
DeepSTABp: A Deep Learning Approach for the Prediction of Thermal Protein Stability

Abstract
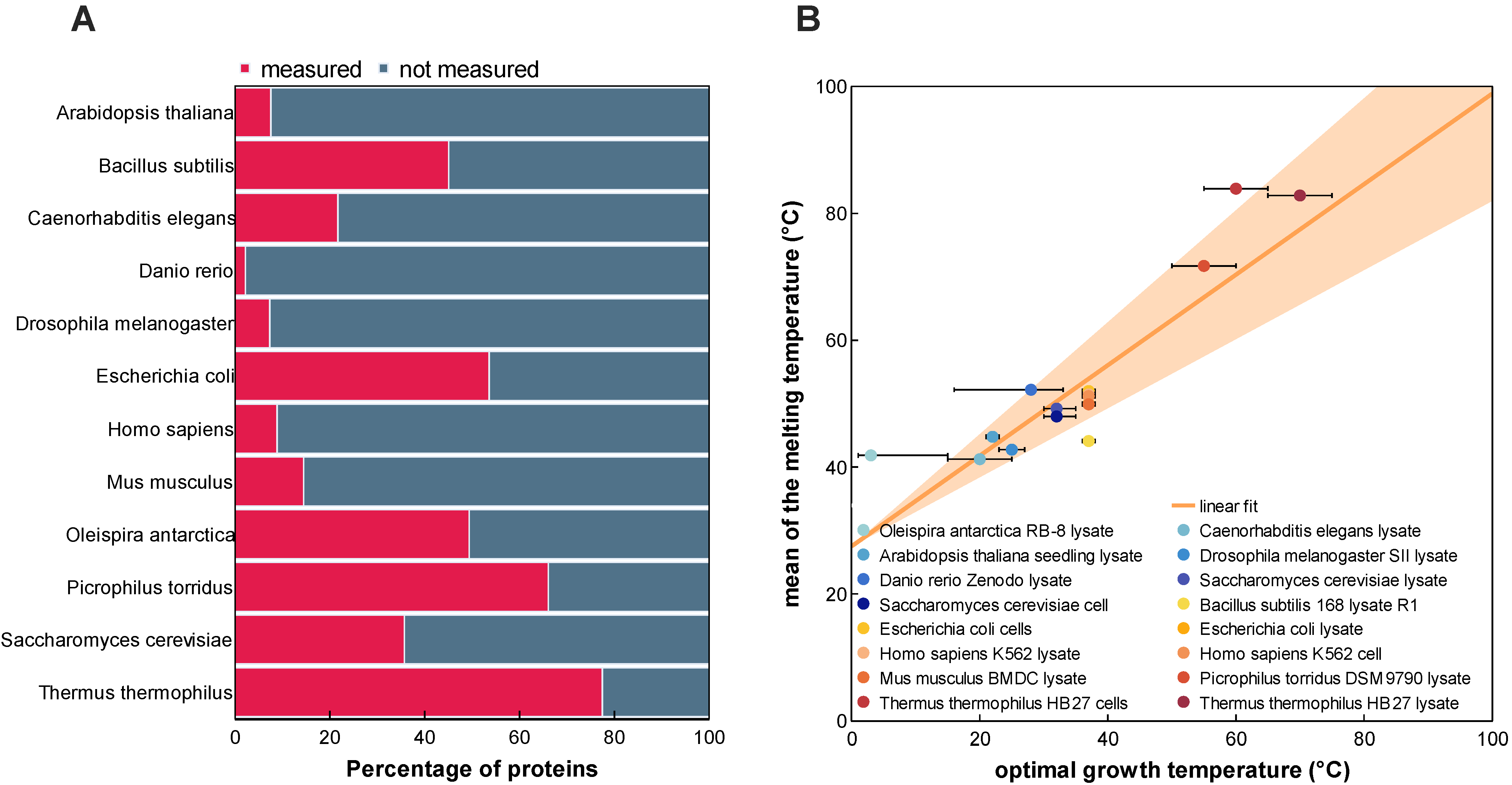
:1. Introduction
2. Results
2.1. Data Driven Design of the DeepSTABp Model
2.2. Evaluation and Comparison of Prediction Performance
2.3. Capture and Representation of Biological Features
2.4. Future Applications of DeepSTABp
2.5. Web-Interface of DeepSTABp
3. Discussion
4. Materials and Methods
4.1. Tm Dataset Assembly and Extraction of Protein Melting Points
4.2. Dataset Augmentation
4.3. Amino Acid Sequence Embedding Using Protein Language Models
4.4. DeepSTABp
4.5. Model Evaluation Metrices
4.6. Variation of Measured and Predicted Tms of Protein Complexes
4.7. Relating Predicted Tms to Secondary Structure Elements
4.8. Software
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deller, M.C.; Kong, L.; Rupp, B. Protein stability: A crystallographer’s perspective. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 72–95. [Google Scholar] [CrossRef]
- Fágáin, C.O. Understanding and increasing protein stability. Biochim. Biophys. Acta 1995, 1252, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Almeida, P. Proteins: Concepts in Biochemistry; Garland Science Taylor & Francis Group: London, UK; New York, NY, USA, 2016; ISBN 9780429258817. [Google Scholar]
- Bischof, J.H.; He, X. Thermal Stability of Proteins. Ann. N. Y. Acad. Sci. 2006, 1066, 12–33. [Google Scholar] [CrossRef] [PubMed]
- Leuenberger, P.; Ganscha, S.; Kahraman, A.; Cappelletti, V.; Boersema, P.J.; von Mering, C.; Claassen, M.; Picotti, P. Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science 2017, 355, eaai7825. [Google Scholar] [CrossRef]
- Savitski, M.M.; Reinhard, F.B.M.; Franken, H.; Werner, T.; Savitski, M.F.; Eberhard, D.; Martinez Molina, D.; Jafari, R.; Dovega, R.B.; Klaeger, S.; et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 2014, 346, 1255784. [Google Scholar] [CrossRef] [PubMed]
- Mateus, A.; Kurzawa, N.; Becher, I.; Sridharan, S.; Helm, D.; Stein, F.; Typas, A.; Savitski, M.M. Thermal proteome profiling for interrogating protein interactions. Mol. Syst. Biol. 2020, 16, e9232. [Google Scholar] [CrossRef]
- Mateus, A.; Määttä, T.A.; Savitski, M.M. Thermal proteome profiling: Unbiased assessment of protein state through heat-induced stability changes. Proteome Sci. 2016, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Jarzab, A.; Kurzawa, N.; Hopf, T.; Moerch, M.; Zecha, J.; Leijten, N.; Bian, Y.; Musiol, E.; Maschberger, M.; Stoehr, G.; et al. Meltome atlas-thermal proteome stability across the tree of life. Nat. Methods 2020, 17, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Horne, J.; Shukla, D. Recent Advances in Machine Learning Variant Effect Prediction Tools for Protein Engineering. Ind. Eng. Chem. Res. 2022, 61, 6235–6245. [Google Scholar] [CrossRef]
- Montanucci, L.; Capriotti, E.; Birolo, G.; Benevenuta, S.; Pancotti, C.; Lal, D.; Fariselli, P. DDGun: An untrained predictor of protein stability changes upon amino acid variants. Nucleic Acids Res. 2022, 50, W222–W227. [Google Scholar] [CrossRef]
- Benevenuta, S.; Pancotti, C.; Fariselli, P.; Birolo, G.; Sanavia, T. An antisymmetric neural network to predict free energy changes in protein variants. J. Phys. D Appl. Phys. 2021, 54, 245403. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, J.; Zeng, L.; Vihinen, M. ProTstab2 for Prediction of Protein Thermal Stabilities. Int. J. Mol. Sci. 2022, 23, 798. [Google Scholar] [CrossRef] [PubMed]
- Ku, T.; Lu, P.; Chan, C.; Wang, T.; Lai, S.; Lyu, P.; Hsiao, N. Predicting melting temperature directly from protein sequences. Comput. Biol. Chem. 2009, 33, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Pucci, F.; Kwasigroch, J.M.; Rooman, M. SCooP: An accurate and fast predictor of protein stability curves as a function of temperature. Bioinformatics 2017, 33, 3415–3422. [Google Scholar] [CrossRef]
- Yang, Y.; Ding, X.; Zhu, G.; Niroula, A.; Lv, Q.; Vihinen, M. ProTstab—Predictor for cellular protein stability. BMC Genom. 2019, 20, 804. [Google Scholar] [CrossRef]
- Zhao, J.; Yan, W.; Yang, Y. DeepTP: A Deep Learning Model for Thermophilic Protein Prediction. Int. J. Mol. Sci. 2023, 24, 2217. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Li, J.; Ma, S.; Jiang, J.; Li, M.; Zou, Q.; Lv, Z. Identification of Thermophilic Proteins Based on Sequence-Based Bidirectional Representations from Transformer-Embedding Features. Appl. Sci. 2023, 13, 2858. [Google Scholar] [CrossRef]
- Gado, J.E.; Beckham, G.T.; Payne, C.M. Improving Enzyme Optimum Temperature Prediction with Resampling Strategies and Ensemble Learning. J. Chem. Inf. Model. 2020, 60, 4098–4107. [Google Scholar] [CrossRef]
- He, H.; Garcia, E.A. Learning from Imbalanced Data. IEEE Trans. Knowl. Data Eng. 2009, 21, 1263–1284. [Google Scholar] [CrossRef]
- Krawczyk, B. Learning from imbalanced data: Open challenges and future directions. Prog. Artif. Intell. 2016, 5, 221–232. [Google Scholar] [CrossRef]
- López, V.; Fernández, A.; García, S.; Palade, V.; Herrera, F. An insight into classification with imbalanced data: Empirical results and current trends on using data intrinsic characteristics. Inf. Sci. 2013, 250, 113–141. [Google Scholar] [CrossRef]
- Tan, C.S.H.; Go, K.D.; Bisteau, X.; Dai, L.; Yong, C.H.; Prabhu, N.; Ozturk, M.B.; Lim, Y.T.; Sreekumar, L.; Lengqvist, J.; et al. Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science 2018, 359, 1170–1177. [Google Scholar] [CrossRef]
- Meldal, B.H.M.; Bye-A-Jee, H.; Gajdoš, L.; Hammerová, Z.; Horácková, A.; Melicher, F.; Perfetto, L.; Pokorný, D.; Lopez, M.R.; Türková, A.; et al. Complex Portal 2018: Extended content and enhanced visualization tools for macromolecular complexes. Nucleic Acids Res. 2019, 47, D550–D558. [Google Scholar] [CrossRef] [PubMed]
- Radestock, S.; Gohlke, H. Protein rigidity and thermophilic adaptation. Proteins 2011, 79, 1089–1108. [Google Scholar] [CrossRef]
- Kumar, S.; Tsai, C.J.; Nussinov, R. Factors enhancing protein thermostability. Protein Eng. 2000, 13, 179–191. [Google Scholar] [CrossRef]
- Vogt, G.; Argos, P. Protein thermal stability: Hydrogen bonds or internal packing? Fold. Des. 1997, 2, S40–S46. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.B.; Manoj, N. A conserved π-helix plays a key role in thermoadaptation of catalysis in the glycoside hydrolase family 4. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140523. [Google Scholar] [CrossRef]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 44, 7112–7127. [Google Scholar] [CrossRef] [PubMed]
- Lamosa, P.; Turner, D.L.; Ventura, R.; Maycock, C.; Santos, H. Protein stabilization by compatible solutes. Effect of diglycerol phosphate on the dynamics of Desulfovibrio gigas rubredoxin studied by NMR. Eur. J. Biochem. 2003, 270, 4606–4614. [Google Scholar] [CrossRef]
- Faria, T.Q.; Knapp, S.; Ladenstein, R.; Maçanita, A.L.; Santos, H. Protein stabilisation by compatible solutes: Effect of mannosylglycerate on unfolding thermodynamics and activity of ribonuclease A. Chembiochem 2003, 4, 734–741. [Google Scholar] [CrossRef]
- Faria, T.Q.; Lima, J.C.; Bastos, M.; Maçanita, A.L.; Santos, H. Protein stabilization by osmolytes from hyperthermophiles: Effect of mannosylglycerate on the thermal unfolding of recombinant nuclease a from Staphylococcus aureus studied by picosecond time-resolved fluorescence and calorimetry. J. Biol. Chem. 2004, 279, 48680–48691. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Gao, Y.; Mih, N.; O’Brien, E.J.; Yang, L.; Palsson, B.O. Thermosensitivity of growth is determined by chaperone-mediated proteome reallocation. Proc. Natl. Acad. Sci. USA 2017, 114, 11548–11553. [Google Scholar] [CrossRef]
- Pak, M.A.; Markhieva, K.A.; Novikova, M.S.; Petrov, D.S.; Vorobyev, I.S.; Maksimova, E.S.; Kondrashov, F.A.; Ivankov, D.N. Using AlphaFold to predict the impact of single mutations on protein stability and function. PLoS ONE 2023, 18, e0282689. [Google Scholar] [CrossRef] [PubMed]
- Volkening, J.D.; Stecker, K.E.; Sussman, M.R. Proteome-wide Analysis of Protein Thermal Stability in the Model Higher Plant Arabidopsis thaliana. Mol. Cell. Proteom. 2019, 18, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Groh, C.; Haberkant, P.; Stein, F.; Filbeck, S.; Pfeffer, S.; Savitski, M.M.; Boos, F.; Herrmann, J.M. Mitochondrial dysfunction rapidly modulates the abundance and thermal stability of cellular proteins. Life Sci. Alliance 2023, 6, e202201805. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Stärk, H.; Dallago, C.; Heinzinger, M.; Rost, B. Light attention predicts protein location from the language of life. Bioinform. Adv. 2021, 1, vbab035. [Google Scholar] [CrossRef]
- Robinson, R.K. (Ed.) Encyclopedia of Food Microbiology, 1st ed.; Academic Press: Cambridge, MA, USA, 1999; ISBN 978-0-12-227070-3. [Google Scholar]
- Balasubramanian, S.; Sureshkumar, S.; Lempe, J.; Weigel, D. Potent induction of Arabidopsis thaliana flowering by elevated growth temperature. PLoS Genet. 2006, 2, e106. [Google Scholar] [CrossRef]
- Hong, E.; Jeong, M.; Kim, T.; Lee, J.; Cho, J.; Lee, K. Development of Coupled Biokinetic and Thermal Model to Optimize Cold-Water Microbial Enhanced Oil Recovery (MEOR) in Homogenous Reservoir. Sustainability 2019, 11, 1652. [Google Scholar] [CrossRef]
- López-Olmeda, J.F.; Sánchez-Vázquez, F.J. Thermal biology of zebrafish (Danio rerio). J. Therm. Biol. 2011, 36, 91–104. [Google Scholar] [CrossRef]
- Mołoń, M.; Dampc, J.; Kula-Maximenko, M.; Zebrowski, J.; Mołoń, A.; Dobler, R.; Durak, R.; Skoczowski, A. Effects of Temperature on Lifespan of Drosophila melanogaster from Different Genetic Backgrounds: Links between Metabolic Rate and Longevity. Insects 2020, 11, 470. [Google Scholar] [CrossRef]
- Noor, R.; Islam, Z.; Munshi, S.K.; Rahman, F. Influence of Temperature on Escherichia coli Growth in Different Culture Media. J. Pure Appl. Microbiol. 2013, 7, 899–904. [Google Scholar]
- Oshima, T.; Imahori, K. Description of Thermus thermophilus (Yoshida and Oshima) comb. nov., a Nonsporulating Thermophilic Bacterium from a Japanese Thermal Spa. Int. J. Syst. Bacteriol. 1974, 24, 102–112. [Google Scholar] [CrossRef]
- Schleper, C.; Puehler, G.; Holz, I.; Gambacorta, A.; Janekovic, D.; Santarius, U.; Klenk, H.P.; Zillig, W. Picrophilus gen. nov., fam. nov.: A novel aerobic, heterotrophic, thermoacidophilic genus and family comprising archaea capable of growth around pH 0. J. Bacteriol. 1995, 177, 7050–7059. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R.; Keijer, J. Not so hot: Optimal housing temperatures for mice to mimic the thermal environment of humans. Mol. Metab. 2012, 2, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.M.; Martin, P.A. Growth of Saccharomyces cerevisiae and Saccharomyces uvarum in a temperature gradient incubator. J. Inst. Brew. 1977, 83, 169–172. [Google Scholar] [CrossRef]
- Yakimov, M.M.; Giuliano, L.; Gentile, G.; Crisafi, E.; Chernikova, T.N.; Abraham, W.-R.; Lünsdorf, H.; Timmis, K.N.; Golyshin, P.N. Oleispira antarctica gen. nov., sp. nov., a novel hydrocarbonoclastic marine bacterium isolated from Antarctic coastal sea water. Int. J. Syst. Evol. Microbiol. 2003, 53, 779–785. [Google Scholar] [CrossRef]
- Zhang, B.; Xiao, R.; Ronan, E.A.; He, Y.; Hsu, A.-L.; Liu, J.; Xu, X.Z.S. Environmental Temperature Differentially Modulates C. elegans Longevity through a Thermosensitive TRP Channel. Cell Rep. 2015, 11, 1414–1424. [Google Scholar] [CrossRef]
- Klambauer, G.; Unterthiner, T.; Mayr, A.; Hochreiter, S. Self-Normalizing Neural Networks. In Proceedings of the 31st Annual Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Ba, J.L.; Kiros, J.R.; Hinton, G.E. Layer Normalization. arXiv 2016, arXiv:1607.06450v1. Available online: https://arxiv.org/pdf/1607.06450 (accessed on 10 March 2023).
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Bergstra, J.; Bardenet, R.; Bengio, Y.; Kégl, B. Algorithms for Hyper-Parameter Optimization. In Proceedings of the 24th International Conference on Neural Information Processing Systems, Granada, Spain, 12–15 December 2011. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.H.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Venn, B.; Mühlhaus, T.; Schneider, K.; Weil, L.; Zimmer, D.; Ziegler, S.; Frey, K.; Ott, J.; Lux, C. fslaborg/FSharp.Stats: Release 0.4.11; Zenodo: Meyrin, Geneva, Switzerland, 2023. [Google Scholar]
- Falcon, W.; The PyTorch Lightning Team. PyTorch Lightning; GitHub: San Francisco, CA, USA, 2019. [Google Scholar]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. arXiv 2019, arXiv:1912.01703v1. Available online: http://arxiv.org/pdf/1912.01703v1 (accessed on 10 March 2023).
- Wolf, T.; Debut, L.; Sanh, V.; Chaumond, J.; Delangue, C. Transformers: State-of-the-Art Natural Language Processing. arXiv 2019, arXiv:1910.03771v5. Available online: http://arxiv.org/pdf/1910.03771v5 (accessed on 10 March 2023).
- The Pandas Development Team. pandas-dev/pandas: Pandas; Zenodo: Meyrin, Geneva, Switzerland, 2023. [Google Scholar]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A Next-generation Hyperparameter Optimization Framework. arXiv 2019, arXiv:1907.10902v1. Available online: http://arxiv.org/pdf/1907.10902v1 (accessed on 10 March 2023).
- Schneider, K.; Weil, L.; Zimmer, D.; Venn, B.; Mühlhaus, T. CSBiology/BioFSharp: 2.0.0-Preview.2; Zenodo: Meyrin, Switzerland; Geneva, Switzerland, 2022. [Google Scholar]
- Plotly Technologies Inc. Collaborative Data Science; Plotly Technologies Inc.: Montreal, QC, Canada, 2015. [Google Scholar]
- Schneider, K.; Venn, B.; Mühlhaus, T. Plotly.NET: A fully featured charting library for .NET programming languages. F1000Research 2022, 11, 1094. [Google Scholar] [CrossRef]
- Xiao, N.; Cao, D.S.; Zhu, M.F.; Xu, Q.S. protr/ProtrWeb: R package and web server for generating various numerical representation schemes of protein sequences. Bioinformatics 2015, 31, 1857–1859. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metrics | Training (DeepSTABp) | Testing | |||
|---|---|---|---|---|---|
| Naïve | Uniform Sampling | Extended Uniform Sampling | DeepSTABp | ProTstab2 | |
| R2 | 0.86 | 0.89 | 0.93 | 0.80 | 0.57 |
| PCC | 0.93 | 0.96 | 0.97 | 0.90 | 0.76 |
| MAE (°C) | 3.20 | 2.43 | 1.81 | 3.22 | 4.95 |
| MSE (°C) | 17.84 | 9.70 | 5.54 | 18.46 | 41.59 |
| RMSE (°C) | 4.22 | 3.11 | 2.35 | 4.30 | 6.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, F.; Frey, K.; Zimmer, D.; Mühlhaus, T. DeepSTABp: A Deep Learning Approach for the Prediction of Thermal Protein Stability. Int. J. Mol. Sci. 2023, 24, 7444. https://doi.org/10.3390/ijms24087444
Jung F, Frey K, Zimmer D, Mühlhaus T. DeepSTABp: A Deep Learning Approach for the Prediction of Thermal Protein Stability. International Journal of Molecular Sciences. 2023; 24(8):7444. https://doi.org/10.3390/ijms24087444
Chicago/Turabian StyleJung, Felix, Kevin Frey, David Zimmer, and Timo Mühlhaus. 2023. "DeepSTABp: A Deep Learning Approach for the Prediction of Thermal Protein Stability" International Journal of Molecular Sciences 24, no. 8: 7444. https://doi.org/10.3390/ijms24087444