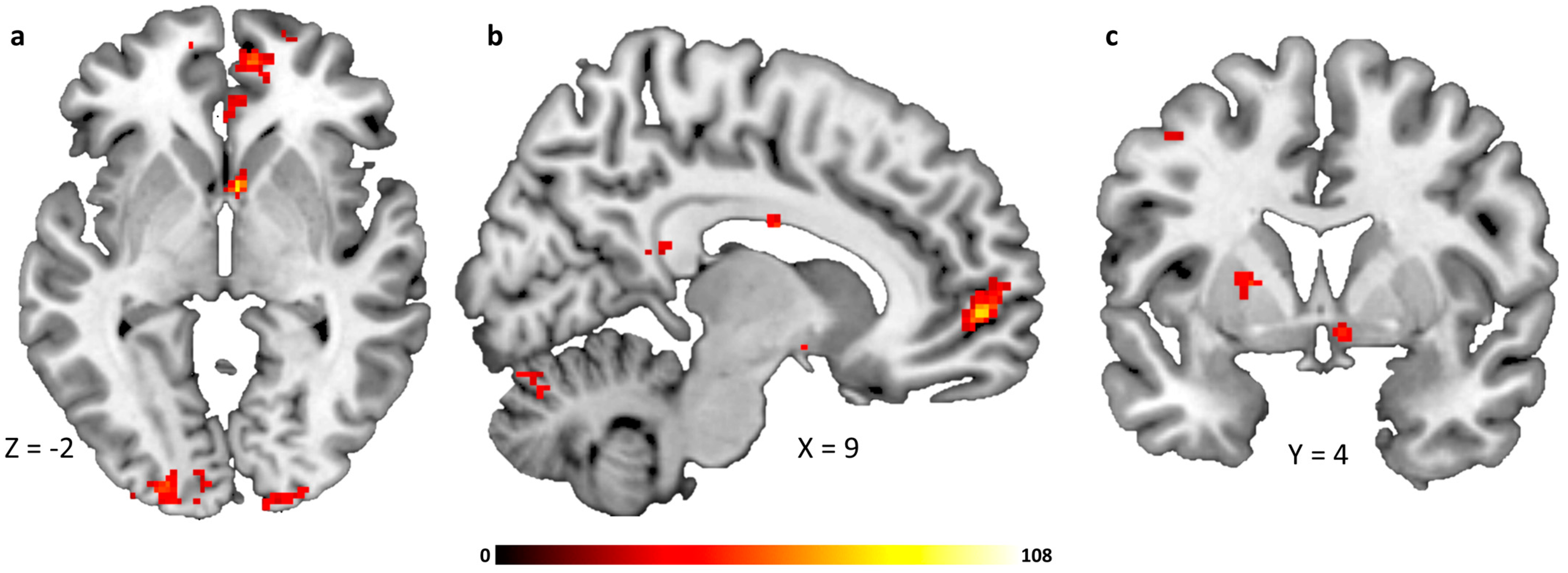
2.3. Infusion-Type Dependent Functional Connectivity from the Hypothalamus to Reward-Related Brain Regions
3. Discussion
3.1. Limitations
3.2. Conclusions
4. Materials and Methods
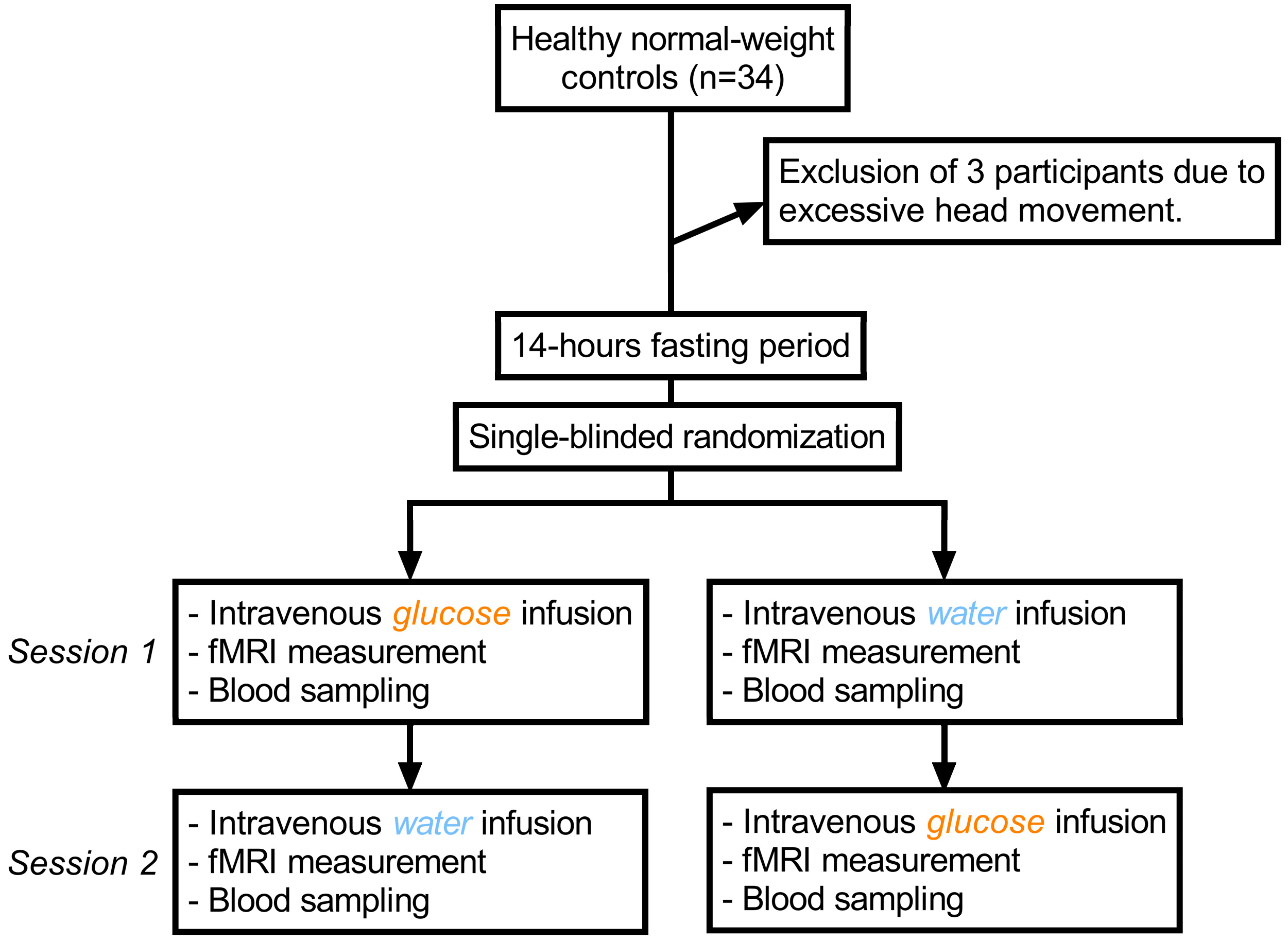
4.1. Study Design and Test Group
4.2. Procedure
4.3. fMRI Acquisition
4.4. fMRI Analysis
4.5. Seed-Based Connectivity Analysis
4.6. Biochemical Analysis of Glucose and Insulin
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berthoud, H.-R. Metabolic and hedonic drives in the neural control of appetite: Who is the boss? Curr. Opin. Neurobiol. 2011, 21, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Kringelbach, M.L.; Stein, A. Cortical mechanisms of human eating. Front. Eat. Weight. Regul. 2010, 63, 164–175. [Google Scholar] [CrossRef]
- Kelley, A.E.; Baldo, B.A.; Pratt, W.E.; Will, M.J. Corticostriatal-hypothalamic circuitry and food motivation: Integration of energy, action and reward. Physiol. Behav. 2005, 86, 773–795. [Google Scholar] [CrossRef]
- Roh, E.; Song, D.K.; Kim, M.S. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp. Mol. Med. 2016, 48, e216. [Google Scholar] [CrossRef] [PubMed]
- DiLeone, R.J.; Georgescu, D.; Nestler, E.J. Lateral hypothalamic neuropeptides in reward and drug addiction. Life Sci. 2003, 73, 759–768. [Google Scholar] [CrossRef] [PubMed]
- DiLeone, R.J.; Taylor, J.R.; Picciotto, M.R. The drive to eat: Comparisons and distinctions between mechanisms of food reward and drug addiction. Nat. Neurosci. 2012, 15, 1330–1335. [Google Scholar] [CrossRef]
- Seitz, J.; Belheouane, M.; Schulz, N.; Dempfle, A.; Baines, J.F.; Herpertz-Dahlmann, B. The Impact of Starvation on the Microbiome and Gut-Brain Interaction in Anorexia Nervosa. Front. Endocrinol. 2019, 10, 41. [Google Scholar] [CrossRef]
- Bliss, E.S.; Whiteside, E. The Gut-Brain Axis, the Human Gut Microbiota and Their Integration in the Development of Obesity. Front. Physiol. 2018, 9, 900. [Google Scholar] [CrossRef]
- Smeets, P.A.; de Graaf, C.; Stafleu, A.; van Osch, M.J.P.; van der Grond, J. Functional MRI of human hypothalamic responses following glucose ingestion. Neuroimage 2005, 24, 363–368. [Google Scholar] [CrossRef]
- Page, K.A.; Chan, O.; Arora, J.; Belfort-DeAguiar, R.; Dzuira, J.; Roehmholdt, B.; Cline, G.W.; Naik, S.; Sinha, R.; Constable, R.T.; et al. Effects of fructose vs. glucose on regional cerebral blood flow in brain regions involved with appetite and reward pathways. JAMA 2013, 309, 63–70. [Google Scholar] [CrossRef]
- Simon, J.J.; Stopyra, M.A.; Monning, E.; Sailer, S.; Lavandier, N.; Kihm, L.P.; Bendszus, M.; Preissl, H.; Herzog, W.; Friederich, H.C. Neuroimaging of hypothalamic mechanisms related to glucose metabolism in anorexia nervosa and obesity. J. Clin. Investig. 2020, 130, 4094–4103. [Google Scholar] [CrossRef]
- Kaye, W.H.; Wierenga, C.E.; Bischoff-Grethe, A.; Berner, L.A.; Ely, A.V.; Bailer, U.F.; Paulus, M.P.; Fudge, J.L. Neural Insensitivity to the Effects of Hunger in Women Remitted From Anorexia Nervosa. Am. J. Psychiatry 2020, 177, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Soussignan, R.; Carrier, E.; Royet, J.P. Dysfunction of the Mesolimbic Circuit to Food Odors in Women With Anorexia and Bulimia Nervosa: A fMRI Study. Front. Hum. Neurosci. 2019, 13, 117. [Google Scholar] [CrossRef]
- Simon, J.J.; Becker, A.; Sinno, M.H.; Skunde, M.; Bendszus, M.; Preissl, H.; Enck, P.; Herzog, W.; Friederich, H.C. Neural Food Reward Processing in Successful and Unsuccessful Weight Maintenance. Obesity 2018, 26, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Haltia, L.T.; Savontaus, E.; Vahlberg, T.; Rinne, J.O.; Kaasinen, V. Acute hormonal changes following intravenous glucose challenge in lean and obese human subjects. Scand. J. Clin. Lab. Investig. 2010, 70, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Belfort-DeAguiar, R.; Seo, D.; Lacadie, C.; Naik, S.; Schmidt, C.; Lam, W.; Hwang, J.; Constable, T.; Sinha, R.; Sherwin, R.S. Humans with obesity have disordered brain responses to food images during physiological hyperglycemia. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E522–E529. [Google Scholar] [CrossRef]
- Smeets, P.A.; Vidarsdottir, S.; de Graaf, C.; Stafleu, A.; van Osch, M.J.; Viergever, M.A.; Pijl, H.; van der Grond, J. Oral glucose intake inhibits hypothalamic neuronal activity more effectively than glucose infusion. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E754–E758. [Google Scholar] [CrossRef]
- Purnell, J.Q.; Klopfenstein, B.A.; Stevens, A.A.; Havel, P.J.; Adams, S.H.; Dunn, T.N.; Krisky, C.; Rooney, W.D. Brain functional magnetic resonance imaging response to glucose and fructose infusions in humans. Diabetes Obes. Metab. 2011, 13, 229–234. [Google Scholar] [CrossRef]
- Schopf, V.; Fischmeister, F.P.; Windischberger, C.; Gerstl, F.; Wolzt, M.; Karlsson, K.A.E.; Moser, E. Effects of individual glucose levels on the neuronal correlates of emotions. Front. Hum. Neurosci. 2013, 7, 212. [Google Scholar] [CrossRef]
- Page, K.A.; Arora, J.; Qiu, M.; Relwani, R.; Constable, R.T.; Sherwin, R.S. Small decrements in systemic glucose provoke increases in hypothalamic blood flow prior to the release of counterregulatory hormones. Diabetes 2009, 58, 448–452. [Google Scholar] [CrossRef]
- Luo, S.; Monterosso, J.R.; Sarpelleh, K.; Page, K.A. Differential effects of fructose versus glucose on brain and appetitive responses to food cues and decisions for food rewards. Proc. Natl. Acad. Sci. USA 2015, 112, 6509–6514. [Google Scholar] [CrossRef] [PubMed]
- Vidarsdottir, S.; Smeets, P.A.; Eichelsheim, D.L.; van Osch, M.J.; Viergever, M.A.; Romijn, J.A.; van der Grond, J.; Pijl, H. Glucose ingestion fails to inhibit hypothalamic neuronal activity in patients with type 2 diabetes. Diabetes 2007, 56, 2547–2550. [Google Scholar] [CrossRef]
- Little, T.J.; McKie, S.; Jones, R.B.; D’Amato, M.; Smith, C.; Kiss, O.; Thompson, D.G.; McLaughlin, J.T. Mapping glucose-mediated gut-to-brain signalling pathways in humans. Neuroimage 2014, 96, 1–11. [Google Scholar] [CrossRef]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Yoon, N.A.; Diano, S. Hypothalamic glucose-sensing mechanisms. Diabetologia 2021, 64, 985–993. [Google Scholar] [CrossRef]
- Timper, K.; Bruning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. Dis. Model. Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef]
- Li, J.; An, R.; Zhang, Y.; Li, X.; Wang, S. Correlations of macronutrient-induced functional magnetic resonance imaging signal changes in human brain and gut hormone responses. Am. J. Clin. Nutr. 2012, 96, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Teeuwisse, W.M.; Widya, R.L.; Paulides, M.; Lamb, H.J.; Smit, J.W.; de Roos, A.; van Buchem, M.A.; Pijl, H.; van der Grond, J. Short-term caloric restriction normalizes hypothalamic neuronal responsiveness to glucose ingestion in patients with type 2 diabetes. Diabetes 2012, 61, 3255–3259. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, J.-H.; Liu, H.-L.; Fox, P.T. The temporal response of the brain after eating revealed by functional MRI. Nature 2000, 405, 1058–1062. [Google Scholar] [CrossRef]
- Althubeati, S.; Avery, A.; Tench, C.R.; Lobo, D.N.; Salter, A.; Eldeghaidy, S. Mapping brain activity of gut-brain signaling to appetite and satiety in healthy adults: A systematic review and functional neuroimaging meta-analysis. Neurosci. Biobehav. Rev. 2022, 136, 104603. [Google Scholar] [CrossRef]
- Plum, L.; Belgardt, B.F.; Bruning, J.C. Central insulin action in energy and glucose homeostasis. J. Clin. Investig. 2006, 116, 1761–1766. [Google Scholar] [CrossRef]
- Figlewicz, D.P.; Evans, S.B.; Murphy, J.; Hoen, M.; Baskin, D.G. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Res. 2003, 964, 107–115. [Google Scholar] [CrossRef]
- Baskin, D.G.; Figlewicz Lattemann, D.; Seeley, R.J.; Woods, S.C.; Porte, D., Jr.; Schwartz, M.W. Insulin and leptin: Dual adiposity signals to the brain for the regulation of food intake and body weight. Brain Res. 1999, 848, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Mebel, D.M.; Wong, J.C.; Dong, Y.J.; Borgland, S.L. Insulin in the ventral tegmental area reduces hedonic feeding and suppresses dopamine concentration via increased reuptake. Eur. J. Neurosci. 2012, 36, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Figlewicz, D.P.; Bennett, J.L.; Naleid, A.M.; Davis, C.; Grimm, J.W. Intraventricular insulin and leptin decrease sucrose self-administration in rats. Physiol. Behav. 2006, 89, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, N.B.; Krebs, L.; Kobiella, A.; Grimm, O.; Vollstädt-Klein, S.; Wolfensteller, U.; Kling, R.; Bidlingmaier, M.; Zimmermann, U.S.; Smolka, M.N. (Still) longing for food: Insulin reactivity modulates response to food pictures. Hum. Brain Mapp. 2013, 34, 2367–2380. [Google Scholar] [CrossRef]
- Wallner-Liebmann, S.; Koschutnig, K.; Reishofer, G.; Sorantin, E.; Blaschitz, B.; Kruschitz, R.; Unterrainer, H.F.; Gasser, R.; Freytag, F.; Bauer-Denk, C.; et al. Insulin and Hippocampus Activation in Response to Images of High-Calorie Food in Normal Weight and Obese Adolescents. Obesity 2010, 18, 1552–1557. [Google Scholar] [CrossRef]
- Belgardt, B.F.; Bruning, J.C. CNS leptin and insulin action in the control of energy homeostasis. Ann. N. Y. Acad. Sci. 2010, 1212, 97–113. [Google Scholar] [CrossRef]
- Campos, A.; Port, J.D.; Acosta, A. Integrative Hedonic and Homeostatic Food Intake Regulation by the Central Nervous System: Insights from Neuroimaging. Brain Sci. 2022, 12, 431. [Google Scholar] [CrossRef]
- Hussain, S.S.; Bloom, S.R. The regulation of food intake by the gut-brain axis: Implications for obesity. Int. J. Obes. 2013, 37, 625–633. [Google Scholar] [CrossRef]
- Arnoni-Bauer, Y.; Bick, A.; Raz, N.; Imbar, T.; Amos, S.; Agmon, O.; Marko, L.; Levin, N.; Weiss, R. Is It Me or My Hormones? Neuroendocrine Activation Profiles to Visual Food Stimuli Across the Menstrual Cycle. J. Clin. Endocrinol. Metab. 2017, 102, 3406–3414. [Google Scholar] [CrossRef] [PubMed]
- Wittchen, H.; Zaudig, M.; Fydrich, T. Structured Clinical Interview for DSM-IV—German Version; Hoegrefe: Göttingen, Germany, 1997. [Google Scholar]
- Lehrl, S. Mehrfachwahl-Wortschatz-Intelligenztest: MWT-B, 5th ed.; Spitta: Balingen, Germany, 2005. [Google Scholar]
- Hilbert, A.; Tuschen-Caffier, B.; Ohms, M. Eating disorder examination: Deutschsprachige Version des strukturierten Essstörungsinterviews. Diagnostica 2004, 50, 98–106. [Google Scholar] [CrossRef]
- Hautzinger, M.; Keller, F.; Kühner, C. BDI-II. Beck-Depressions-Inventar Revision—Manual; Harcourt Test Services: Frankfurt, Germany, 2006. [Google Scholar]
- Mond, J.M.; Hay, P.J.; Rodgers, B.; Owen, C.; Beumont, P. Validity of the Eating Disorder Examination Questionnaire (EDE-Q) in screening for eating disorders in community samples. Behav. Res. Ther. 2004, 42, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Meule, A. Reconsidering the use of cut-off scores for the Eating Disorder Examination-Questionnaire. Eat. Disord. 2021, 29, 480–484. [Google Scholar] [CrossRef]
- Beck, A.; Steer, R.; Brown, G. (Eds.) Manual for the Beck Depression Inventory—Second Edition (BDI-II); Psychological Corporation: San Antonio, TX, USA, 1996. [Google Scholar]
- Satzger, W.; Fessmann, H.; Engel, R. The equivalence of three German vocabulary tests and the German Version of the Wechsler Adult Intelligence Scale-Revised (HAWIE-R). Z. Fur Differ. Diagn. Psychol. 2002, 23, 159–170. [Google Scholar]
- DeFronzo, R.A.; Tobin, J.D.; Andres, R. Glucose clamp technique: A method for quantifying insulin secretion and resistance. Am. J. Physiol. 1979, 237, E214–E223. [Google Scholar] [CrossRef] [PubMed]
- Mosteller, R.D. Simplified calculation of body-surface area. N. Engl. J. Med. 1987, 317, 1098. [Google Scholar] [CrossRef]
- Robinson, S.; Windischberger, C.; Rauscher, A.; Moser, E. Optimized 3 T EPI of the amygdalae. Neuroimage 2004, 22, 203–210. [Google Scholar] [CrossRef]
- Karlsson, K.A.; Windischberger, C.; Gerstl, F.; Mayr, W.; Siegel, J.M.; Moser, E. Modulation of hypothalamus and amygdalar activation levels with stimulus valence. Neuroimage 2010, 51, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.D.; Pripfl, J.; Bauer, H.; Moser, E. The impact of EPI voxel size on SNR and BOLD sensitivity in the anterior medio-temporal lobe: A comparative group study of deactivation of the Default Mode. Mater. Phys. Biol. Med. 2008, 21, 279–290. [Google Scholar] [CrossRef]
- Kwong, K.K.; Belliveau, J.W.; Chesler, D.A.; Goldberg, I.E.; Weisskoff, R.M.; Poncelet, B.P.; Kennedy, D.N.; Hoppel, B.E.; Cohen, M.S.; Turner, R. Dynamic magnetic resonance imaging of human brain activity during primary sensory stimulation. Proc. Natl. Acad. Sci. USA 1992, 89, 5675–5679. [Google Scholar] [CrossRef]
- Cho, Z.H.; Son, Y.D.; Kang, C.K.; Han, J.Y.; Wong, E.K.; Bai, S.J. Pain dynamics observed by functional magnetic resonance imaging: Differential regression analysis technique. J. Magn. Reson. Imaging 2003, 18, 273–283. [Google Scholar] [CrossRef]
- Matsuda, M.; Liu, Y.; Mahankali, S.; Pu, Y.; Mahankali, A.; Wang, J.; DeFronzo, R.A.; Fox, P.T.; Gao, J.H. Altered hypothalamic function in response to glucose ingestion in obese humans. Diabetes 1999, 48, 1801–1806. [Google Scholar] [CrossRef]
- Whitfield-Gabrieli, S.; Nieto-Castanon, A. Conn: A functional connectivity toolbox for correlated and anticorrelated brain networks. Brain Connect. 2012, 2, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Whitfield-Gabrieli, S.; Nieto-Castanon, A.; Ghosh, S. Artifact Detection Tools (ART); Release Version: Cambridge, MA, USA, 2011; Volume 7, p. 11. [Google Scholar]
- Power, J.D.; Mitra, A.; Laumann, T.O.; Snyder, A.Z.; Schlaggar, B.L.; Petersen, S.E. Methods to detect, characterize, and remove motion artifact in resting state fMRI. Neuroimage 2014, 84, 320–341. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Castanon, A. Handbook of Functional Connectivity Magnetic Resonance Imaging Methods in CONN; Hilbert Press: Boston, MA, USA, 2020. [Google Scholar]
- Hallquist, M.N.; Hwang, K.; Luna, B. The nuisance of nuisance regression: Spectral misspecification in a common approach to resting-state fMRI preprocessing reintroduces noise and obscures functional connectivity. Neuroimage 2013, 82, 208–225. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.J.; Castañón, A.N.; Öngür, D.; Whitfield-Gabrieli, S. Anticorrelations in resting state networks without global signal regression. Neuroimage 2012, 59, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Behzadi, Y.; Restom, K.; Liau, J.; Liu, T.T. A component based noise correction method (CompCor) for BOLD and perfusion based fMRI. Neuroimage 2007, 37, 90–101. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean ± SD (MIN–MAX) | |
|---|---|
| Female/male | 17/14 (N = 31) |
| Age | 24.1 ± 4.7 |
| BMI (kg/m2) | 21.9 ± 1.8 |
| EDE-Q | 0.3 ± 0.4 (0–1.9) |
| BDI-II | 2.5 ± 2.6 (0–11) |
| MWT-B | 30.1 ± 3.2 (24–36) |
| Region | Hemisphere | x | y | z | k | F-Value | p-Value |
|---|---|---|---|---|---|---|---|
| Anterior Cerebellum | R | 22 | −32 | −24 | 39 | 108 | <0.001 |
| Parahippocampal Gyrus | R | 14 | 0 | −16 | 79 | 83 | Id. |
| Caudate Head | R | 4 | 8 | −2 | Id. | 75 | Id. |
| Medial Frontal/Orbitofrontal Cortex | R | 8 | 54 | 0 | 165 | 78 | Id. |
| Middle Occipital Gyrus | R | 34 | −94 | −6 | 220 | 76 | Id. |
| Posterior Cerebellum | R | 6 | −76 | −16 | 36 | 76 | Id. |
| Anterior Cingulate Cortex | R | 2 | 40 | −6 | 54 | 74 | Id. |
| Middle Occipital Gyrus | L | −26 | −100 | 2 | 218 | 72 | Id. |
| Hippocampus | R | 40 | −14 | −20 | 30 | 71 | Id. |
| Fusiform Gyrus | L | −36 | −74 | −18 | 58 | 67 | Id. |
| Posterior Cingulate Cortex | R | 4 | −50 | 14 | 77 | 66 | Id. |
| Precuneus | L | −8 | −64 | 42 | 23 | 56 | Id. |
| Superior Temporal Gyrus | L | −42 | −34 | 12 | 27 | 55 | Id. |
| Ansiform Lobule | R | 18 | −86 | −20 | 26 | 54 | Id. |
| Anterior Cerebellum | R | 24 | −46 | −28 | 28 | 52 | Id. |
| Vermis | R | 32 | −70 | −40 | 49 | 51 | Id. |
| Anterior Medial Prefrontal Cortex | L | −8 | 52 | 2 | 45 | 50 | Id. |
| Inferior Occipital Gyrus | R | 10 | −100 | −10 | 33 | 50 | Id. |
| Inferior Parietal Lobule | R | 40 | −42 | 52 | 30 | 50 | Id. |
| Putamen | L | −20 | 4 | 6 | 35 | 50 | Id. |
| Precentral Gyrus | L | −44 | −12 | 48 | 57 | 44 | Id. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, J.J.; Lang, P.M.; Rommerskirchen, L.; Bendszus, M.; Friederich, H.-C. Hypothalamic Reactivity and Connectivity following Intravenous Glucose Administration. Int. J. Mol. Sci. 2023, 24, 7370. https://doi.org/10.3390/ijms24087370
Simon JJ, Lang PM, Rommerskirchen L, Bendszus M, Friederich H-C. Hypothalamic Reactivity and Connectivity following Intravenous Glucose Administration. International Journal of Molecular Sciences. 2023; 24(8):7370. https://doi.org/10.3390/ijms24087370
Chicago/Turabian StyleSimon, Joe J., Pia M. Lang, Lena Rommerskirchen, Martin Bendszus, and Hans-Christoph Friederich. 2023. "Hypothalamic Reactivity and Connectivity following Intravenous Glucose Administration" International Journal of Molecular Sciences 24, no. 8: 7370. https://doi.org/10.3390/ijms24087370
APA StyleSimon, J. J., Lang, P. M., Rommerskirchen, L., Bendszus, M., & Friederich, H.-C. (2023). Hypothalamic Reactivity and Connectivity following Intravenous Glucose Administration. International Journal of Molecular Sciences, 24(8), 7370. https://doi.org/10.3390/ijms24087370