
Characterization of Intrinsic Radiation Sensitivity in a Diverse Panel of Normal, Cancerous and CRISPR-Modified Cell Lines

Abstract
1. Introduction
2. Results
2.1. X-Ray Radiosensitivity of CRISPR-Modified Cell Lines
2.2. Correlation between Cellular Features and Mean Inactivation Dose (MID) in CRISPR-Modified Cells
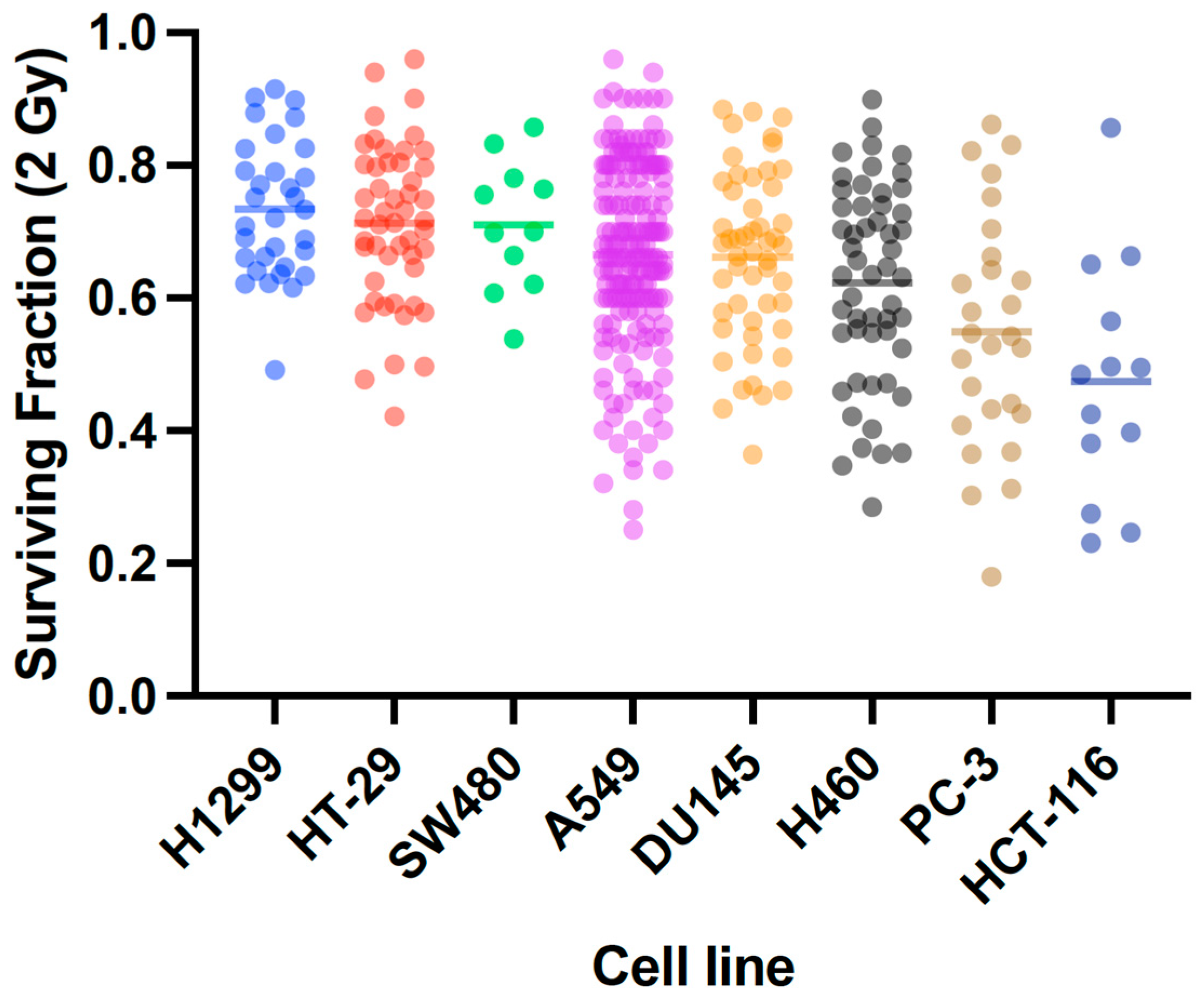
2.3. X-Ray Radiosensitivity of Cancer and Normal Human Cells
2.4. DNA DSB Repair Rates
2.5. Baseline Cell Cycle Profile and Ploidy
2.6. Correlation between Cellular Features and Mean Inactivation Dose (MID)
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Establishment of DNA Repair Deficient CRISPR/Cas9 Stable Cell Lines
4.3. Knock-Out Validation by Western Blot
4.4. Clonogenic Survival Assay
4.5. DNA Damage Immunofluorescence Assay
4.6. Cell Cycle Profile and Ploidy Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- West, C.M.L.; Davidson, S.E.; Roberts, S.A.; Hunter, R.D. The independence of intrinsic radiosensitivity as a prognostic factor for patient response to radiotherapy of carcinoma of the cervix. Br. J. Cancer 1997, 76, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- West, C.M.L. Intrinsic radiosensitivity as a predictor of patient response to radiotherapy. Br. J. Radiol. 2014, 68, 827–837. [Google Scholar] [CrossRef] [PubMed]
- West, C.M.L.; Davidson, S.E.; Roberts, S.A.; Hunter, R.D. Intrinsic radiosensitivity and prediction of patient response to radiotherapy for carcinoma of the cervix. Br. J. Cancer 1993, 68, 819–823. [Google Scholar] [CrossRef]
- Webb, S.; Nahum, A.E. A model for calculating tumour control probability in radiotherapy including the effects of inhomogeneous distributions of dose and clonogenic cell density. Phys. Med. Biol. 1993, 38, 653–666. [Google Scholar] [CrossRef]
- Nuryadi, E.; Permata, T.B.M.; Komatsu, S.; Oike, T.; Nakano, T. Inter-assay precision of clonogenic assays for radiosensitivity in cancer cell line A549. Oncotarget 2018, 9, 13706–13712. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Oike, T.; Komatsu, Y.; Kubota, Y.; Sakai, M.; Matsui, T.; Nuryadi, E.; Permata, T.B.M.; Sato, H.; Kawamura, H.; et al. Deep learning-assisted literature mining for in vitro radiosensitivity data. Radiother. Oncol. 2019, 139, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Banáth, J.P.; Klokov, D.; MacPhail, S.H.; Banuelos, C.A.; Olive, P.L. Residual γH2AX foci as an indication of lethal DNA lesions. BMC Cancer 2010, 10, 4. [Google Scholar] [CrossRef]
- Taneja, N.; Davis, M.; Choy, J.S.; Beckett, M.A.; Singh, R.; Kron, S.J.; Weichselbaum, R.R. Histone H2AX Phosphorylation as a Predictor of Radiosensitivity and Target for Radiotherapy. J. Biol. Chem. 2004, 279, 2273–2280. [Google Scholar] [CrossRef]
- Menegakis, A.; Yaromina, A.; Eicheler, W.; Drfler, A.; Beuthien-Baumann, B.; Thames, H.D.; Baumann, M.; Krause, M. Prediction of clonogenic cell survival curves based on the number of residual DNA double strand breaks measured by γh2AX staining. Int. J. Radiat. Biol. 2009, 85, 1032–1041. [Google Scholar] [CrossRef]
- Prise, K.M.; Davies, S.; Michael, B.D. The relationship between radiation-induced DNA double-strand breaks and cell kill in hamster V79 fibroblasts irradiated with 250 kVp X-rays, 2.3 MeV neutrons or 238Pu alpha-particles. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1987, 52, 893–902. [Google Scholar] [CrossRef]
- Dunne, A.L.; Price, M.E.; Mothersill, C.; McKeown, S.R.; Robson, T.; Hirst, D.G. Relationship between clonogenic radiosensitivity, radiation-induced apoptosis and DNA damage/repair in human colon cancer cells. Br. J. Cancer 2003, 89, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Mahrhofer, H.; Bürger, S.; Oppitz, U.; Flentje, M.; Djuzenova, C.S. Radiation induced DNA damage and damage repair in human tumor and fibroblast cell lines assessed by histone H2AX phosphorylation. Int. J. Radiat. Oncol. Biol. Phys. 2006, 64, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Bush, C.; McMillan, T.J. Micronucleus formation in human tumour cells: Lack of correlation with radiosensitivity. Br. J. Cancer 1993, 106, 102–106. [Google Scholar] [CrossRef]
- Heaven, C.J.; Wanstall, H.C.; Henthorn, N.T.; Warmenhoven, J.W.; Ingram, S.P.; Chadwick, A.L.; Santina, E.; Honeychurch, J.; Schmidt, C.K.; Kirkby, K.J.; et al. The suitability of micronuclei as markers of relative biological effect. Mutagenesis 2022, 37, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, M.; Cho, T.; Álvarez-Quilón, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020, 182, 481–496.e21. [Google Scholar] [CrossRef]
- Amundson, S.A.; Do, K.T.; Vinikoor, L.C.; Lee, R.A.; Koch-Paiz, C.A.; Ahn, J.; Reimers, M.; Chen, Y.; Scudiero, D.A.; Weinstein, J.N.; et al. Integrating Global Gene Expression and Radiation Survival Parameters across the 60 Cell Lines of the National Cancer Institute Anticancer Drug Screen. Cancer Res. 2008, 68, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Classen, S.; Elena, R.; Jungwirth, J.; Albers, N.; Hebestreit, L.; Zielinski, A.; Poole, L.; Groth, M.; Koch, P.; Liehr, T.; et al. Partial Reduction in BRCA1 Gene Dose Modulates DNA Replication Stress Level and Thereby Contributes to Sensitivity or Resistance. Int. J. Mol. Sci. 2022, 23, 13363. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- West, C.M.L.; Davidson, S.E.; Burt, P.A.; Hunter, R.D. The intrinsic radiosensitivity of cervical carcinoma: Correlations with clinical data. Int. J. Radiat. Oncol. Biol. Phys. 1995, 31, 841–846. [Google Scholar] [CrossRef]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef]
- Schwartz, J.L. The Radiosensitivity of the Chromosomes of the Cells of Human Squamous Cell Carcinoma Cell Lines. Radiat. Res. 1992, 129, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.S.; Iype, R.; Senra, J.; Taylor, J.; Armenoult, L.; Oguejiofor, K.; Li, Y.; Stratford, I.; Stern, P.L.; O’Connor, M.J.; et al. Investigation of radiosensitivity gene signatures in cancer cell lines. PLoS ONE 2014, 9, e86329. [Google Scholar] [CrossRef]
- McMahon, S.J.; Prise, K.M. A Mechanistic DNA Repair and Survival Model (Medras): Applications to Intrinsic Radiosensitivity, Relative Biological Effectiveness and Dose-Rate. Front. Oncol. 2021, 11, 689112. [Google Scholar] [CrossRef]
- Kim, M.K.; Kim, T.J.; Sung, C.O.; Choi, C.H.; Lee, J.W.; Kim, B.G.; Bae, D.S. High expression of mTOR is associated with radiation resistance in cervical cancer. J. Gynecol. Oncol. 2010, 21, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.S.; O’Prey, J.; Cardaci, S.; Barthet, V.J.A.; Sakamaki, J.I.; Beaumatin, F.; Roseweir, A.; Gay, D.M.; Mackay, G.; Malviya, G.; et al. Mannose impairs tumour growth and enhances chemotherapy. Nature 2018, 563, 719–723. [Google Scholar] [CrossRef]
- Zhang, R.; Yang, Y.; Dong, W.; Lin, M.; He, J.; Zhang, X.; Tian, T.; Yang, Y.; Chen, K.; Lei, Q.Y.; et al. D-mannose facilitates immunotherapy and radiotherapy of triple-negative breast cancer via degradation of PD-L1. Proc. Natl. Acad. Sci. USA 2022, 119, e2114851119. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Puck, T.; Marcus, P. Action of X-rays on Mammalian Cells. J. Exp. Med. 1956, 103, 653–666. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Ploidy | Cell-Cycle (%) | PE (%) | SF2 | MID | Baseline DSBs | Induced DSBs | Residual DSBs | ||
|---|---|---|---|---|---|---|---|---|---|---|
| G1 | S | G2/M | ||||||||
| RPE-1 | 1.94 | 58.0 | 13.3 | 28.7 | 32.5 | 0.39 | 1.95 | 0.9 | 26.2 | 2.4 |
| RPE-1 LIG4−/− | 1.94 | 60.5 | 17.5 | 22.0 | 21.4 | 0.05 | 0.65 | 1.7 | 30.5 | 15.3 |
| RPE-1 ATM−/− | 1.94 | 52.2 | 19.0 | 28.8 | 33.0 | 0.11 | 0.96 | 1.2 | 28.7 | 9.3 |
| RPE-1 PRKDC−/− | 1.94 | 62.0 | 15.3 | 22.7 | 27.4 | 0.26 | 1.45 | 2.5 | 30.3 | 7.6 |
| RPE-1 DCLRE1C−/− | 1.94 | 60.2 | 15.9 | 23.9 | 33.2 | 0.33 | 1.63 | 1.9 | 26.4 | 2.8 |
| RPE-1 TP53/BRCA1−/− | 1.94 | 54.6 | 18.4 | 27.0 | 26.1 | 0.31 | 1.67 | 4.4 | 27.4 | 7.6 |
| RPE-1 FANCD2−/− | 1.94 | 58.8 | 16.5 | 24.7 | 32.8 | 0.40 | 2.02 | 2.3 | 25.1 | 2.9 |
| RPE-1 BAX−/− | 1.94 | 60.7 | 15.8 | 23.5 | 30.7 | 0.41 | 2.05 | 0.9 | 26.3 | 1.9 |
| RPE-1 TP53−/− | 1.94 | 58.9 | 11.7 | 29.3 | 37.3 | 0.48 | 2.36 | 2.7 | 27.1 | 3.1 |
| Cell Line | Ploidy | Cell-Cycle (%) | PE (%) | SF2 | MID | Baseline DSBs | Induced DSBs | Residual DSBs | ||
|---|---|---|---|---|---|---|---|---|---|---|
| G1 | S | G2/M | ||||||||
| H441 | 2.33 | 57.1 | 9.1 | 33.8 | 10.0 | 0.27 | 1.54 | 6.3 | 30.1 | 2.8 |
| H23 | 3.05 | 59.0 | 11.1 | 29.9 | 26.1 | 0.53 | 2.44 | 2.1 | 25.5 | 0.7 |
| H1792 | 2.50 | 59.5 | 13.7 | 26.8 | 34.9 | 0.56 | 2.72 | 2.9 | 29.5 | 1.1 |
| H460 | 2.32 | 64.4 | 12.5 | 23.1 | 65.3 | 0.63 | 2.79 | 3.8 | 28.1 | 0.1 |
| SW1573 | 3.29 | 59.4 | 14.8 | 25.8 | 58.4 | 0.71 | 3.21 | 3.3 | 27.2 | 1.9 |
| A549 | 2.57 | 62.9 | 11.7 | 25.4 | 63.3 | 0.67 | 3.40 | 1.2 | 30.2 | 1.2 |
| C42B | 2.76 | 64.3 | 10.1 | 25.6 | 9.9 | 0.37 | 1.88 | 6.0 | 30.5 | 0.8 |
| PC-3 | 3.95 | 62.4 | 13.9 | 23.7 | 46.3 | 0.51 | 2.31 | 5.6 | 30.7 | 0.8 |
| 22RV1 | 2.12 | 48.5 | 13.5 | 38.0 | 45.1 | 0.59 | 2.65 | 2.5 | 21.2 | 0.5 |
| LNCAP | 3.79 | 66.3 | 10.5 | 23.2 | 4.3 | 0.67 | 2.93 | 5.8 | 23.1 | 1.9 |
| DU145 | 2.33 | 60.6 | 13.2 | 26.2 | 40.4 | 0.65 | 3.12 | 6.7 | 22.6 | 0.9 |
| MRC5 | 2.06 | 63.0 | 10.3 | 26.7 | 13.9 | 0.22 | 1.31 | 1.7 | 28.1 | 1.5 |
| AGO1522 | 2.03 | 65.6 | 9.4 | 25.0 | 27.5 | 0.37 | 1.80 | 2.3 | 23.8 | 1.6 |
| RPE-1 | 1.94 | 58.0 | 13.3 | 28.7 | 32.5 | 0.39 | 1.95 | 0.9 | 26.2 | 2.3 |
| MCF10A | 2.05 | 58.7 | 10.2 | 31.1 | 11.6 | 0.42 | 2.27 | 5.4 | 25.8 | 2.0 |
| PNT1A | 2.06 | 62.2 | 12.1 | 25.7 | 30.5 | 0.54 | 2.55 | 3.6 | 26.5 | 2.9 |
| RWPE1 | 2.20 | 69.2 | 11.7 | 19.1 | 20.6 | 0.56 | 2.57 | 5.0 | 23.4 | 0.6 |
| WPMY-1 | 2.98 | 49.2 | 15.1 | 35.7 | 20.8 | 0.69 | 3.22 | 6.3 | 29.7 | 2.0 |
| PNT2 | 2.18 | 47.7 | 12.4 | 39.9 | 44.4 | 0.65 | 3.43 | 1.5 | 27.0 | 1.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liberal, F.D.C.G.; McMahon, S.J. Characterization of Intrinsic Radiation Sensitivity in a Diverse Panel of Normal, Cancerous and CRISPR-Modified Cell Lines. Int. J. Mol. Sci. 2023, 24, 7861. https://doi.org/10.3390/ijms24097861
Liberal FDCG, McMahon SJ. Characterization of Intrinsic Radiation Sensitivity in a Diverse Panel of Normal, Cancerous and CRISPR-Modified Cell Lines. International Journal of Molecular Sciences. 2023; 24(9):7861. https://doi.org/10.3390/ijms24097861
Chicago/Turabian StyleLiberal, Francisco D. C. Guerra, and Stephen J. McMahon. 2023. "Characterization of Intrinsic Radiation Sensitivity in a Diverse Panel of Normal, Cancerous and CRISPR-Modified Cell Lines" International Journal of Molecular Sciences 24, no. 9: 7861. https://doi.org/10.3390/ijms24097861
APA StyleLiberal, F. D. C. G., & McMahon, S. J. (2023). Characterization of Intrinsic Radiation Sensitivity in a Diverse Panel of Normal, Cancerous and CRISPR-Modified Cell Lines. International Journal of Molecular Sciences, 24(9), 7861. https://doi.org/10.3390/ijms24097861