
A Pseudomonas Lysogenic Bacteriophage Crossing the Antarctic and Arctic, Representing a New Genus of Autographiviridae

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
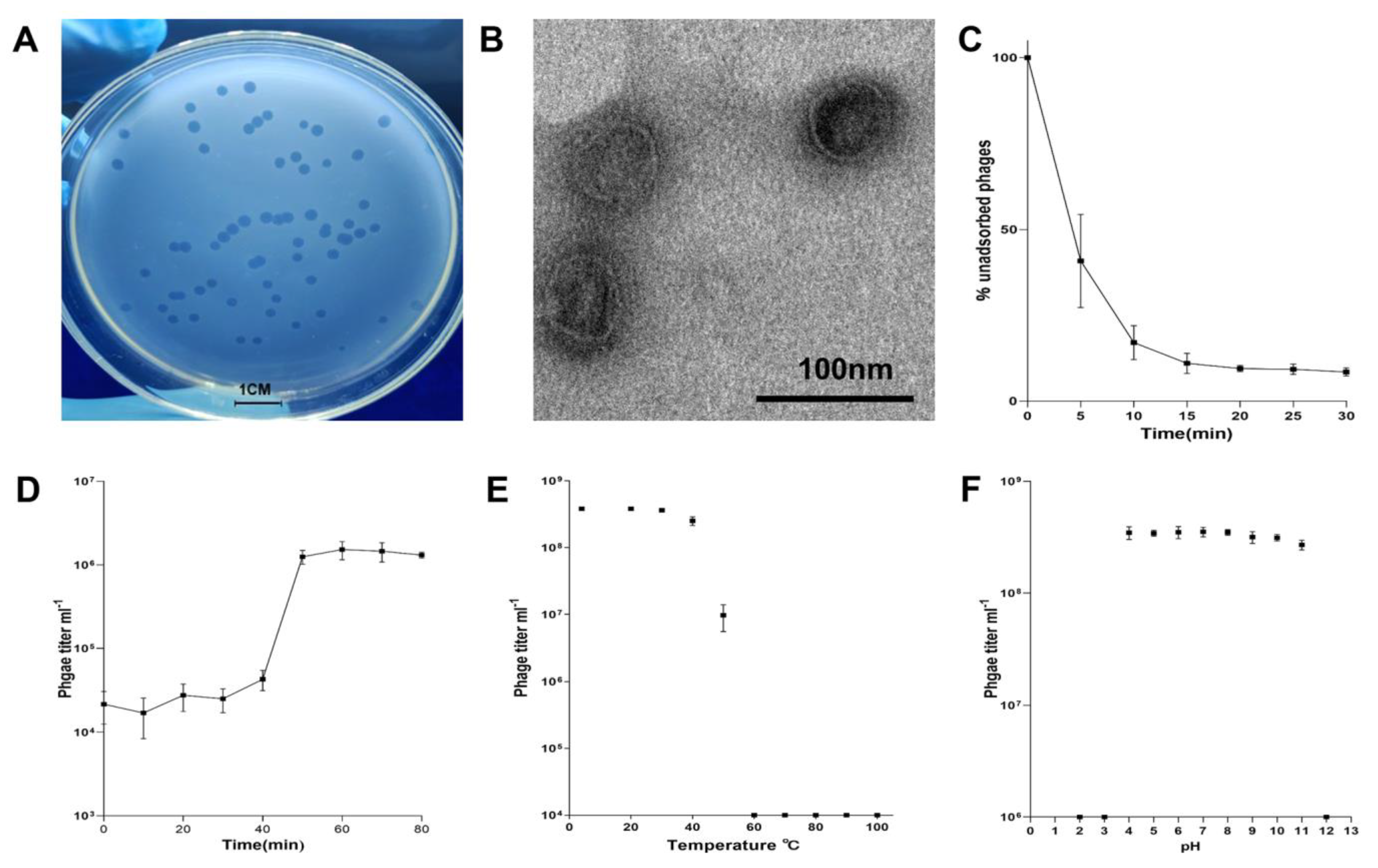
2.1. Biological Characteristics of Pseudomonas Phage vB_PaeM-G11
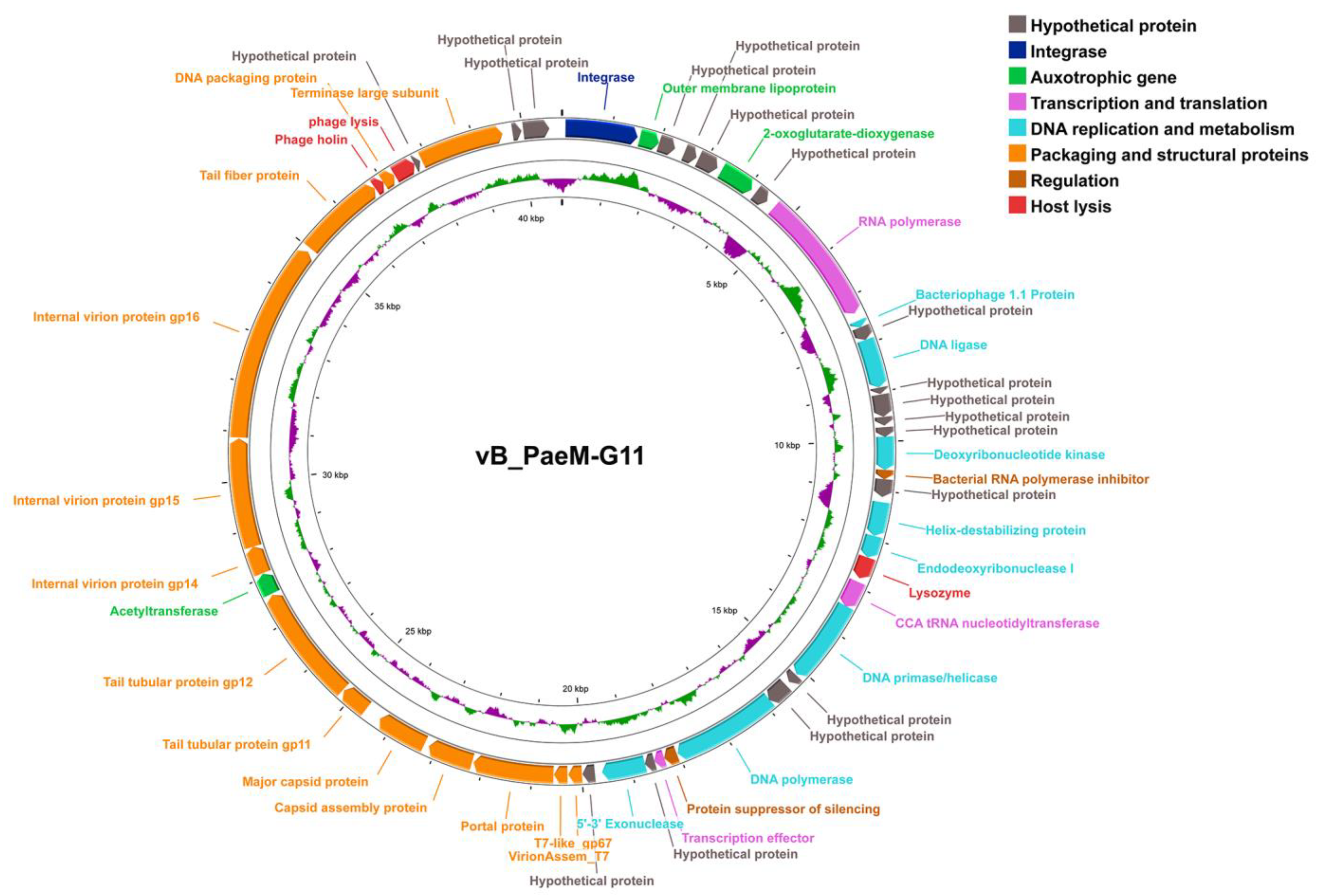
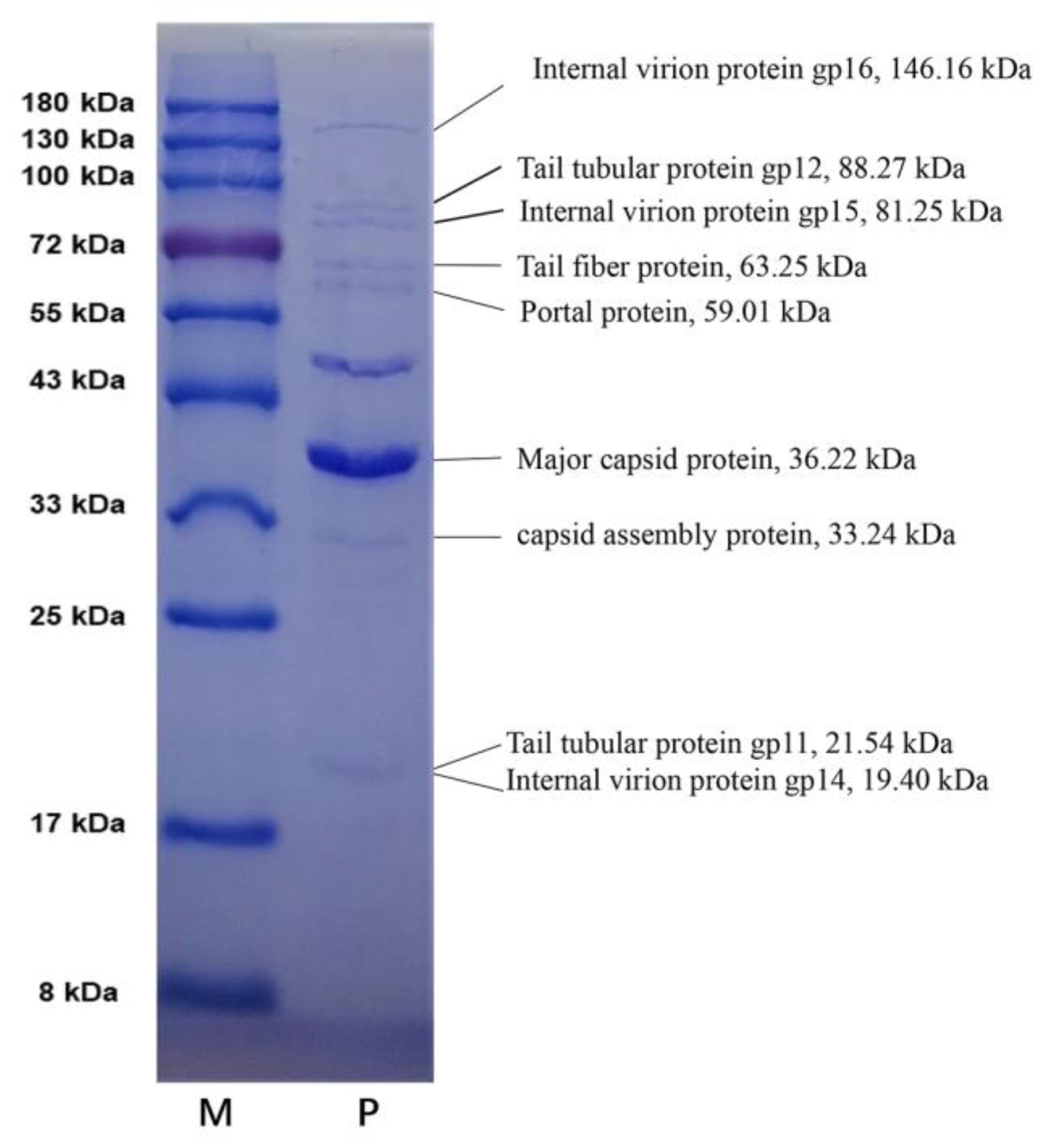
2.2. Genomic and Structural Proteome Characteristics of vB_PaeM-G11
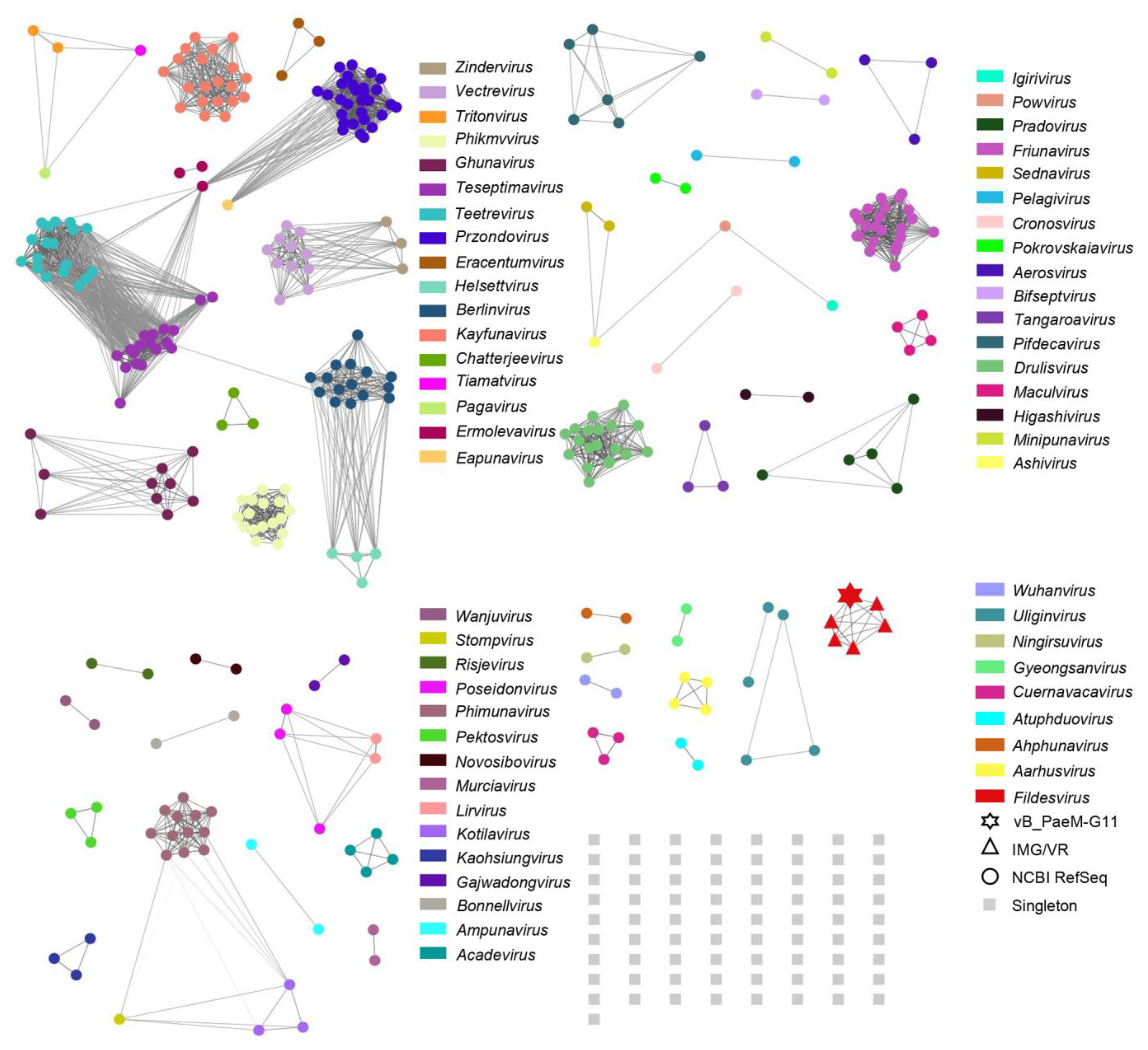
2.3. Phylogenetic and Comparative Genomic Analyses
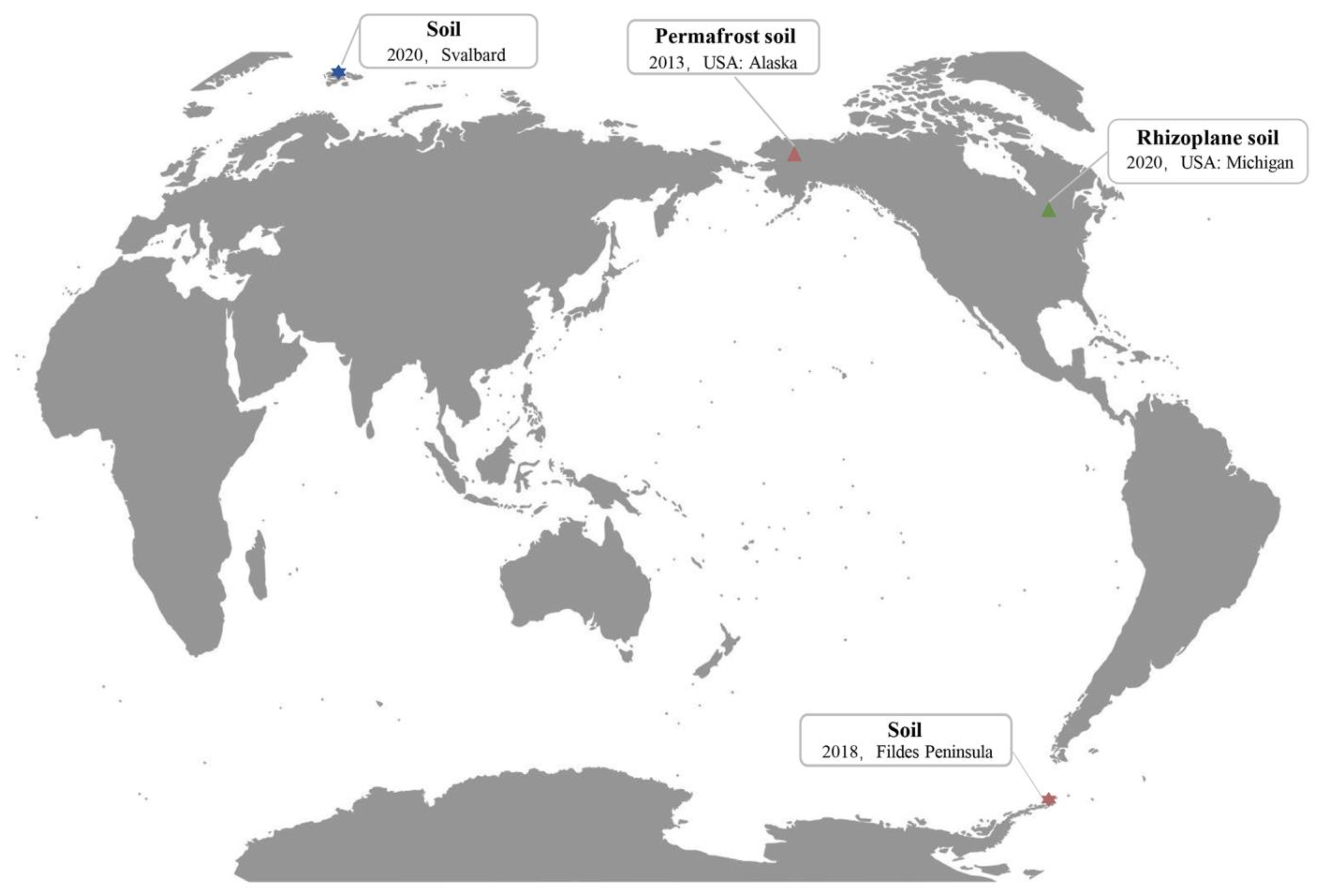
2.4. The Global Distribution Range of Phage vB_PaeM-G11
3. Materials and Methods
3.1. Host Bacteria Culture and Phage Induction
3.2. Phage Isolation, Amplification, and Purification
3.3. Transmission Electron Microscopy
3.4. Phage Biology Experiments
3.4.1. Phage Adsorption
3.4.2. One-Step Growth
3.4.3. Temperature and pH Tolerance Range of Phage
3.5. Phage and Bacterial DNA Extraction and Sequencing
3.6. Genomic and Structural Proteome Characteristics
3.7. Phylogenetic and Comparative Genomic Analysis
3.8. The Global Distribution of vB_PaeM-G11 Phage
3.9. Nucleotide Sequence Accession Numbers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yau, S.; Seth-Pasricha, M. Viruses of Polar Aquatic Environments. Viruses 2019, 11, 189. [Google Scholar] [CrossRef] [PubMed]
- Cowan, D.A.; Tow, L.A. Endangered Antarctic environments. Annu. Rev. Microbiol. 2004, 58, 649–690. [Google Scholar] [CrossRef] [PubMed]
- Ghiglione, J.F.; Galand, P.E.; Pommier, T.; Pedros-Alio, C.; Maas, E.W.; Bakker, K.; Bertilson, S.; Kirchman, D.L.; Lovejoy, C.; Yager, P.L.; et al. Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc. Natl. Acad. Sci. USA 2012, 109, 17633–17638. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.L.; Wang, Z.B.; Cha, Q.Q.; Liu, S.S.; Ren, X.B.; Fu, H.H.; Sun, M.L.; Zhao, D.L.; McMinn, A.; Chen, Y.; et al. Biogeography of culturable marine bacteria from both poles reveals that ‘everything is not everywhere’ at the genomic level. Environ. Microbiol. 2022, 24, 98–109. [Google Scholar] [CrossRef] [PubMed]
- De Smet, J.; Hendrix, H.; Blasdel, B.G.; Danis-Wlodarczyk, K.; Lavigne, R. Pseudomonas predators: Understanding and exploiting phage-host interactions. Nat. Rev. Microbiol. 2017, 15, 517–530. [Google Scholar] [CrossRef]
- Romaniuk, K.; Styczynski, M.; Decewicz, P.; Buraczewska, O.; Uhrynowski, W.; Fondi, M.; Wolosiewicz, M.; Szuplewska, M.; Dziewit, L. Diversity and Horizontal Transfer of Antarctic Pseudomonas spp. Plasmids. Genes 2019, 10, 850. [Google Scholar] [CrossRef]
- Snopkova, K.; Cejkova, D.; Dufkova, K.; Sedlacek, I.; Smajs, D. Genome sequences of two Antarctic strains of Pseudomonas prosekii: Insights into adaptation to extreme conditions. Arch. Microbiol. 2020, 202, 447–454. [Google Scholar] [CrossRef]
- Imam, M.; Alrashid, B.; Patel, F.; Dowah, A.S.A.; Brown, N.; Millard, A.; Clokie, M.R.J.; Galyov, E.E. vB_PaeM_MIJ3, a Novel Jumbo Phage Infecting Pseudomonas aeruginosa, Possesses Unusual Genomic Features. Front. Microbiol. 2019, 10, 2772. [Google Scholar] [CrossRef]
- Moreno, R.; Rojo, F. Features of pseudomonads growing at low temperatures: Another facet of their versatility. Environ. Microbiol. Rep. 2014, 6, 417–426. [Google Scholar] [CrossRef]
- Aguirre de Cárcer, D.; López-Bueno, A.; Pearce, D.A.; Alcamí, A. Biodiversity and distribution of polar freshwater DNA viruses. Sci. Adv. 2015, 1, e1400127. [Google Scholar] [CrossRef]
- Suttle, C.A. Marine viruses--major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, P.; Fabijan, A.P.; Gavric, D.; Pejic, J.; Doffkay, Z.; Rakhely, G. Phages from Genus Bruynoghevirus and Phage Therapy: Pseudomonas Phage Delta Case. Viruses 2021, 13, 1965. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.M.; Touchon, M.; Rocha, E.P.C. Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. USA 2014, 111, 12127–12132. [Google Scholar] [CrossRef]
- Anesio, A.M.; Bellas, C.M. Are low temperature habitats hot spots of microbial evolution driven by viruses? Trends Microbiol. 2011, 19, 52–57. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Varsani, A.; Tong, Y.G.; Simmonds, P.; Sabanadzovic, S.; Rubino, L.; Roux, S.; Munoz, A.R.; Lood, C.; Lefkowitz, E.J.; et al. Perspective on taxonomic classification of uncultivated viruses. Curr. Opin. Virol. 2021, 51, 1–9. [Google Scholar] [CrossRef]
- Gregory, A.C.; Zayed, A.A.; Conceicao-Neto, N.; Temperton, B.; Bolduc, B.; Alberti, A.; Ardyna, M.; Arkhipova, K.; Carmichael, M.; Cruaud, C.; et al. Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell 2019, 177, 1109–1123.e14. [Google Scholar] [CrossRef]
- Demina, T.A.; Luhtanen, A.M.; Roux, S.; Oksanen, H.M. Virus-Host Interactions and Genetic Diversity of Antarctic Sea Ice Bacteriophages. Mbio 2022, 13, e00651-22. [Google Scholar] [CrossRef]
- Wang, J.; Xiao, J.; Zhu, Z.; Wang, S.Y.; Zhang, L.; Fan, Z.J.; Deng, Y.L.; Hu, Z.H.; Peng, F.; Shen, S.; et al. Diverse viromes in polar regions: A retrospective study of metagenomic data from Antarctic animal feces and Arctic frozen soil in 2012–2014. Virol. Sin. 2022, 37, 883–893. [Google Scholar] [CrossRef]
- Bezuidt, O.K.I.; Lebre, P.H.; Pierneef, R.; Leon-Sobrino, C.; Adriaenssens, E.M.; Cowan, D.A.; Van de Peer, Y.; Makhalanyane, T.P. Phages Actively Challenge Niche Communities in Antarctic Soils. Msystems 2020, 5, e00234-20. [Google Scholar] [CrossRef] [PubMed]
- Labbe, M.; Girard, C.; Vincent, W.F.; Culley, A.I. Extreme Viral Partitioning in a Marine-Derived High Arctic Lake. Msphere 2020, 5, e00334-20. [Google Scholar] [CrossRef] [PubMed]
- Sommers, P.; Fontenele, R.S.; Kringen, T.; Kraberger, S.; Porazinska, D.L.; Darcy, J.L.; Schmidt, S.K.; Varsani, A. Single-Stranded DNA Viruses in Antarctic Cryoconite Holes. Viruses 2019, 11, 1022. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Unraveling the viral dark matter through viral metagenomics. Front. Immunol. 2022, 13, 1005107. [Google Scholar] [CrossRef] [PubMed]
- Labbe, M.; Thaler, M.; Pitot, T.M.; Rapp, J.Z.; Vincent, W.F.; Culley, A.I. Climate-Endangered Arctic Epishelf Lake Harbors Viral Assemblages with Distinct Genetic Repertoires. Appl. Environ. Microb. 2022, 88, e00228-22. [Google Scholar] [CrossRef]
- Moon, K.; Cho, J.C. Metaviromics coupled with phage-host identification to open the viral ‘black box’. J. Microbiol. 2021, 59, 311–323. [Google Scholar] [CrossRef]
- Yu, Z.C.; Chen, X.L.; Shen, Q.T.; Zhao, D.L.; Tang, B.L.; Su, H.N.; Wu, Z.Y.; Qin, Q.L.; Xie, B.B.; Zhang, X.Y.; et al. Filamentous phages prevalent in Pseudoalteromonas spp. confer properties advantageous to host survival in Arctic sea ice. ISME J. 2015, 9, 871–881. [Google Scholar] [CrossRef]
- Borriss, M.; Helmke, E.; Hanschke, R.; Schweder, T. Isolation and characterization of marine psychrophilic phage-host systems from Arctic sea ice. Extremophiles 2003, 7, 377–384. [Google Scholar] [CrossRef]
- Holovan, V.; Andriichuk, O.; Budzanivska, I.; Zelena, P.; Kondratiuk, T.; Shevchenko, O. Bacteriophages and their microbial hosts in terrestrial biotopes of Antarctica. Antarct. Sci. 2022, 34, 120–136. [Google Scholar] [CrossRef]
- Gong, Z.; Liang, Y.T.; Wang, M.; Jiang, Y.; Yang, Q.W.; Xia, J.; Zhou, X.H.; You, S.Y.; Gao, C.; Wang, J.; et al. Viral Diversity and Its Relationship With Environmental Factors at the Surface and Deep Sea of Prydz Bay, Antarctica. Front. Microbiol. 2018, 9, 2981. [Google Scholar] [CrossRef]
- Groth, A.C.; Calos, M.P. Phage integrases: Biology and applications. J. Mol. Biol. 2004, 335, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Fogg, P.C.M.; Colloms, S.; Rosser, S.; Stark, M.; Smith, M.C.M. New Applications for Phage Integrases. J. Mol. Biol. 2014, 426, 2703–2716. [Google Scholar] [CrossRef] [PubMed]
- Leverrier, P.; Declercq, J.P.; Denoncin, K.; Vertommen, D.; Hiniker, A.; Cho, S.H.; Collet, J.F. Crystal Structure of the Outer Membrane Protein RcsF, a New Substrate for the Periplasmic Protein-disulfide Isomerase DsbC. J. Biol. Chem. 2011, 286, 16734–16742. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome. Biol. 2001, 2, research0007.1. [Google Scholar] [CrossRef] [PubMed]
- Reidl, C.; Majorek, K.A.; Dang, J.; Tran, D.; Jew, K.; Law, M.; Payne, Y.; Minor, W.; Becker, D.P.; Kuhn, M.L. Generating enzyme and radical-mediated bisubstrates as tools for investigating Gcn5-related N-acetyltransferases. Febs. Lett. 2017, 591, 2348–2361. [Google Scholar] [CrossRef]
- Vetting, M.W.; de Carvalho, L.P.S.; Yu, M.; Hegde, S.S.; Magnet, S.; Roderick, S.L.; Blanchard, J.S. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 2005, 433, 212–226. [Google Scholar] [CrossRef]
- Wang, W.Y.; Li, Y.W.B.; Wang, Y.Q.; Shi, C.; Li, C.M.; Li, Q.; Linhardt, R.J. Bacteriophage T7 transcription system: An enabling tool in synthetic biology. Biotechnol. Adv. 2018, 36, 2129–2137. [Google Scholar] [CrossRef]
- Deutscher, M.P. 7 tRNA Nucleotidyltransferase. In The Enzymes; Boyer, P.D., Ed.; Academic Press: Cambridge, MA, USA, 1982; Volume 15, pp. 183–215. [Google Scholar]
- Xiong, Y.; Steitz, T.A. Mechanism of transfer RNA maturation by CCA-adding enzyme without using an oligonucleotide template. Nature 2004, 430, 640–645. [Google Scholar] [CrossRef]
- Zhu, B.; Lee, S.J.; Tan, M.; Wang, E.D.; Richardson, C.C. Gene 5.5 protein of bacteriophage T7 in complex with Escherichia coli nucleoid protein H-NS and transfer RNA masks transfer RNA priming in T7 DNA replication. Proc. Natl. Acad. Sci. USA 2012, 109, 8050–8055. [Google Scholar] [CrossRef]
- Saito, H.; Tabor, S.; Tamanoi, F.; Richardson, C.C. Replication of Bacteriophage-T7 Deoxyribonucleic-Acid.18. Nucleotide-Sequence of the Primary Origin of Bacteriophage-T7 DNA-Replication—Relationship to Adjacent Genes and Regulatory Elements. Porc. Natl. Acad. Sci. USA 1980, 77, 3917–3921. [Google Scholar] [CrossRef]
- Mikoulinskaia, G.V.; Gubanov, S.I.; Zimin, A.A.; Kolesnikov, I.V.; Feofanov, S.A.; Miroshnikov, A.I. Purification and characterization of the deoxynucleoside monophosphate kinase of bacteriophage T5. Protein. Expres. Purif. 2003, 27, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Hollis, T.; Stattel, J.M.; Walther, D.S.; Richardson, C.C.; Ellenberger, T. Structure of the gene 2.5 protein, a single-stranded DNA binding protein encoded by bacteriophage T7. Proc. Natl. Acad. Sci. USA 2001, 98, 9557–9562. [Google Scholar] [CrossRef] [PubMed]
- Hadden, J.M.; Declais, A.C.; Phillips, S.E.V.; Lilley, D.M.J. Metal ions bound at the active site of the junction-resolving enzyme T7 endonuclease I. Embo J. 2002, 21, 3505–3515. [Google Scholar] [CrossRef] [PubMed]
- Bae, B.; Davis, E.; Brown, D.; Campbell, E.A.; Wigneshweraraj, S.; Darst, S.A. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of sigma(70) domain 1.1. Proc. Natl. Acad. Sci. USA 2013, 110, 19772–19777. [Google Scholar] [CrossRef]
- Savalia, D.; Robins, W.; Nechaev, S.; Molineux, I.; Severinov, K. The Role of the T7 Gp2 Inhibitor of Host RNA Polymerase in Phage Development. J. Mol. Biol. 2010, 402, 118–126. [Google Scholar] [CrossRef]
- Ali, S.S.; Beckett, E.; Bae, S.J.; Navarre, W.W. The 5.5 Protein of Phage T7 Inhibits H-NS through Interactions with the Central Oligomerization Domain. J. Bacteriol. 2011, 193, 4881–4892. [Google Scholar] [CrossRef]
- Kemp, P.; Garcia, L.R.; Molineux, I.J. Changes in bacteriophage T7 virion structure at the initiation of infection. Virology 2005, 340, 307–317. [Google Scholar] [CrossRef]
- Cuervo, A.; Fabrega-Ferrer, M.; Machon, C.; Conesa, J.J.; Fernandez, F.J.; Perez-Luque, R.; Perez-Ruiz, M.; Pous, J.; Vega, M.C.; Carrascosa, J.L.; et al. Structures of T7 bacteriophage portal and tail suggest a viral DNA retention and ejection mechanism. Nat. Commun. 2019, 10, 3746. [Google Scholar] [CrossRef]
- Chen, W.Y.; Xiao, H.; Wang, L.; Wang, X.R.; Tan, Z.X.; Han, Z.; Li, X.W.; Yang, F.; Liu, Z.H.; Song, J.D.; et al. Structural changes in bacteriophage T7 upon receptor-induced genome ejection. Proc. Natl. Acad. Sci. USA 2021, 118, e2102003118. [Google Scholar] [CrossRef]
- Moak, M.; Molineux, I.J. Role of the Gp16 lytic transglycosylase motif in bacteriophage T7 virions at the initiation of infection. Mol. Microbiol. 2000, 37, 345–355. [Google Scholar] [CrossRef]
- Nobrega, F.L.; Vlot, M.; de Jonge, P.A.; Dreesens, L.L.; Beaumont, H.J.E.; Lavigne, R.; Dutilh, B.E.; Brouns, S.J.J. Targeting mechanisms of tailed bacteriophages. Nat. Rev. Microbiol. 2018, 16, 760–773. [Google Scholar] [CrossRef]
- Hamada, K.; Fujisawa, H.; Minagawa, T. Purification and Properties of Gene-18 Product of Bacteriophage-T3. Virology 1984, 139, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V.A. Development of Phage Lysins as Novel Therapeutics: A Historical Perspective. Viruses 2018, 10, 310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Young, R. Complementation and characterization of the nested Rz and Rz1 reading frames in the genome of bacteriophage lambda. Mol. Gen. Genet. 1999, 262, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Goker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic. Acids. Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Bayzid, M.S. Machine learning based imputation techniques for estimating phylogenetic trees from incomplete distance matrices. BMC Genom. 2020, 21, 497. [Google Scholar] [CrossRef]
- De Seixas, M.M.M.; de Araujo, J.; Krauss, S.; Fabrizio, T.; Walker, D.; Ometto, T.; Thomazelli, L.M.; Vanstreels, R.E.T.; Hurtado, R.F.; Kruger, L.; et al. H6N8 avian influenza virus in Antarctic seabirds demonstrates connectivity between South America and Antarctica. Transbound. Emerg. Dis. 2022, 69, E3436–E3446. [Google Scholar] [CrossRef]
- Prakash, E.A.; Hromadkova, T.; Jabir, T.; Vipindas, P.V.; Krishnan, K.P.; Hatha, A.A.M.; Briedis, M. Dissemination of multidrug resistant bacteria to the polar environment—Role of the longest migratory bird Arctic tern (Sterna paradisaea). Sci. Total Environ. 2022, 815, 152727. [Google Scholar] [CrossRef]
- Egevang, C.; Stenhouse, I.J.; Phillips, R.A.; Petersen, A.; Fox, J.W.; Silk, J.R.D. Tracking of Arctic terns Sterna paradisaea reveals longest animal migration. Proc. Natl. Acad. Sci. USA 2010, 107, 2078–2081. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Colombet, J.; Robin, A.; Lavie, L.; Bettarel, Y.; Cauchie, H.M.; Sime-Ngando, T. Virioplankton ‘pegylation’: Use of PEG (polyethylene glycol) to concentrate and purify viruses in pelagic ecosystems. J. Microbiol. Meth. 2007, 71, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Kwiatek, M.; Parasion, S.; Rutyna, P.; Mizak, L.; Gryko, R.; Niemcewicz, M.; Olender, A.; Lobocka, M. Isolation of bacteriophages and their application to control Pseudomonas aeruginosa in planktonic and biofilm models. Res. Microbiol. 2017, 168, 194–207. [Google Scholar] [CrossRef]
- Liu, Y.D.; Zheng, K.Y.; Liu, B.H.; Liang, Y.T.; You, S.Y.; Zhang, W.J.; Zhang, X.R.; Jie, Y.Q.; Shao, H.B.; Jiang, Y.; et al. Characterization and Genomic Analysis of Marinobacter Phage vB_MalS-PS3, Representing a New Lambda-Like Temperate Siphoviral Genus Infecting Algae-Associated Bacteria. Front. Microbiol. 2021, 12, 726074. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome. Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome analysis using the Kraken software suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zheng, K.Y.; Zou, X.; Liang, Y.T.; Liu, Y.D.; Li, X.; Shao, H.B.; Sung, Y.Y.; Mok, W.J.; Wong, L.L.; et al. Characterization and Genomic Analysis of the First Podophage Infecting Shewanella, Representing a Novel Viral Cluster. Front. Microbiol. 2022, 13, 853973. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic. Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic. Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Boulanger, P. Purification of bacteriophages and SDS-PAGE analysis of phage structural proteins from ghost particles. Methods Mol. Biol. 2009, 502, 227–238. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking Virus Genomes with Host Taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC-A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef]
- Jang, H.B.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Camargo, A.P.; Nayfach, S.; Chen, I.M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Ritter, S.J.; Reddy, T.B.K.; Mukherjee, S.; Schulz, F.; et al. IMG/VR v4: An expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata. Nucleic. Acids Res. 2022, 51, D733–D743. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic. Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Gao, L.; Dong, J.H.; Yang, X.F. Detecting Overlapping Protein Complexes by Rough-Fuzzy Clustering in Protein-Protein Interaction Networks. PLoS ONE 2014, 9, e91856. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome. Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Goker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Goker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Yu, G. Using ggtree to Visualize Data on Tree-Like Structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Micr. 2016, 66, 1100–1103. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Goker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORF | Size (aa) | Prediction Function | Conserved Domains | E-Values |
|---|---|---|---|---|
| 1 | 486 | Integrase | COG4973 | 1.40 × 10−26 |
| 2 | 131 | Outer membrane lipoprotein | 6T1W_C (PDB) | 1.20 × 10−17 |
| 6 | 238 | 2-oxoglutarate-dioxygenase | 6FXR_A (PDB) | 1.00 × 10−17 |
| 8 | 897 | RNA polymerase | 1MSW_D (PDB) | 3.30 × 10−157 |
| 9 | 47 | Bacteriophage 1.1 Protein | PF08200.14 (Pfam) | 1.70 × 10−9 |
| 11 | 331 | DNA ligase | 1A0I_A (PDB) | 9.30 × 10−36 |
| 16 | 222 | Deoxyribonucleotide kinase | 1DEK_A (PDB) | 7.40 × 10−17 |
| 17 | 63 | Bacterial RNA polymerase inhibitor | 4LLG_N (PDB) | 2.60 × 10−25 |
| 19 | 230 | Helix-destabilizing proteins | 1JE5_B (PDB) | 5.30 × 10−34 |
| 20 | 149 | Endodeoxyribonuclease I | 1M0D_B (PDB) | 2.90 × 10−28 |
| 21 | 148 | Lysozyme | 1LBA_A (PDB) | 1.90 × 10−13 |
| 22 | 184 | CCA tRNA nucleotidyltransferase | KOG2159 | 2.40 × 10−22 |
| 23 | 563 | DNA primase/helicase | 6N7I_C (PDB) | 1.20 × 10−64 |
| 26 | 715 | DNA polymerase | 1X9M_A (PDB) | 1.00 × 10−68 |
| 27 | 88 | Protein suppressor of silencing | PF11247.11 (Pfam) | 1.20 × 10−29 |
| 28 | 69 | Transcription effector | 5LGM_A (PDB) | 1.50 × 10−38 |
| 30 | 292 | 5′-3′ exonuclease | 6C33_A (PDB) | 3.80 × 10−28 |
| 32 | 90 | T7-like_gp67; | PF17570.5 (Pfam) | 6.00 × 10−23 |
| 33 | 89 | VirionAssem_T7 | PF11653.11 (Pfam) | 1.40 × 10−28 |
| 34 | 541 | Portal protein | 6R21_J (PDB) | 5.50 × 10−60 |
| 35 | 306 | capsid assembly protein | PF05396.14 (Pfam) | 9.00 × 10−28 |
| 36 | 341 | Major capsid protein | 3J7W_D (PDB) | 5.00 × 10−30 |
| 37 | 192 | Tail tubular protein gp11 | 6R21_X (PDB) | 2.00 × 10−41 |
| 38 | 794 | Tail tubular protein gp12 | 6R21_b (PDB) | 1.00 × 10−105 |
| 39 | 145 | Acetyltransferase | 3KKW_A (PDB) | 9.00 × 10−14 |
| 40 | 188 | Internal virion protein gp14 | 7EY7_A (PDB) | 8.50 × 10−31 |
| 41 | 734 | Internal virion protein gp15 | 7K5C_I (PDB) | 6.30 × 10−117 |
| 42 | 1349 | Internal virion protein gp16 | 7K5C_G (PDB) | 5.60 × 10−122 |
| 43 | 589 | Tail fiber protein | 7EY9_r (PDB) | 7.30 × 10−47 |
| 44 | 67 | Phage holin | PF10746.12 (Pfam) | 9.70 × 10−21 |
| 45 | 85 | DNA packaging protein | PF11123.11 (Pfam) | 3.50 × 10−22 |
| 46 | 150 | Phage lysis | PF03245.16 (Pfam) | 3.70 × 10−16 |
| 48 | 569 | Terminase large subunit | 8DGC_G (PDB) | 5.30 × 10−55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Jiang, W.; Kim, C.; Peng, X.; Fan, C.; Wu, Y.; Xie, Z.; Peng, F. A Pseudomonas Lysogenic Bacteriophage Crossing the Antarctic and Arctic, Representing a New Genus of Autographiviridae. Int. J. Mol. Sci. 2023, 24, 7662. https://doi.org/10.3390/ijms24087662
Liu Z, Jiang W, Kim C, Peng X, Fan C, Wu Y, Xie Z, Peng F. A Pseudomonas Lysogenic Bacteriophage Crossing the Antarctic and Arctic, Representing a New Genus of Autographiviridae. International Journal of Molecular Sciences. 2023; 24(8):7662. https://doi.org/10.3390/ijms24087662
Chicago/Turabian StyleLiu, Zhenyu, Wenhui Jiang, Cholsong Kim, Xiaoya Peng, Cong Fan, Yingliang Wu, Zhixiong Xie, and Fang Peng. 2023. "A Pseudomonas Lysogenic Bacteriophage Crossing the Antarctic and Arctic, Representing a New Genus of Autographiviridae" International Journal of Molecular Sciences 24, no. 8: 7662. https://doi.org/10.3390/ijms24087662