
Genetic Etiology of Nonsyndromic Hearing Loss in Hungarian Patients
 , , , , , and
, , , , , and
Abstract
:1. Introduction
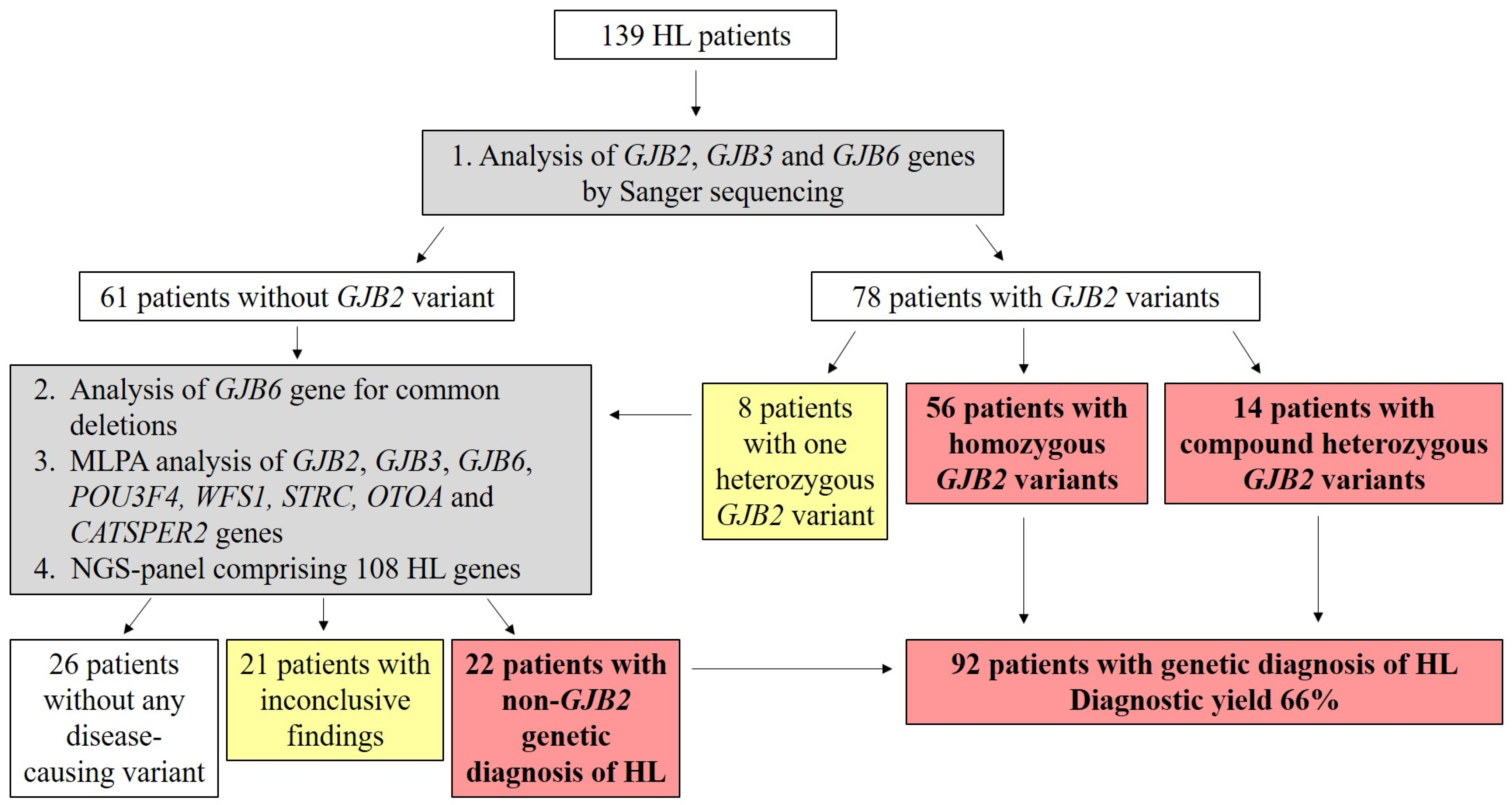
2. Results
Genetic Screening
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Clinical Characteristics of the Patients
4.3. Genetic Analysis
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Toriello, H.V.; Shelley, D. Smith Hereditary Hearing Loss and Its Syndromes; Oxford University Press: New York, NY, USA, 2004. [Google Scholar]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J. Hereditary Hearing Loss and Deafness Overview. 1999 February 14 [Updated 2023 April 6]. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1434/ (accessed on 22 January 2023).
- Teek, R.; Kruustük, K.; Žordania, R.; Joost, K.; Kahre, T.; Tõnisson, N.; Nelis, M.; Zilina, O.; Tranebjaerg, L.; Reimand, T.; et al. Hearing Impairment in Estonia: An Algorithm to Investigate Genetic Causes in Pediatric Patients. Adv. Med. Sci. 2013, 58, 419–428. [Google Scholar] [CrossRef] [PubMed]
- del Castillo, I.; Morín, M.; Domínguez-Ruiz, M.; Moreno-Pelayo, M.A. Genetic Etiology of Non-Syndromic Hearing Loss in Europe. Hum. Genet. 2022, 141, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Tóth, T.; Kupka, S.; Haack, B.; Riemann, K.; Braun, S.; Fazakas, F.; Zenner, H.-P.; Muszbek, L.; Blin, N.; Pfister, M.; et al. GJB2 Mutations in Patients with Non-Syndromic Hearing Loss from Northeastern Hungary: Mutations in Brief. Hum. Mutat. 2004, 23, 631–632. [Google Scholar] [CrossRef]
- Kecskeméti, N.; Szönyi, M.; Gáborján, A.; Küstel, M.; Milley, G.M.; Süveges, A.; Illés, A.; Kékesi, A.; Tamás, L.; Molnár, M.J.; et al. Analysis of GJB2 Mutations and the Clinical Manifestation in a Large Hungarian Cohort. Eur. Arch. Otorhinolaryngol. 2018, 275, 2441–2448. [Google Scholar] [CrossRef]
- Hoefsloot, L.H.; Roux, A.-F.; Bitner-Glindzicz, M. EMQN Best Practice Guidelines for Diagnostic Testing of Mutations Causing Non-Syndromic Hearing Impairment at the DFNB1 Locus. Eur. J. Hum. Genet. 2013, 21, 1325–1329. [Google Scholar] [CrossRef]
- Tóth, T.; Kupka, S.; Haack, B.; Fazakas, F.; Muszbek, L.; Blin, N.; Pfister, M.; Sziklai, I. Coincidence of Mutations in Different Connexin Genes in Hungarian Patients. Int. J. Mol. Med. 2007, 20, 315–321. [Google Scholar] [CrossRef]
- Houseman, M.J.; Ellis, L.A.; Pagnamenta, A.; Di, W.-L.; Rickard, S.; Osborn, A.H.; Dahl, H.-H.M.; Taylor, G.R.; Bitner-Glindzicz, M.; Reardon, W.; et al. Genetic Analysis of the Connexin-26 M34T Variant: Identification of Genotype M34T/M34T Segregating with Mild-Moderate Non-Syndromic Sensorineural Hearing Loss. J. Med. Genet. 2001, 38, 20–25. [Google Scholar] [CrossRef]
- Rabionet, R.; Gasparini, P.; Estivill, X. Molecular Genetics of Hearing Impairment Due to Mutations in Gap Junction Genes Encoding Beta Connexins. Hum. Mutat. 2000, 16, 190–202. [Google Scholar] [CrossRef]
- Engel-Yeger, B.; Zaaroura, S.; Zlotogora, J.; Shalev, S.; Hujeirat, Y.; Carrasquillo, M.; Barges, S.; Pratt, H. The E¡ects of a Connexin 26 Mutation–35delG–on Oto-Acoustic Emissions and Brainstem Evoked Potentials: Homozygotes and Carriers. Hear. Res. 2003, 175, 140–151. [Google Scholar] [CrossRef] [PubMed]
- del Castillo, F.J. A Novel Deletion Involving the Connexin-30 Gene, Del(GJB6-D13s1854), Found in Trans with Mutations in the GJB2 Gene (Connexin-26) in Subjects with DFNB1 Non-Syndromic Hearing Impairment. J. Med. Genet. 2005, 42, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive Genetic Testing in the Clinical Evaluation of 1119 Patients with Hearing Loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Sahin-Calapoglu, N.; Calapoglu, M.; Karaguzel, A. Non-Syndromic Recessive Hearing Loss Linkaged TMPRSS3 Gene in the Turkish Population. SDÜ Tıp. Fakültesi. Derg. 2005, 12, 31–35. [Google Scholar]
- Reardon, W. Enlarged Vestibular Aqueduct: A Radiological Marker of Pendred Syndrome, and Mutation of the PDS Gene. QJM 2000, 93, 99–104. [Google Scholar] [CrossRef]
- Pan, B.; Géléoc, G.S.; Asai, Y.; Horwitz, G.C.; Kurima, K.; Ishikawa, K.; Kawashima, Y.; Griffith, A.J.; Holt, J.R. TMC1 and TMC2 Are Components of the Mechanotransduction Channel in Hair Cells of the Mammalian Inner Ear. Neuron 2013, 79, 504–515. [Google Scholar] [CrossRef]
- Sırmacı, A.; Duman, D.; Öztürkmen-Akay, H.; Erbek, S.; İncesulu, A.; Öztürk-Hişmi, B.; Arıcı, Z.S.; Yüksel-Konuk, E.B.; Taşır-Yılmaz, S.; Tokgöz-Yılmaz, S.; et al. Mutations in TMC1 Contribute Significantly to Nonsyndromic Autosomal Recessive Sensorineural Hearing Loss: A Report of Five Novel Mutations. Int. J. Pediatr. Otorhinolaryngol. 2009, 73, 699–705. [Google Scholar] [CrossRef]
- Mašindová, I.; Šoltýsová, A.; Varga, L.; Mátyás, P.; Ficek, A.; Hučková, M.; Sůrová, M.; Šafka-Brožková, D.; Anwar, S.; Bene, J.; et al. MARVELD2 (DFNB49) Mutations in the Hearing Impaired Central European Roma Population—Prevalence, Clinical Impact and the Common Origin. PLoS ONE 2015, 10, e0124232. [Google Scholar] [CrossRef]
- Wattenhofer, M.; Di Iorio, M.; Rabionet, R.; Dougherty, L.; Pampanos, A.; Schwede, T.; Montserrat-Sentis, B.; Arbones, M.; Iliades, T.; Pasquadibisceglie, A.; et al. Mutations in the TMPRSS3 Gene Are a Rare Cause of Childhood Nonsyndromic Deafness in Caucasian Patients. J. Mol. Med. 2002, 80, 124–131. [Google Scholar] [CrossRef]
- Gharanei, S.; Zatyka, M.; Astuti, D.; Fenton, J.; Sik, A.; Nagy, Z.; Barrett, T.G. Vacuolar-Type H+-ATPase V1A Subunit Is a Molecular Partner of Wolfram Syndrome 1 (WFS1) Protein, Which Regulates Its Expression and Stability. Hum. Mol. Genet. 2013, 22, 203–217. [Google Scholar] [CrossRef]
- Takei, D.; Ishihara, H.; Yamaguchi, S.; Yamada, T.; Tamura, A.; Katagiri, H.; Maruyama, Y.; Oka, Y. WFS1 Protein Modulates the Free Ca2+ Concentration in the Endoplasmic Reticulum. FEBS Lett. 2006, 580, 5635–5640. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Tranebjærg, L.; Gupta, R.; Rendtorff, N.D.; Williams, D.; Wright, B.; Dias, R. WFS1 Spectrum Disorder. 24 February 2009 [updated 1 December 2022]. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2023. [Google Scholar] [PubMed]
- Rendtorff, N.D.; Lodahl, M.; Boulahbel, H.; Johansen, I.R.; Pandya, A.; Welch, K.O.; Norris, V.W.; Arnos, K.S.; Bitner-Glindzicz, M.; Emery, S.B.; et al. Identification of p.A684V Missense Mutation in the WFS1 Gene as a Frequent Cause of Autosomal Dominant Optic Atrophy and Hearing Impairment. Am. J. Med. Genet. 2011, 155, 1298–1313. [Google Scholar] [CrossRef] [PubMed]
- Cryns, K.; Sivakumaran, T.A.; Van den Ouweland, J.M.W.; Pennings, R.J.E.; Cremers, C.W.R.J.; Flothmann, K.; Young, T.-L.; Smith, R.J.H.; Lesperance, M.M.; Camp, G.V. Mutational Spectrum of TheWFS1 Gene in Wolfram Syndrome, Nonsyndromic Hearing Impairment, Diabetes Mellitus, and Psychiatric Disease. Hum. Mutat. 2003, 22, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Aloi, C.; Salina, A.; Pasquali, L.; Lugani, F.; Perri, K.; Russo, C.; Tallone, R.; Ghiggeri, G.M.; Lorini, R.; d’Annunzio, G. Wolfram Syndrome: New Mutations, Different Phenotype. PLoS ONE 2012, 7, e29150. [Google Scholar] [CrossRef] [PubMed]
- Weil, D.; Levy, G.; Sahly, I.; Levi-Acobas, F.; Blanchard, S.; El-Amraoui, A.; Crozet, F.; Philippe, H.; Abitbol, M.; Petit, C. Human Myosin VIIA Responsible for the Usher 1B Syndrome: A Predicted Membrane-Associated Motor Protein Expressed in Developing Sensory Epithelia. Proc. Natl. Acad. Sci. USA 1996, 93, 3232–3237. [Google Scholar] [CrossRef] [PubMed]
- Joo, S.Y.; Na, G.; Kim, J.A.; Yoo, J.E.; Kim, D.H.; Kim, S.J.; Jang, S.H.; Yu, S.; Kim, H.-Y.; Choi, J.Y.; et al. Clinical Heterogeneity Associated with MYO7A Variants Relies on Affected Domains. Biomedicines 2022, 10, 798. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- McKusick, V.A. Mendelian Inheritance in Man and Its Online Version, OMIM. Am. J. Hum. Genet. 2007, 80, 588–604. [Google Scholar] [CrossRef]
- De Kok, Y.J.M.; van der Maarel, S.M.; Bitner-Glindzicz, M.; Huber, I.; Monaco, A.P.; Malcolm, S.; Pembrey, M.E.; Ropers, H.-H.; Cremers, F.P.M. Association Between X-Linked Mixed Deafness and Mutations in the POU Domain Gene POU3F4. Sci. New Ser. 1995, 267, 685–688. [Google Scholar] [CrossRef]
- Song, M.; Lee, H.; Choi, J.; Kim, S.; Bok, J.; Kim, U.-K. Clinical Evaluation of DFN3 Patients with Deletions in the POU3F4 Locus and Detection of Carrier Female Using MLPA. Clin. Genet. 2010, 78, 524–532. [Google Scholar] [CrossRef]
- Shearer, A.; Kolbe, D.L.; Azaiez, H.; Sloan, C.M.; Frees, K.L.; Weaver, A.E.; Clark, E.T.; Nishimura, C.J.; Black-Ziegelbein, E.; Smith, R.J.H. Copy Number Variants Are a Common Cause of Non-Syndromic Hearing Loss. Genome Med. 2014, 6, 37. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.T.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD®): 2003 Update: HGMD 2003 UPDATE. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Vooren, S.V.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Variant—HGVS | MAF | ACMG | Number of Cases (Homozygous/Compound Heterozygous/Heterozygous) |
|---|---|---|---|
| c.35delG; p.Gly12ValfsTer2 | 0.00597 | Pathogenic a | 56/11/4 |
| c.71G>A; p.Trp24Ter | 0.00058 | Pathogenic a | 0/0//1 |
| c.101T>C; p.Met34Thr | 0.00868 | Pathogenic a | 0/1//2 |
| c.109G>T; p.Val37Phe | 0 | Pathogenic a | 0/2/0 |
| c.119C>A; p.Ala40Glu | 0 | Pathogenic b | 0/0//1 |
| c.139G>T; p.Glu47Ter | 0.00013 | Pathogenic a | 0/1/0 |
| c.167delT; p.Leu56ArgfsTer26 | 0.00089 | Pathogenic a | 0/3/0 |
| c.269T>C; p.Leu90Pro | 0.00065 | Pathogenic a | 0/1/0 |
| c.313_326del; p.Lys105GlyfsTer5 | 0.00013 | Pathogenic a | 0/2/0 |
| c.427C>T; p.Arg143Trp | 0.00012 | Pathogenic | 0/2/0 |
| c.439G>A; p.Glu147Lys | 0.00001 | Pathogenic b | 0/1/0 |
| c.551G>C; p.Arg184Pro | 0.00006 | Pathogenic | 0/1/0 |
| c.−23+1G>A (IVS1+1G>A) | 0.00019 | Pathogenic a | 0/3/0 |
| Gene | Transcript (hg19) | Variant—HGVS (cDNA; Protein) | MAF | ACMG Classification | Inherit. | No of Alleles | No of Cases (hmz/Compound htz/htz) |
|---|---|---|---|---|---|---|---|
| CDH23 | NM_022124 | c.6204del; p.Phe2069LeufsTer11 | 0 | LP | AR | 1 | 0/0/1 |
| CDH23 | NM_022124 | c.1349_1350del; p.Leu450HisfsTer3 | 0 | LP | AR | 1 | 0/1/0 |
| CDH23 | NM_022124 | c.4846-2A>G; p.? | 0 | P | AR | 1 | 0/1/0 |
| COL11A2 | NM_080680.3 | c.3403G>C; p.Gly1135Arg | 0 | VUS/LP | AR/AD | 1 | 0/1/0 |
| COL11A2 | NM_080680.3 | c.1297C>T; p.Arg433Ter | 0 | LP | AR/AD | 1 | 0/1/0 |
| KCNQ4 | NM_004700 | c.647G>C; p.Arg216Pro | 0 | VUS | AD | 1 | 0/0/1 |
| KCNQ4 | NM_004700 | c.1031_1040del; p.Asn344ArgfsTer11 | 0 | P | AD | 1 | 0/0/1 |
| MARVELD2 | NM_001038603 | c.1331+2T>C; p.? | 0.0000398 | P | AR | 4 | 2/0/0 |
| MYO15A | NM_016239 | c.2493_2505del; p.Arg832ProfsTer27 | 0 | LP | AR | 1 | 0/1/0 |
| MYO15A | NM_016239 | c.2677C>T; p.Arg893Ter | 0.0000211 | P | AR | 1 | 0/1/0 |
| MYO15A | NM_016239 | c.4030C>T; p.Gln1344Ter | 0 | P | AR | 1 | 0/1/0 |
| MYO15A | NM_016239 | c.8183G>A; p.Arg2728His | 0.000189 | P | AR | 1 | 0/1/0 |
| MYO15A | NM_016239 | c.8153T>C; p.Leu2718Pro | 0 | VUS/LP | AR | 1 | 0/1/0 |
| MYO15A | NM_016239 | c.8548C>T; p.Arg2850Ter | 0.00002 | P | AR | 1 | 0/1/0 |
| MYO7A | NM_000260 | c.268C>T; p.Arg90Trp | 0.0000438 | VUS/LP | AD/AR | 1 | 0/0/1 |
| MYO7A | NM_000260 | c.770G>T; p.Cys257Phe | 0 | VUS | AD/AR | 1 | 0/0/1 |
| MYO7A | NM_000260 | c.4087G>A; p.Ala1363Thr | 0 | VUS | AD/AR | 1 | 0/0/1 |
| OSBPL2 | NM_001363878 | c.313C>T; p.His105Tyr | 0.0000159 | VUS | AD | 1 | 0/0/1 |
| OTOF | NM_194248.3 | c.2665del; p.Leu889SerfsTer111 | 0 | LP | AR | 1 | 0/0/1 |
| POU3F4 | NM_000307 | c.446G>T; p.Gly149Val | 0 | VUS | XR | 1 | 0/0/1 |
| POU4F3 | NM_002700 | c.868G>C; p.Glu290Gln | 0.00000398 | VUS/LP | AD | 1 | 0/0/1 |
| PNPT1 | NM_033109.5 | c.79delG; p.Asp27IlefsTer25 | 0 | LP | AR | 1 | 0/0/1 |
| PTPRQ | NM_001145026 | c.5959C>T; p.Gln1987Ter | 0 | LP | AR | 5 | 2/0/1 |
| SLC26A4 | NM_000441 | c.349C>T; p.Leu117Phe | 0.000326 | LP | AR | 1 | 0/1/0 |
| SLC26A4 | NM_000441 | c.1204G>T; p.Val402Leu | 0 | P | AR | 1 | 0/1/0 |
| SLC26A4 | NM_000441 | c.1670G>T; p.Gly557Val | 0.00000399 | LP | AR | 2 | 1/0/0 |
| TECTA | NM_005422 | c.6094G>T; p.Asp2032Tyr | 0.00000398 | VUS/LP | AD/AR | 1 | 0/0/1 |
| TJP2 | NM_001170416 | c.53T>A; p.Leu18Ter | 0 | VUS/LP | AD | 1 | 0/0/1 |
| TMC1 | NM_138691 | c.312_325del; p.Val106MetfsTer8 | 0 | LP | AD/AR | 1 | 0/1/0 |
| TMC1 | NM_138691 | c.2030T>C; p.Ile677Tyr | 0.0000119 | VUS/LP | AD/AR | 2 | 1/0/0 |
| TMC1 | NM_138691 | c.2050G>A; p.Asp684Asn | 0.0000239 | VUS/LP | AD/AR | 1 | 0/1/0 |
| TMPRSS3 | NM_001256317 | c.208delC; p.His70ThrfsTer19 | 0.000489 | P | AR | 3 | 1/1/0 |
| TMPRSS3 | NM_001256317 | c.646C>T; p.Arg216Cys | 0.00002387 | P | AR | 1 | 0/1/0 |
| USH1C | NM_153676.4 | c.241C>T; p.Arg81Cys | 0.00002001 | VUS | AR | 1 | 0/0/1 |
| WFS1 | NM_006005 | c.958C>T; p.Pro320Ser | 0 | VUS | AD/AR | 1 | 0/0/1 |
| WFS1 | NM_006005 | c.1181A>T; p.Glu394Val | 0.0001273 | LP | AD/AR | 1 | 0/0/1 |
| WFS1 | NM_006005 | c.2051C>T; p.Ala684Val | 0 | P | AD/AR | 1 | 0/0/1 |
| WFS1 | NM_006005 | c.2527A>G; p.Lys843Glu | 0 | VUS/LP | AD/AR | 1 | 0/0/1 |
| WFS1 | NM_006005 | c.2575C>T; p.Arg859Trp | 0.0000319 | VUS | AD/AR | 1 | 0/0/1 |
| STRC | NM_153700 | Contiguous gene deletion, including the CKMT1B, STRC and CATSPER2 genes | P | AR | 1 | 0/0/1 | |
| OTOA | NM_170664 | Contiguous gene duplication, including the METTL9 and OTOA genes | VUS | AR | 1 | 0/0/1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pál, M.; Nagy, D.; Neller, A.; Farkas, K.; Leprán-Török, D.; Nagy, N.; Füstös, D.; Nagy, R.; Németh, A.; Szilvássy, J.; et al. Genetic Etiology of Nonsyndromic Hearing Loss in Hungarian Patients. Int. J. Mol. Sci. 2023, 24, 7401. https://doi.org/10.3390/ijms24087401
Pál M, Nagy D, Neller A, Farkas K, Leprán-Török D, Nagy N, Füstös D, Nagy R, Németh A, Szilvássy J, et al. Genetic Etiology of Nonsyndromic Hearing Loss in Hungarian Patients. International Journal of Molecular Sciences. 2023; 24(8):7401. https://doi.org/10.3390/ijms24087401
Chicago/Turabian StylePál, Margit, Dóra Nagy, Alexandra Neller, Katalin Farkas, Dóra Leprán-Török, Nikoletta Nagy, Dalma Füstös, Roland Nagy, Adrienne Németh, Judit Szilvássy, and et al. 2023. "Genetic Etiology of Nonsyndromic Hearing Loss in Hungarian Patients" International Journal of Molecular Sciences 24, no. 8: 7401. https://doi.org/10.3390/ijms24087401